
Abstract
Dysregulation of the kynurenine pathway (KP) is implicated in many human diseases and disorders, from immunological, metabolic, neurodegenerative, and neuropsychiatric conditions to cancer, and represents an appealing target for new therapeutic approaches. In this intricate scenario, invertebrates, like Lymnaea stagnalis (LS), provide a flexible tool to unravel the complexity of the KP. Starting from the available LS genome and transcriptome, we identified putative transcripts of all KP enzymes containing an ORF; each predicted protein possessed a high degree of sequence conservation to known orthologues of other invertebrate and vertebrate model organisms. Sequences were confirmed by qualitative PCR and sequencing. At the same time, the qRT-PCR analysis revealed that Lym IDO-like, Lym TDO-like, Lym AFMID-like, Lym KMO-like, Lym AADAT-like, Lym KYAT I/III-like, Lym KYNU-like, Lym HAAO-like, and Lym ACMSD-like showed widespread tissue expression. Then, tryptophan, kynurenine, kynurenic acid, anthranilic acid, 3-hydroxy-kynurenine, xanthurenic acid, picolinic acid, and quinolinic acid were identified in the hemolymph of LS by UHPLC-Q exactive mass spectrometer. Our study provides the most thorough characterization to date of the KP in an invertebrate model, supporting the value of LS for future functional studies of this pathway at the cellular, synaptic, and behavioral levels.
Similar content being viewed by others


Introduction
The kynurenine pathway (KP) is crucial for peripheral and central catabolism of L-tryptophan (L-TRP), the essential amino acid precursor of serotonin and melatonin. The KP, a complex multi-step cascade with biologically active metabolites, catabolizes more than 95% of dietary tryptophan 1 (Fig. 1).

Predicted kynurenine pathway in Lymnaea stagnalis. IDO Indoleamine-2,3-dioxygenase, TDO Tryptophan-2,3-dioxygenase, AFMID Kynurenine formamidase, AADAT Aminoadipate aminotransferase, KYAT Kynurenine-oxoglutarate transaminase, KMO Kynurenine 3-monooxygenase, KYNU Kynureninase, HAAO 3-hydroxyanthranilate 3,4-dioxygenase, ACMSD 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase, NAD Nicotinamide-Adenine-Dinucleotide.
Physiologically, it leads to the production of the redox cofactor oxidized nicotinamide adenine dinucleotide (NAD+) and contributes to the establishment and maintenance of the immune privilege in several sites of the organism (including the brain, eye, and colon) 2. Moreover, many kynurenines have neuro-modulatory properties, regulating neuroplasticity and neurotoxicity 3, such as the N-methyl-d-aspartate (NMDA) receptor agonist quinolinic acid (QUINA), the NMDA antagonist kynurenic acid (KYNA), and the neuroactive 3-hydroxy-kynurenine (3HK) and the immunosuppressive 3-hydroxyanthranilic acid (3HANA) 2.
The kynurenine pathway starts with the oxidative cleavage of TRP by tryptophan 2,3-dioxygenase (TDO), which in humans and rodents is mainly expressed in the liver and brain, or by indoleamine-2,3-dioxygenase (IDO). Both these enzymes convert TRP to N-formylkynurenine 4,5, which is then metabolized by the enzyme kynurenine formamidase (AFMID) into the pivotal KP intermediate L-kynurenine (KYN). KYN in turn can be metabolized by at least three different enzymes: i) kynurenine 3-monooxygenase (KMO), forming 3-hydroxy-kynurenine; ii) four kynurenine aminotransferase (KYAT I-IV) isoenzymes that catalyze the transamination of KYN into KYNA; and iii) kynureninase (KYNU), developing anthranilic acid (ANA) 1 (Fig. 1).
Under normal conditions, the KMO branch seems to be the major metabolic route in the KP since this enzyme has the highest affinity for KYN, while metabolism via the KYNU branch only occurs when KYN concentrations are elevated 2,3,4. Continuing along the KMO-branch, 3-hydroxy-kynurenine is metabolized by KYNU into 3HANA, which has a 20-fold higher affinity for 3HK than for KYN, favoring the generation of 3HANA over ANA. 3-hydroxyanthranilate 3,4-dioxygenase (HAAO) then catalyzes the conversion of 3HANA to an unstable intermediate, 2-amino-3-carboxymuconic semialdehyde, which is then preferentially converted into QUINA by non-enzymatic cyclization 6. The balance between the excitotoxin, quinolinic acid, and neuroprotective picolinic acid (PICA) is maintained by the enzyme 2-amino 3-carboxymuconate 6-semialdehyde decarboxylase (ACMSD). QUINA is the endogenous source of nicotinamide and NAD+ 3.
The relative balance of the KP metabolites generated in peripheral tissues and the central nervous system (CNS) appears to be regulated at different interdependent levels 2. Moreover, several lines of evidence propose an essential role of this pathway in physiological and pathological conditions. Alterations and imbalances in the rheostat of neuroactive KP metabolites have been implied in a wide range of diseases and disorders, including inflammation/immune disorders, endocrine/metabolic conditions, Alzheimer’s disease, amyotrophic lateral sclerosis, Huntington’s disease, cancer, depression, and schizophrenia 7,8,9,10.
Thus, the KP has become an appealing target for developing new therapeutic approaches. Unfortunately, up to date, pharmacological inhibitors of different KP enzymes have led to mixed results in both preclinical and clinical studies 11.
In this complex scenario, invertebrates represent a flexible tool to study fundamental and conserved mechanisms of CNS physiology and pathology, such as the kynurenine pathway 12,13. The kynurenine metabolism is highly conserved throughout the eukaryotic lineage from yeast to humans 1. Until now, the most common organisms used to investigate the KP during immune activation and different human conditions have been primates and rodents.
Research using invertebrate models has been associated with significant experimental efficiency due to the reduced time needed for experiments and the low costs required for animal care thanks to their short generation times, numerous offspring, and the fact that they can be more easily manipulated experimentally. While many studies and breakthroughs in our understanding of neurodegenerative diseases have used D. melanogaster and C. elegans, their role in the study of the KP pathway has not been established yet. In fact, while TDO has been identified in both C. elegans and D. melanogaster, IDO has not been detected in either species 5. Furthermore, no IDO genes have been found in Arthropods yet, whereas some species of Nematodes (e.g., Brugia malayi and Necator americanus) have proven to carry the IDO gene, no corresponding gene was found in C. elegans 5,14,15,16.
Although invertebrates can never replace mammal models in preclinical studies, a full characterization of the KP in one of these models may be beneficial to unraveling its complexity while translating results to mammals. An interesting candidate model system is the gastropod mollusk Lymnaea stagnalis (L. stagnalis, Linnaeus, 1758), which has a relatively long average life span and, like other gastropods such as Aplysia californica, Biomphalaria glabrata, and Pomacea canaliculata 17, has proven to be a valuable tool in gaining a deeper understanding of the functioning of the nervous system. L. stagnalis belongs to the phylum Mollusca class Gastropoda and has been widely used to investigate genetic and epigenetic processes 18,19,20, aging 21,22, learning and memory 23,24,25, and for ecotoxicological studies 26,27. The CNS of L. stagnalis consists of approximately 20,000 readily identifiable neurons, organized in a ring of interconnected ganglia, offering a relatively large amount of biological material that could be molecularly, physiologically, and morphologically analyzed 13.
Considering the importance of the kynurenine pathway in different fields from neuroscience to immunology and pharmacology and the different fields in which our versatile model has been and could be adopted, in this study we aimed to identify and characterize the kynurenine pathway in L. stagnalis. Starting from the L. stagnalis genome and transcriptional profiles, we first identified and annotated putative transcripts coding for the KP enzymes in the CNS of L. stagnalis. Then, we identified the KP metabolites in the hemolymph of L. stagnalis using a UHPLC-Q exactive mass spectrometer, one of the most reliable techniques for metabolite identification and characterization.
Overall, our results indicate that L. stagnalis may represent an appropriate model organism to study the involvement of the kynurenine pathway in neuropsychiatric disorders and immune diseases and further corroborate its use as a model for translational neuroscience.
Results
Identification and characterization of putative transcripts of the kynurenine pathway in L. stagnalis
To identify putative KP genes in L. stagnalis, we first aligned with BLASTx all 328,378 contigs of L. stagnalis genome to 36,675 protein sequences annotated with RefSeq ID of B. glabrata, i.e., one of the L. stagnalis phylogenetically closest organisms for which annotated proteins are available, and to 76,216 and 110,386 annotated protein sequences of M. musculus and H. sapiens, respectively. At an E-value cutoff of 1E-6, BLASTx returned 81,845 contigs having a hit in B. glabrata, 30,478 contigs having a hit in M. musculus, and 30,001 contigs having a hit in H. sapiens. Using the RefSeq ID of protein sequences, we annotated contigs with at least one hit with the B. glabrata, mouse, and human gene symbols and with Gene Ontology (GO) and KEGG pathway terms (Supplementary Table 1). We quantified contigs’ gene expression levels using publicly available raw RNA-sequencing (RNA-seq) data from L. stagnalis central nervous system 18. We assembled the transcriptome with Cufflinks aligning the RNA-seq reads to the L. stagnalis genome with TopHat. Before assembly, we trimmed the original 81,851,004 reads and filtered them for quality obtaining 78,245,030 reads, 64.1% of which (50,124,678 reads) were mapped to contigs by TopHat. The gene expression level of each contig was then quantified, converting into TPM the counts of 44,576,148 uniquely mapping reads (see Methods for details; Supplementary Table 1).
Using the contig annotation table, we identified 20 L. stagnalis contigs annotated to the nine genes of the KP pathway in B. glabrata, with the IDO-like orthologue and the two putative isoforms of aminotransferases named as myoglobin-like, kynurenine-oxoglutarate transaminase 1/3-like, and 2-aminoadipate transaminase-like, respectively (Supplementary Table 2). To identify transcripts in these contigs, we first manually extracted the putative exonic sequences visualizing the RNA-seq reads with the Integrative Genomics Viewer (IGV). Then, we retrieved the precise mRNA sequence aligning these putative exonic sequences to the transcriptome shotgun assembly of L. stagnalis. This procedure allowed the identification of one specific transcript per each KP enzyme, except for tryptophan 2,3-dioxygenase-like, which was associated with three small transcripts (FX225637.1, FX224560.1, and FX206683.1; Supplementary Table 2). All transcripts resulted to be expressed (TPM > 0) in the publicly available L. stagnalis CNS transcriptome. To confirm whether these transcripts were expressed in the L. stagnalis CNS, we performed qPCR on each transcript using the set of primers listed in Supplementary Table 3 and displayed in Supplementary Fig. 1. The PCR products were purified, sequenced, and aligned to FX190660.1 (Lym IDO-like), FX191423.1 (Lym AFMID-like), FX185910.1 (Lym KMO-like), FX183988.1 (Lym AADAT-like), FX191915.1 (Lym KYAT-like), FX188572.1 (Lym KYNU-like), and FX187039.1 (Lym ACMSD-like). All sequences, except for Lym TDO-like and Lym HAAO-like, uniquely matched their respective templates. For Lym TDO-like, we used a combination of primers on FX225637.1, FX224560.1, and FX206683.1 and amplified and sequenced a single PCR product resulting from the variety of the three transcripts with a 15 bp gap (TATTCCTCTATAAGG) between the first two (Supplementary Fig. 1). Moreover, the amplified sequence of Lym HAAO-like missed an A in position 422 and presented a T to C mutation in position 447 (Supplementary Fig. 1). All transcripts contained at least one ORF (Fig. 2), whose identity was further confirmed by aligning its sequence with the amino acid sequences of the corresponding enzyme from H. sapiens, M. musculus, R. norvegicus, A. californica, B. glabrata, P. canaliculata, and C. elegans 28 (Table 1). In C. elegans, we identified six orthologs that had an average homology of about 48% with the corresponding sequence of L. stagnalis. A similar degree of homology was found with mammalian sequences, while we observed a higher degree of homology with mollusk sequences (i.e., 63% for P. canaliculata, 64% for A. californica, and 70% for B. glabrata). AADAT and ACMSD were the less (30% average homology) and the most (average homology of 71%) conserved genes, respectively (Table 1). This conservation pattern was also confirmed using multiple sequence alignment between each putative enzyme identified in L. stagnalis and orthologues from other species (Fig. 3). Phylogenetic analyses revealed a predictable pattern in the relatedness of L. stagnalis sequences to those of other Mollusca (Fig. 4). Finally, the PFAM analysis confirmed that all the KP-predicted enzymes had the expected domains (Supplementary Fig. S2).

Nucleotide and deduced amino acid sequence of the putative enzymes of the KP pathway. (A) Lym IDO-like, (B) Lym TDO-like, (C) Lym AFMID-like, (D) Lym KMO-like, (E) Lym AADAT-like, (F) Lym KYAT I/III-like, (G) Lym KYNU-like, (H) Lym HAAO-like, (I) Lym ACMSD-like. Differences between the amplified and the deposited sequences are bold and underlined.

Multiple sequence alignments of each putative KP enzyme in L. stagnalis with the amino acid sequences of the corresponding enzyme from H. sapiens, M. musculus, R. norvegicus, A. californica, B. glabrata, P. canaliculata, and C. elegans. (A) Lym IDO-like, (B) Lym TDO-like, (C) Lym AFMID-like, (D) Lym KMO-like, (E) Lym AADAT-like, (F) Lym KYAT I/III-like, (G) Lym KYNU-like, (H) Lym HAAO-like, (I) Lym ACMSD-like. Box shade alignment displays show conserved residues (identical black, similar grey).

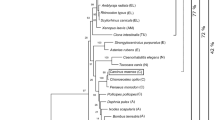
Molecular phylogenetic tree of the putative KP enzymes in L. stagnalis. (A) Lym IDO-like, (B) Lym TDO-like, (C) Lym AFMID-like, (D) Lym KMO-like, (E) Lym AADAT-like, (F) Lym KYAT I/III-like, (G) Lym KYNU-like, (H) Lym HAAO-like, (I) Lym ACMSD-like. The tree is drawn to scale, with branch lengths in the same units as the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Poisson correction method and are in the units of the number of amino acid substitutions per site. This analysis involved 7–8 amino acid sequences. Evolutionary analyses were conducted in MEGA X. The GenBank accession numbers of proteins used are indicated and listed in Table 1.
Primer specificity, efficiency, and validation
qRT-PCR was used to evaluate the expression levels of the identified L. stagnalis KP-enzymes. All gene-specific primers for the analysis are shown in Supplementary Table S3. All primers produced consistent results without amplifying off-target products or generating primer dimers. Following amplification, each primer pair produced amplicons that yielded single bands at the correct size after electrophoresis in 2% agarose gels (Supplementary Fig. S3). Additionally, no amplification was observed in cDNA from another pulmonated gastropod P. canaliculata or controls that lacked reverse transcriptase in the RT-PCR. Primer specificity was also checked by melt curve analysis. A single sharp peak with no primer-dimer was observed for all used primer pairs (Supplementary Fig. S4).
Results showed that the PCR efficiency was between 94.82 and 125.30%, and the R2 of primers was greater than 0.981. In addition, the mean Cq of all KP-enzymes ranged from 25.41 to 30.02 for 20 ng of cDNA with moderately abundant mRNA levels in the ganglia of L. stagnalis (Supplementary Table S3).
Comparison of expression levels of KP-like enzymes among several tissues of L. stagnalis
We next evaluated the levels of L. stagnalis KP transcripts in the CNS and other tissues of the snail, such as the gut (i.e., digestive gland), stomach, muscle (foot), penis, and hemocytes (n = 4 pools of 4 snails per each tissue). All KP-enzymes were ubiquitously transcribed in the L. stagnalis tissues with different relative abundance patterns (Fig. 5). Specifically, we found that Lym TDO-like was expressed at higher levels in the gut with respect to all the other tissues [F(5; 23) = 175.671; p < 0.0001]. Tukey’s post hoc test also revealed that the levels of Lym TDO-like mRNA in the muscle were higher than in SNC, stomach, and hemocytes. In contrast, Lym IDO-like was not differentially expressed in the considered tissues [F(5; 23) = 2.510; p = 0.068 with one-way ANOVA]. Lym AFMID-like mRNA levels in the gut and penis were significantly higher than in the stomach and hemocytes. No other difference was detected in any other tissue [F(5; 23) = 6.063; p = 0.002). The expression levels of Lym KMO-like in the CNS were significantly higher than those in the penis, gut, muscle, and hemocytes [F(5;23) = 29.639; p < 0.001]. Moreover, post-hoc analyses showed that the expression of this gene was significantly higher in the muscle than in all other tissues, while mRNA levels in the stomach were significantly higher than those in the hemocytes. The expression of Lym HAAO-like and Lym KYAT-like was significantly higher in the gut with respect to all the other tissues [F(5;23) = 66.715; p < 0.0001 and F(5;23) = 91.652; p < 0.0001, respectively]. We also found that the expression levels of Lym AADAT-like were significantly higher in the gut than in all other tissues considered [F(5;23) = 70.261; p < 0.0001]. Post-hoc analyses revealed that the mRNA levels of this gene in the hemocytes were significantly lower with respect to all the other areas, and its expression in the penis was significantly lower than in the muscle. The mRNA levels of Lym KYNU-like in the CNS were significantly lower compared to those in the penis, gut, and hemocytes [F(5;23) = 6.923; p < 0.001], and its expression in the stomach was significantly lower than that in the penis. Finally, we found that the mRNA levels of Lym ACMSD-like were significantly higher in the muscle with respect to all the other tissues [F(5;23) = 70.850; p < 0.0001]. Post-hoc tests also revealed that the expression levels of this gene in the hemocytes were significantly lower than that in the penis, gut, stomach, and muscle.

Comparison of mRNA expression levels of putative KP enzymes in different tissues of L. stagnalis. (A) Lym IDO-like, (B) Lym TDO-like, (C) Lym AFMID-like, (D) Lym KMO-like, (E) Lym AADAT-like, (F) Lym KYAT I/III-like, (G) Lym KYNU-like, (H) Lym HAAO-like, (I) Lym ACMSD-like mRNA expression levels were analyzed in snail CNS (white bars), gut (yellow bars), stomach (light grey bars), foot (i.e., muscle—dark grey bars), penis (black bars), and hemocytes (light-blue bars). Relative mRNA expression levels were determined by qRT-PCR (2−ΔΔCt), with the geometric mean of the reference genes (Lym TUB and Lym EF1a) as endogenous control and CNS as calibrator. N = 4 pools of 4 snails for each tissue. Data are expressed as means ± S.E.M and were analyzed with one-way ANOVA followed by Tukey's post hoc test: ***p < 0.001, **p < 0.01, *p < 0.05 versus gut; °°°p < 0.001, °°p < 0.01, °p < 0.05 versus muscle; ###p < 0.001, ##p < 0.01, #p < 0.05 versus penis; §§§p < 0.001, §§p < 0.01, §p < 0.05 versus hemocytes.
Identification of KP metabolites in the hemolymph.
As the KP transcripts were successfully identified in all the tissues evaluated, we aimed to detect KP metabolites in LS hemolymph utilizing UHPLC-MS. Each target metabolite was revealed as a chromatographic peak, eluting at its specific retention time, using mass range chromatograms with theoretical m/z ± five ppm values of putative [M + H]+ molecular ions. For other identity confirmation, peak areas were used to compare each potential metabolite in hemolymph with those observed in hemolymph spiked with a mixture of standards (1 μM final concentration) (Fig. 6). Subsequently, the MS2 fragmentation behavior was compared with that of the standard mix using the Q-Exactive PRM mode (Fig. 7). TRP, KYN, KYNA, ANA, 3HK, XANA, PICA, and QUINA were accurately defined by comparing retention time, accurate mass, and MS2 data. With the sole exception of 3HANA (not shown), for each compound, we observed a single peak in hemolymph that was increased in the spiked sample (Fig. 6).

Extracted ion chromatogram of Tryptophan (Trp), kynurenine (KYN), kynurenic acid (KYNA), anthranilic acid (ANA), 3-hydroxy-kynurenine (3HK), xanthurenic acid (XANA), picolinic acid (PICA), and quinolinic acid (QUINA) in hemolymph (red) or hemolymph spiked with a mixture of internal standard (black). RT: retention time.

M2 mass spectra in positive ion mode for Tryptophan (Trp), kynurenine (KYN), kynurenic acid (KYNA), anthranilic acid (ANA), 3-hydroxy-kynurenine (3HK), xanthurenic acid (XANA), picolinic acid (PICA), and quinolinic acid (QUINA) in hemolymph compared with the corresponding M2 spectra of the standard mix (0.01 μM).
The maximum mass errors between the theoretical and measured ones were between − 0.67/1.18 ppm (Supplementary Table S4).
In hemolymph, TRP eluted at the retention time of 11.42 min and showed characteristic product ions at m/z 188.0705, 146.0599, and 118.0653 matching the pattern of the reference standard.
KYN eluted in hemolymph at 8.05 min showed a protonated molecular ion at 209.0294 and product ions at 94.0656, 192.0656, 146.0600, 136.0756, like those of the standard mix.
Kynurenic acid had a retention time in the hemolymph of 12.64 min and yielded an m/z of 190.0499 as [M + H]+ ion and other fragmented ions at 162.0549 and 116.0498 similar to the pattern of the reference standard.
The fragmentation of ANA in hemolymph, eluted at 10.37 min, with ions at 120.0446 and 92.0500, matched the corresponding standard. In hemolymph, 3HK eluted at the retention time of 5.31 min and showed characteristic product ions at m/z 110.0603, 208.0603, 136.0392, and 162.0549 matching the pattern of the reference standard.
XANA eluted in hemolymph at 14.14 min showed a protonated molecular ion at 206.0447 and product ions at 178.0498 and 132.0444 like those of the standard mix.
In our samples, PICA and QUINA were eluted at 2.65 and 2.21 min, respectively, and both showed a fragmented pattern like that of their corresponding standard. PICA exhibited a m/z of 124.0395 as [M + H]+ ion and other fragmented ions at 96.0448 and 80.0501, while the fragmentation of QUINA provided several fragments at 150.0185, 124.0394, and 96.0449.
Discussion
In this study, we identified transcripts of putative enzymes and metabolites of the KP in the pond snail L. stagnalis (LS), a promising candidate for translational neuroscience research 20.
The KP possesses a high level of conservation between vertebrates and invertebrates, is pharmacologically inducible, and is especially relevant to neuroscience 4,29. Under physiological conditions, the KP is the primary route for tryptophan catabolism and the starting point for synthesizing the ubiquitous co-enzyme nicotinamide adenine dinucleotide, which, in turn, fulfills the cellular energy requirements 10. As the KP plays a crucial role in immune function and energy metabolism, aberrations of its molecular cascade have been identified in many human diseases and disorders, including inflammation/immune disorders, endocrine/metabolic conditions, Alzheimer's disease, amyotrophic lateral sclerosis, Huntington's disease, cancer, depression, and schizophrenia 10,30,31,32.
The KP has been studied only in two invertebrate models: the worm C. elegans and the fly D. melanogaster. In C. elegans, alterations in the KP cause neurodegeneration and aging 33, whereas in D. melanogaster a correlation between elevated levels of 3-hydroxy-kynurenine and synaptic plasticity has been reported 34. To our knowledge, apart from these two studies, this is the first complete characterization of the KP in a molluscan model.
In the last few years, an increasing effort has been made to produce comprehensive L. stagnalis transcriptomic and proteomic databases 21,35,36,37,38. Still, along with the identification of conserved genes and transcripts, the lack of their functional and pharmacological characterization has limited molecular studies in this model to date.
Given this, instead of generating a new transcriptome, here we compared one of the latest assemblies of L. stagnalis genome with annotated proteins of H. sapiens, M. musculus, and B. glabrata; contigs were also combined with transcripts from the Sadamoto et al., (2012) dataset to provide an estimation of transcript abundance 18. Thanks to this approach, we identified contigs of L. stagnalis matching with the genes coding for the enzymes of the KP in the annotated genome of B. glabrata. All the enzymes were identified univocally, in B. glabrata the IDO-like orthologue is named myoglobin-like since it is possible that in molluscs it functions in their buccal mass as myoglobin, not as an IDO 39, and two putative isoforms of kynurenine aminotransferases, KYAT I-III and AADAT (KYAT II), were included in the analysis. In fact, human and rodent genomes encode four aminotransferases (KYAT I-IV) 40, KYAT I and KYAT III share the highest sequence identity and are phylogenetically distant from AADAT 41.
These contigs then matched univocally (except TDO) with putative transcripts in L. stagnalis CNS. All putative transcripts identified contained an ORF; the predicted protein had several amino acids very close to those of the human protein and the corresponding orthologue of several organisms extensively employed as a standard models in preclinical studies. Consistent with previous studies, our phylogenetic analyses revealed a high degree of sequence conservation between L. stagnalis and other gastropods and model organisms 1,29.
The transcripts predicted in silico by the computational analysis were validated using Sanger sequencing and the expression patterns of the KP enzymes were quantified in different tissues of L. stagnalis. Studies from mammals and humans demonstrated that the KP metabolites act as versatile mediators in inter-cell or inter-organ cross-talk, as they can be exchanged among tissues and be either metabolized or exert a biological effect 1. In particular, considerable evidence demonstrates a strong link between the KP, proinflammatory cytokines, and the nervous and immune systems 30. Moreover, some KP metabolites can be further processed, leading to the production of NAD+ 32, which plays a crucial role in energy production and is extremely important in organs like the brain and muscles 42,43. Furthermore, studies from rodents and humans, underlined the importance of the tryptophan metabolism through the KP in the gut-brain axis communication and the link between depression and bowel inflammation 44. Finally, the KP controls the immune and inflammatory environment of the epididymis in mice 45,46.
Given the importance of the KP in these body areas, in this study, we compared the mRNA expression levels in the L. stagnalis CNS, gut (i.e., digestive gland), stomach, muscle (i.e., foot), penis, and hemocytes (i.e., invertebrate immune system cell). To our knowledge, we provide the first evidence that the enzymes of the KP are widely expressed in the whole body of L. stagnalis, suggesting that the KP is active in various tissues in this organism as it does in mammals 47. Specifically, we found that Lym TDO-like is expressed mainly in the gut and muscle, this is consistent with data from vertebrates showing that the TDO enzyme is particularly active in hepatocytes, which are part of the alimentary system and the gut 47,48.
IDO has been identified in vertebrates, lower vertebrates, several invertebrates, fungi, and bacteria. In vertebrates were identified two distinct IDO paralogues, IDO1 and IDO2, originated from a gene duplication event that possibly occurred before the divergence of vertebrates. Many of the isoforms of IDO enzyme identified in invertebrates 5,49 show low catalytic efficiency/affinity for TRP, similar to vertebrate IDO2s 49. Here we identify a single IDO transcript ubiquitously distributed in all tissues, these data are consistent with those from mammals, where IDO1 and IDO2 are widespread throughout the body and are expressed in almost every type of cell.
Furthermore, we found that Lym HAAO-like, Lym AADAT-like, and Lym KYAT-like are also mainly expressed in the gut. In rodents, KYAT-I and KYAT-III share similar expression patterns: their mRNA levels are much higher in the liver and neuroendocrine tissues than in the CNS 40,50. This trend is reported also in LS for Lym KYAT-like. AADAT expression is higher in the rodent brain where it is mostly located in astrocytes, while in the periphery it is detected as well in most tissues with the sole exception of the skeletal muscle. In L. stagnalis we demonstrated that its expression levels in the foot muscle are like those observed in the CNS.
Our results support a strong connection between the KP and the digestive gland of L. stagnalis. In mollusks this organ known also as hepatopancreas is involved, among others, in the secretion of digestive enzymes and food and nutrients absorption. From a translational point of view, this is in line with the established role proposed for KP in regulating intestinal (patho) physiology in humans and rodents. Alterations of the TRP metabolism can give rise to gastrointestinal dysfunction 51 and in turn the gut microbiota can influence the KP.
Consequently, changes in gut biodiversity may cause increased gut permeability and systemic and CNS inflammation 52. Thus, future studies may investigate whether similar results can be reproduced in L. stagnalis by changing the composition of the snails’ microbiota to further understand the complex communication between GI microbes and tryptophan catabolism and identify possible targets for the treatment of various human diseases.
To complete the biochemical characterization of the KP, we also performed UHPLC-Q Exactive Mass Spectrometry to identify KP metabolites in hemolymph and hemocytes of L. stagnalis. This analysis led to the identification of tryptophan, kynurenine, kynurenic acid, anthranilic acid, 3-hydroxy-kynurenine, xanthurenic acid, picolinic acid, and quinolinic acid.
In this context, studying the molecular and behavioral effects induced by the manipulation of the KP in L. stagnalis represents an appealing strategy both to assist the study of current unsolved questions and to drive the field forward. This model not only can help discover additional physiological and pathological roles for kynurenines but also may contribute to the development of new therapies based on these roles.
Finally, our results not only constitute new evidence on the potentialities of the pond snail L. stagnalis as a powerful model for neuroscience but also open numerous scenarios in the use of this model in translational neuroscience and the efficient study and characterization of other molecular pathways.
Methods
Identification of putative KP genes in L. stagnalis genome
To identify putative KP genes in L. stagnalis, we first downloaded its genome assembly in contigs from the NCBI Genome database (https://www.ncbi.nlm.nih.gov/genome/?term=Lymnea+stagnalis). Then, we used the program BLASTx of the NCBI BLAST + suite (release 2.2.28) to align all contigs to the protein sequences annotated with RefSeq IDs of i) B. glabrata (36,675 sequences), ii) M. musculus (76,216 protein sequences) and iii) H. sapiens (110,386 sequences). All protein sequences have been downloaded from https://ftp.ncbi.nih.gov/genomes/refseq/, and BLASTx results have been filtered, setting an e-value cutoff of 1E-6. Finally, starting from the RefSeq ID of each aligned protein sequence, we used the queryMany function of the mygene R package (https://www.bioconductor.org/packages/release/bioc/html/mygene.html) to annotate contigs with B. glabrata, mouse, and human gene symbols and with Gene Ontology (GO) and KEGG pathway terms.
To determine whether homologs of KP genes are indeed expressed transcripts in L. stagnalis, we downloaded the raw RNA-sequencing (RNA-seq) data of the L. stagnalis central nervous system from the NCBI SRA database (https://www.ncbi.nlm.nih.gov/sra/DRX001464) 18. We then assembled L. stagnalis transcriptome with Cufflinks (2.0.2; http://cole-trapnell-lab.github.io/cufflinks/), aligning RNA-seq reads to its genome with TopHat (2.1.0; https://ccb.jhu.edu/software/tophat/index.shtml). Before assembly, all reads have been trimmed and filtered for quality using Trimmomatic (http://www.usadellab.org/cms/?page=trimmomatic). Specifically, we trimmed the read tails when the read quality was lower than 20 and removed nine nucleotides from the start of each read (HEADCROP: 9 in Trimmomatic). The gene expression level of each contig was then quantified by converting the counts of uniquely mapping reads into TPM (Transcripts Per Million), as follows:
where Xiand li are the read count and the length of contig i, respectively.
To identify the exact sequence of transcripts annotated to the kynurenine pathway, we manually extracted putative exonic sequences visualizing RNA-seq reads on the L. stagnalis genome with Integrative Genomics Viewer (IGV, http://www.broadinstitute.org/igv/). Then, we aligned these sequences to the transcriptome shotgun assembly of L. stagnalis using BLASTn to retrieve the precise mRNA sequence.
Sequence analysis
We predicted the amino acid sequences using the Open Reading Frame (ORF) Finder tool of NCBI (https://www.ncbi.nlm.nih.gov/orffinder/). Alignments of nucleotide and translated protein sequences were performed with http://nadv.herokuapp.com/. ORF sequences were fed into the protein family database PFAM (https://pfam.xfam.org/) to identify the major conserved characteristic domains belonging to each previously organized sequence group 53.
Phylogenetic analysis
To determine sequence evolutionary organization and distribution, as well as to provide further evidence of the presence of putative proteins in L. stagnalis, we performed a phylogenetic analysis using the UPGMA method 54 of Molecular Evolutionary Genetic Analysis (MEGA)-X tool (https://www.megasoftware.net/). Specifically, we used the amino acid sequences of deuterostomes and protostomes organisms such as H. sapiens, M. musculus, R. norvegicus, C. elegans, and mollusks such as A. californica and B. glabrata, obtained from NCBI. The evolutionary distances were computed using the Poisson correction method and are in the units of the number of amino acid substitutions per site. The predicted amino acid sequences of L. stagnalis KP enzymes were aligned with those of H. sapiens, M. musculus, R. norvegicus, C. elegans, A. californica, and B. glabrata molluscan using T-Coffee tool (http://tcoffee.crg.cat/apps/tcoffee/do:regular) 55 and visualized with BoxShade (v3.21; https://bio.tools/BoxShade).
Ethics statement
Pond snails L. stagnalis are abundant on earth northern hemisphere and are not endangered or a protected species. Experiments on pond snails are not subject to the approval of our ethics committee. Nonetheless, every effort was made to maximize the well-being of the snails during the behavioral procedures.
Snails and colony maintenance
Laboratory-reared L. stagnalis, originally derived from a stock generously donated by the Vrije University in Amsterdam (The Netherlands), were used in this study. Animals were maintained in aquaria at the University of Modena, and Reggio Emilia (Italy) at 21–23 °C in well-aerated dechlorinated tap water on a 12/12 h light/dark cycle (lights on at 08:00 a.m.). Six-month-old snails having shell lengths of 20–25 mm were used in these experiments. Animals were fed pesticide-free lettuce twice a week.
Hemolymph collection
The mechanical hemolymph collection was carried out by tickling the foot sole of the snail with the tip of a micropipette; as a reflex mechanism, the hemolymph is extruded through the hemal pore. The hemolymph was then gently collected in polypropylene tubes and stored at − 80 °C before analysis.
RNA extraction and retrotranscription
Following hemolymph collection, animals were anesthetized on ice for 10 min, and the central ring ganglia (buccal ganglia were excluded), digestive gland, stomach, foot muscle, and penis were dissected and stored at -80 °C before analysis. Hemocytes were isolated by centrifuging 500 μL of hemolymph for 10 min at 1,000 × g at 4 °C. The supernatant was discarded, and the pellet containing hemocytes was resuspended with 1 mL of Trizol (Merck KGaA; Darmstadt, Germany). Total RNA extraction and DNAse treatment were performed on four replicates of a pool of 4 animals using GenElute Total RNA Miniprep Kit and DNASE70-On-Column DNase I Digestion Set (Merck KGaA; Darmstadt, Germany) as previously described 56,57. Four hundred ng of total RNA was reverse transcribed with a High-Capacity cDNA Reverse Transcription Kit (Life Technologies Corporation) in 20 µL of the reaction mix.
Qualitative PCR analysis and sequencing
Qualitative PCR was performed using Dream Taq DNA polymerase (Thermo Scientific, Waltham, Massachusetts, United States) under the general 3-step amplification of 95 °C for 5 min, followed by 35 cycles of 95 °C for 30 s, 55 °C for 30 s; 72 °C for 30 s, and final extension of 72 °C for 7 min. Primer sequences were designed by NCBI Primer-BLAST software (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and were synthesized by Merck KGaA (Darmstadt, Germany). PCR products were electrophoresed on agarose gel (2%), and DNA fragments were visualized by UV illumination to confirm the correct amplicon size. PCR products (600–800 bp) were purified using the High Pure PCR Product Purification Kit (Roche Diagnostics Corporation, USA) following the manufacturer’s instructions and were directly sequenced using the Sanger sequencing method. Sequencing was performed using ABI PRISM 3130xl Genetic Analyzer (Applied Biosystems, California, USA) and BigDye Terminator v1.1 Cycle Sequencing Kit (Life Technologies Corporation, Massachusetts, USA). The sequence analysis of the PCR fragments was performed using Sequence Scanner Software 2.0 (Applied Biosystems, California, USA), and sequences were compared with the contigs of L. stagnalis using the online version of BLASTn (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&BLAST_SPEC=&LINK_LOC=blasttab&LAST_PAGE=blastn).
Design, validation, and optimization of primers for quantitative PCR analysis
Candidate primers for quantitative Real-Time PCR (qRT-PCR) were designed with NCBI Primer-BLAST software (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and synthesized by Merck KGaA (Darmstadt, Germany). Primers were designed to have a length of 19–23 nucleotides, a melting temperature between 58 and 62 °C, a GC content between 40 and 60%, and generating an amplicon between 100 and 200 bp. First, primer specificity was assessed by qualitative PCR as previously described; each experiment contained two biological replicates of cDNA from central ring ganglia of L. stagnalis and P. canaliculata (kindly provided by Prof. Davide Malagoli, UNIMORE), minus reverse transcription (RT) controls to assess the genomic DNA, and non-template controls. PCR products were run as previously described. Then primer efficiency was evaluated in Bio-Rad CFX Connect. Curves were generated from four-fold serial dilutions of cDNA run in triplicate. Amplification of all genes was detected with SyBR Green dye which generates fluorescence based on the synthesis of double-stranded DNA. The reactions contained 5 µL of cDNA with 10 µL of Bio-Rad SsoAdvanced Universal SyBR Mix, 300 nM forward and reverse primer concentration, and topped to 20 µL with ddH2O. Each point was run in triplicate. The qPCR reactions occurred in a Bio-Rad CFX Connect thermocycler running a custom program. The custom qPCR program consisted of 95 °C for 30 s, 40 cycles of 95 °C for 15 s, and 60 °C for 30 s. The machine read the plate to measure fluorescence at the end of each cycle.
Statistical analysis
Stability values were calculated for each candidate housekeeping gene elongation factor 1α– Lym EF1α and β–tubulin – Lym βTUB) using NormFinder (https://moma.dk/normfinder-software) 58, taking into account intra and intergroup variation. The geometric mean of Cqs of the reference genes was used as calibrator. For an appropriate application of the comparative ΔΔCt method, it was demonstrated that the amplification efficiency of the target genes and endogenous control gene were approximately equal. Extreme outliers were excluded before the statistical analysis using the boxplot tool in SPSS (more than 3 × the interquartile range outside the end of the interquartile box). Analyses were conducted using SPSS for Windows v.28 (SPSS Inc., Chicago, USA).
Chemicals and materials
LC–MS grade acetonitrile and formic acid were purchased from Merck. Deionized water was obtained from MilliQ IQ 7000 system (Merck Millipore). Tryptophan (Trp), kynurenine (KYN), kynurenic acid (KYNA), anthranilic acid (ANA), 3-hydroxy-kynurenine (3HK), xanthurenic acid (XANA), picolinic acid (PICA), and quinolinic acid (QUINA) were of analytical grade (Merck KGaA (Darmstadt, Germany)).
Hemolymph preparation for metabolite analysis
Two hundred microliters of hemolymph were dounce homogenized to disrupt hemocytes, then KP metabolites were extracted as previously described 59: an equal volume of ice-cold 1 M perchloric acid (Merck KGaA) fortified with deuterated tryptophan, kynurenine, kynurenic acid and quinolinic acid (Buchem B.V., Apeldoorn, The Netherlands) as internal standard (final concentration 1 µM) was added to the homogenized sample that was then centrifuged (15,000 × g, 10 min at 4 °C) and the supernatants were collected for direct injection into the LC–MS/MS.
Instrument (UHPLC-Q exactive mass spectrometer) conditions
The analyses of KP metabolites were performed using ultra-high-performance liquid chromatography (UHPLC) coupled with mass spectrometry using heated electrospray ionization (HESI) in positive ionization mode. Chromatographic separations were carried out on a UHPLC Ultimate 3000 system (Thermo Electron Corporation, San Jose, CA, United States) equipped with an UltiMate 3000 HPG-3400RS High Pressure Mixing Biocompatible Gradient Pump with a Discovery HS F5 HPLC Column (Merck KGaA) (L × I.D. 25 cm × 4.6 mm, 5 μm) thermostated at 25 °C. The mobile phase comprised 0.1% formic acid in water (eluent A) and acetonitrile (eluent B). The gradient elution program was set as follows: 0–0.2 min 2% B; 0.2–18.2 min 38% B; 18.2–24.9 min 98% B; finally, the column was equilibrated with 2% B for 12 min. The flow rate was 1.5 mL/min. The sampler (UltiMate 3000 WPS-3000TRS) was kept at 15 °C. The injection volume was 40 µL. High-resolution MS and MS2 spectra were obtained on a Q Exactive Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Electron Corporation, San Jose, CA, United States); HESI optimized parameters were set as follows: sheath gas flow rate, 37 arbitrary unit; auxiliary gas flow rate, 28 arbitrary unit; sweep gas flow rate, 2 arbitrary unit; spray voltage, 3.20 kV; capillary temperature, 320 °C; S-lens RF level, 55.0; aux gas heater temperature, 290 °C. Targeted selected ion monitoring (tSIM) acquisition was performed using an inclusion list (based on m/z of each target and the expected retention time; Supplementary Table X) at a mass resolution of 70,000, an isolation window of 1.0 m/z, AGC target of 2e5, and Maximum IT of 247 ms. Parallel reaction monitoring (PRM) mode was carried out at a mass resolution of 17,500, an isolation window of 1.0 m/z, AGC target of 5e5, Maximum IT of 120 ms, normalized collisional energies (NCE) of 15, 28, 70 were employed for fragmentation. The mass spectrum of each metabolite was obtained by injecting a standard mix at a concentration of 0.01 μM.
Raw data were acquired and processed using the Xcalibur software (Version 4.2.4, Thermo Electron Corporation, San Jose, CA, United States).
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Savitz, J. The kynurenine pathway: a finger in every pie. Mol. Psychiatry 25, 131–147 (2020).
Schwarcz, R., Bruno, J. P., Muchowski, P. J. & Wu, H.-Q. Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 13, 465–477 (2012).
Campbell, B. M., Charych, E., Lee, A. W. & Möller, T. Kynurenines in CNS disease: regulation by inflammatory cytokines. Front. Neurosci. 8, 12 (2014).
Yuasa, H. J. et al. Evolution of vertebrate indoleamine 2,3-dioxygenases. J. Mol. Evol. 65, 705–714 (2007).
Yuasa, H. J. A comprehensive comparison of the metazoan tryptophan degrading enzymes. Biochim. Biophys. Acta (BBA) Proteins Proteom. 1868, 140247 (2020).
Li, T. et al. Alpha-amino-beta-carboxymuconic-epsilon-semialdehyde decarboxylase (ACMSD) is a new member of the amidohydrolase superfamily. Biochemistry 45, 6628–6634 (2006).
Brunello, N. et al. Acetylsalicylic acid accelerates the antidepressant effect of fluoxetine in the chronic escape deficit model of depression. Int. Clin. Psychopharmacol. 21, 219–225 (2006).
Benatti, C. et al. Central effects of a local inflammation in three commonly used mouse strains with a different anxious phenotype. Behav. Brain Res. 224, 23–34 (2011).
Benatti, C. et al. Disease-induced neuroinflammation and depression. CNS Neurol. Disord. Drug Targets 15, 414–433 (2016).
Braidy, N. & Grant, R. Kynurenine pathway metabolism and neuroinflammatory disease. Neural Regen. Res. 12, 39 (2017).
Stone, T. W. & Darlington, L. G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. https://doi.org/10.1038/nrd870 (2002).
Neri, C. Value of invertebrate genetics and biology to develop neuroprotective and preventive medicine in Huntington’s disease. in Neurobiology of Huntington’s Disease: Applications to Drug Discovery (eds. Lo, D. C. & Hughes, R. E.) (CRC Press/Taylor and Francis, 2011).
Tascedda, F. et al. Molluscs as models for translational medicine. Med. Sci. Monit. Basic Res. 21, 96–99 (2015).
Campesan, S. et al. The kynurenine pathway modulates neurodegeneration in a drosophila model of Huntington’s disease. Curr. Biol. 21, 961–966 (2011).
Roberts Buceta, P. M. et al. The kynurenine pathway is essential for rhodoquinone biosynthesis in Caenorhabditis elegans. J. Biol. Chem. 294, 11047–11053 (2019).
Zhuravlev, A. V., Vetrovoy, O. V., Ivanova, P. N. & Savvateeva-Popova, E. V. 3-Hydroxykynurenine in regulation of drosophila behavior: The novel mechanisms for cardinal phenotype manifestations. Front. Physiol. 11, 971 (2020).
Ottaviani, E. et al. Epigenetic modification in neurons of the mollusc Pomacea canaliculata after immune challenge. Brain Res. 1537, 18–26 (2013).
Sadamoto, H. et al. De novo sequencing and transcriptome analysis of the central nervous system of mollusc Lymnaea stagnalis by deep RNA sequencing. PLoS ONE 7, e42546 (2012).
Fodor, I., Hussein, A. A., Benjamin, P. R., Koene, J. M. & Pirger, Z. The unlimited potential of the great pond snail, Lymnaea stagnalis. Elife 9, e56962 (2020).
Rivi, V. et al. Lymnaea stagnalis as model for translational neuroscience research: From pond to bench. Neurosci. Biobehav. Rev. 108, 602–616 (2020).
Fodor, I., Urbán, P., Kemenes, G., Koene, J. M. & Pirger, Z. Aging and disease-relevant gene products in the neuronal transcriptome of the great pond snail (Lymnaea stagnalis): a potential model of aging, age-related memory loss, and neurodegenerative diseases. Invert. Neurosci. 20, 9 (2020).
Kron, N. S., Schmale, M. C. & Fieber, L. A. Changes in metabolism and proteostasis drive aging phenotype in aplysia californica sensory neurons. Front. Aging Neurosci. 12, 573764 (2020).
Agranoff, B. W., Cotman, C. W. & Uhler, M. D. Invertebrate learning and memory. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. 6th edition (1999).
Benjamin, P. R. & Kemenes, G. Invertebrate Models to Study Learning and Memory: Lymnaea. in Encyclopedia of Neuroscience (ed. Squire, L. R.) 197–204 (Academic Press, 2009). https://doi.org/10.1016/B978-008045046-9.00804-4.
Rivi, V. et al. What can we teach Lymnaea and what can Lymnaea teach us?. Biol. Rev. Camb. Philos. Soc. 96, 1590–1602 (2021).
Lagadic, L. & Caquet, T. Invertebrates in testing of environmental chemicals: are they alternatives?. Environ. Health Perspect. 106, 593–611 (1998).
Amorim, J. et al. Lymnaea stagnalis as a freshwater model invertebrate for ecotoxicological studies. Sci. Total Environ. 669, 11–28 (2019).
Jaronen, M. & Quintana, F. J. Immunological relevance of the coevolution of IDO1 and AHR. Front. Immunol. https://doi.org/10.3389/fimmu.2014.00521 (2014).
Gaertner, F. H. & Shetty, A. S. Kynureninase-type enzymes and the evolution of the aerobic tryptophan-to-nicotinamide adenine dinucleotide pathway. Biochim. Biophys. Acta 482, 453–460 (1977).
Dantzer, R. Role of the Kynurenine Metabolism Pathway in Inflammation-Induced Depression: Preclinical Approaches. in Inflammation-Associated Depression: Evidence, Mechanisms and Implications (eds. Dantzer, R. & Capuron, L.) vol. 31, 117–138 (Springer International Publishing, 2016).
Garrison, A. M. et al. Kynurenine pathway metabolic balance influences microglia activity: Targeting kynurenine monooxygenase to dampen neuroinflammation. Psychoneuroendocrinology 94, 1–10 (2018).
Castro-Portuguez, R. & Sutphin, G. L. Kynurenine pathway, NAD+ synthesis, and mitochondrial function: Targeting tryptophan metabolism to promote longevity and healthspan. Exp. Gerontol. 132, 110841 (2020).
van der Goot, A. T. et al. Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc. Natl. Acad. Sci. U. S. A. 109, 14912–14917 (2012).
Savvateeva, E. et al. Age-dependent memory loss, synaptic pathology and altered brain plasticity in the Drosophila mutant cardinal accumulating 3-hydroxykynurenine. J. Neural Transm. (Vienna) 107, 581–601 (2000).
Feng, Z.-P. et al. Transcriptome analysis of the central nervous system of the mollusc Lymnaea stagnalis. BMC Genomics 10, 451 (2009).
Rosato, M., Hoelscher, B., Lin, Z., Agwu, C. & Xu, F. Transcriptome analysis provides genome annotation and expression profiles in the central nervous system of Lymnaea stagnalis at different ages. BMC Genomics 22, 637 (2021).
Seppälä, O. et al. Transcriptome profiling of Lymnaea stagnalis (Gastropoda) for ecoimmunological research. BMC Genomics 22(1), 1–16 (2021).
Wooller, S. et al. A combined bioinformatics and LC-MS-based approach for the development and benchmarking of a comprehensive database of Lymnaea CNS proteins. J. Exp. Biol. 225, jeb243753 (2022).
Yuasa, H. J. & Ball, H. J. Efficient tryptophan-catabolizing activity is consistently conserved through evolution of TDO enzymes, but not IDO enzymes. J. Exp. Zool. B Mol. Dev. Evol. 324, 128–140 (2015).
Rossi, F., Miggiano, R., Ferraris, D. M. & Rizzi, M. The synthesis of kynurenic acid in mammals: An updated kynurenine aminotransferase structural KATalogue. Front. Mol. Biosci. https://doi.org/10.3389/fmolb.2019.00007 (2019).
Han, Q., Cai, T., Tagle, D. A. & Li, J. Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. Cell. Mol. Life Sci. 67, 353–368. https://doi.org/10.1007/s00018-009-0166-4 (2010).
Cuenoud, B. et al. Brain NAD Is Associated With ATP Energy Production and Membrane Phospholipid Turnover in Humans. Front. Aging Neurosci. 12, (2020).
Goody, M. F. & Henry, C. A. A need for NAD+ in muscle development, homeostasis, and aging. Skelet Muscle 8, 9 (2018).
Chen, L.-M. et al. Tryptophan-kynurenine metabolism: a link between the gut and brain for depression in inflammatory bowel disease. J. Neuroinflammation 18, 135 (2021).
Sordillo, P. P., Sordillo, L. A. & Helson, L. The Kynurenine Pathway: A Primary Resistance Mechanism in Patients with Glioblastoma. Anticancer Res. 37, 2159–2171 (2017).
Badawy, A.A.-B. Kynurenine pathway of tryptophan metabolism: Regulatory and functional aspects. Int. J. Tryptophan. Res. 10, 1178646917691938 (2017).
Huang, M. et al. Longitudinal association of inflammation with depressive symptoms: A 7-year cross-lagged twin difference study. Brain Behav. Immun. 75, 200–207 (2019).
Lestage, J., Verrier, D., Palin, K. & Dantzer, R. The enzyme indoleamine 2,3-dioxygenase is induced in the mouse brain in response to peripheral administration of lipopolysaccharide and superantigen. Brain Behav. Immun. 16, 596–601 (2002).
Yuasa, H. J., Mizuno, K. & Ball, H. J. Low efficiency IDO2 enzymes are conserved in lower vertebrates, whereas higher efficiency IDO1 enzymes are dispensable. FEBS J. 282, 2735–2745 (2015).
Zhen, D., Liu, J., Zhang, X. D. & Song, Z. Kynurenic acid acts as a signaling molecule regulating energy expenditure and is closely associated with metabolic diseases. Front. Endocrinol. (Lausanne) 13, 847611 (2022).
Keszthelyi, D., Troost, F. J. & Masclee, A. A. M. Understanding the role of tryptophan and serotonin metabolism in gastrointestinal function. Neurogastroenterol. Motil. 21, 1239–1249 (2009).
Kaur, H., Bose, C. & Mande, S. S. Tryptophan metabolism by gut microbiome and gut-brain-axis: An in silico analysis. Front. Neurosci. 13, 1365 (2019).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Di Tommaso, P. et al. Cloud-Coffee: implementation of a parallel consistency-based multiple alignment algorithm in the T-Coffee package and its benchmarking on the Amazon Elastic-Cloud. Bioinformatics 26, 1903–1904 (2010).
Benatti, C., Colliva, C., Blom, J. M. C., Ottaviani, E. & Tascedda, F. Transcriptional effect of serotonin in the ganglia of Lymnaea stagnalis. Invertebr. Surv. J. 14(1), 251–258 (2017).
Rivi, V. et al. To eat or not to eat: A Garcia effect in pond snails (Lymnaea stagnalis). J. Comp. Physiol. A 207, 479–495 (2021).
Wang, B. et al. Validation of reference genes for accurate normalization of gene expression with quantitative real-time PCR in Haloxylon ammodendron under different abiotic stresses. Physiol. Mol. Biol. Plants 24, 455–463 (2018).
Borsini, A. et al. Rescue of IL-1β-induced reduction of human neurogenesis by omega-3 fatty acids and antidepressants. Brain Behav. Immun. 65, 230–238 (2017).
Acknowledgements
We would like to thank Dr Diego Pinetti (CIGS-UNIMORE) for the help in setting up the UHPLC/MS method and Dr. Sonia Costa and Dr. Maria Luisa Simone (UNIMORE) for the assistance in the sequencing process.
Funding
This research was supported by Regione Emilia-Romagna "L’invertebrato L. stagnalis quale modello per la Medicina Traslazionale" L.R. N. 20/2002 PROGETTI DI RICERCA SUI METODI ALTERNATIVI ALL'UTILIZZO DI ANIMALI; and FAR 2016 Dipartimento Scienze della Vita, Università degli Studi di Modena e Reggio Emilia. These sources of funding were not involved in the study design, data collection, analysis and interpretation, or in the decision to submit the paper for publication.
Author information
Authors and Affiliations
Contributions
C.B., S.A., and F.T. were responsible for the study concept. C.B., V.R., and S.A. carried out the molecular analysis. A.G., S.C., C.B., and S.B. performed the bioinformatic analysis. C.B. and V.R. drafted the article. S.A., J.M.C.B., S.B., L.P., N.B., and F.T. edited and provided feedback on the manuscript. F.T. and N.B. provided resources. All authors have reviewed the manuscript and approved the final version submitted for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cristina, B., Veronica, R., Silvia, A. et al. Identification and characterization of the kynurenine pathway in the pond snail Lymnaea stagnalis. Sci Rep 12, 15617 (2022). https://doi.org/10.1038/s41598-022-19652-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-19652-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
