
Abstract
Proteins with amino acid homorepeats have the potential to be detrimental to cells and are often associated with human diseases. Why, then, are homorepeats prevalent in eukaryotic proteomes? In yeast, homorepeats are enriched in proteins that are essential and pleiotropic and that buffer environmental insults. The presence of homorepeats increases the functional versatility of proteins by mediating protein interactions and facilitating spatial organization in a repeat-dependent manner. During evolution, homorepeats are preferentially retained in proteins with stringent proteostasis, which might minimize repeat-associated detrimental effects such as unregulated phase separation and protein aggregation. Their presence facilitates rapid protein divergence through accumulation of amino acid substitutions, which often affect linear motifs and post-translational-modification sites. These substitutions may result in rewiring protein interaction and signaling networks. Thus, homorepeats are distinct modules that are often retained in stringently regulated proteins. Their presence facilitates rapid exploration of the genotype–phenotype landscape of a population, thereby contributing to adaptation and fitness.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout









Similar content being viewed by others

References
La Spada, A.R. & Taylor, J.P. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 11, 247–258 (2010).
Moumné, L. et al. Differential aggregation and functional impairment induced by polyalanine expansions in FOXL2, a transcription factor involved in cranio-facial and ovarian development. Hum. Mol. Genet. 17, 1010–1019 (2008).
Gatchel, J.R. & Zoghbi, H.Y. Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 6, 743–755 (2005).
Tsuda, H. et al. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell 122, 633–644 (2005).
Cortes, C.J. et al. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat. Neurosci. 17, 1180–1189 (2014).
Monks, D.A. et al. Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc. Natl. Acad. Sci. USA 104, 18259–18264 (2007).
Nasrallah, I.M., Minarcik, J.C. & Golden, J.A. A polyalanine tract expansion in Arx forms intranuclear inclusions and results in increased cell death. J. Cell Biol. 167, 411–416 (2004).
Gemayel, R., Vinces, M.D., Legendre, M. & Verstrepen, K.J. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 44, 445–477 (2010).
Stevens, K.E. & Mann, R.S. A balance between two nuclear localization sequences and a nuclear export sequence governs extradenticle subcellular localization. Genetics 175, 1625–1636 (2007).
Wolf, A. et al. The polyserine domain of the lysyl-5 hydroxylase Jmjd6 mediates subnuclear localization. Biochem. J. 453, 357–370 (2013).
Salichs, E., Ledda, A., Mularoni, L., Albà, M.M. & de la Luna, S. Genome-wide analysis of histidine repeats reveals their role in the localization of human proteins to the nuclear speckles compartment. PLoS Genet. 5, e1000397 (2009).
Lee, C. et al. Protein aggregation behavior regulates cyclin transcript localization and cell-cycle control. Dev. Cell 25, 572–584 (2013).
Galant, R. & Carroll, S.B. Evolution of a transcriptional repression domain in an insect Hox protein. Nature 415, 910–913 (2002).
Gerber, H.P. et al. Transcriptional activation modulated by homopolymeric glutamine and proline stretches. Science 263, 808–811 (1994).
Michael, T.P. et al. Simple sequence repeats provide a substrate for phenotypic variation in the Neurospora crassa circadian clock. PLoS One 2, e795 (2007).
Fondon, J.W. III & Garner, H.R. Molecular origins of rapid and continuous morphological evolution. Proc. Natl. Acad. Sci. USA 101, 18058–18063 (2004).
Gidalevitz, T., Ben-Zvi, A., Ho, K.H., Brignull, H.R. & Morimoto, R.I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311, 1471–1474 (2006).
Karlin, S., Brocchieri, L., Bergman, A., Mrazek, J. & Gentles, A.J. Amino acid runs in eukaryotic proteomes and disease associations. Proc. Natl. Acad. Sci. USA 99, 333–338 (2002).
Albà, M.M. & Guigó, R. Comparative analysis of amino acid repeats in rodents and humans. Genome Res. 14, 549–554 (2004).
Faux, N.G. et al. Functional insights from the distribution and role of homopeptide repeat-containing proteins. Genome Res. 15, 537–551 (2005).
Faux, N.G. et al. RCPdb: an evolutionary classification and codon usage database for repeat-containing proteins. Genome Res. 17, 1118–1127 (2007).
Délot, E., King, L.M., Briggs, M.D., Wilcox, W.R. & Cohn, D.H. Trinucleotide expansion mutations in the cartilage oligomeric matrix protein (COMP) gene. Hum. Mol. Genet. 8, 123–128 (1999).
Ahn, Y.Y., Bagrow, J.P. & Lehmann, S. Link communities reveal multiscale complexity in networks. Nature 466, 761–764 (2010).
Koch, E.N. et al. Conserved rules govern genetic interaction degree across species. Genome Biol. 13, R57 (2012).
Kemmeren, P. et al. Large-scale genetic perturbations reveal regulatory networks and an abundance of gene-specific repressors. Cell 157, 740–752 (2014).
Munroe, D. & Jacobson, A. mRNA poly(A) tail, a 3′ enhancer of translational initiation. Mol. Cell. Biol. 10, 3441–3455 (1990).
Jackson, R.J., Hellen, C.U. & Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11, 113–127 (2010).
Wen, J.D. et al. Following translation by single ribosomes one codon at a time. Nature 452, 598–603 (2008).
Gingold, H. & Pilpel, Y. Determinants of translation efficiency and accuracy. Mol. Syst. Biol. 7, 481 (2011).
van der Lee, R. et al. Intrinsically disordered segments affect protein half-life in the cell and during evolution. Cell Rep. 8, 1832–1844 (2014).
Glotzer, M., Murray, A.W. & Kirschner, M.W. Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138 (1991).
Gsponer, J., Futschik, M.E., Teichmann, S.A. & Babu, M.M. Tight regulation of unstructured proteins: from transcript synthesis to protein degradation. Science 322, 1365–1368 (2008).
Pfleger, C.M. & Kirschner, M.W. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 14, 655–665 (2000).
Rogers, S., Wells, R. & Rechsteiner, M. Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 234, 364–368 (1986).
Gsponer, J. & Babu, M.M. Cellular strategies for regulating functional and nonfunctional protein aggregation. Cell Rep. 2, 1425–1437 (2012).
Woodsmith, J., Kamburov, A. & Stelzl, U. Dual coordination of post translational modifications in human protein networks. PLoS Comput. Biol. 9, e1002933 (2013).
Mateo, F. et al. Degradation of cyclin A is regulated by acetylation. Oncogene 28, 2654–2666 (2009).
Qian, M.X. et al. Acetylation-mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 153, 1012–1024 (2013).
Tyers, M., Tokiwa, G., Nash, R. & Futcher, B. The Cln3-Cdc28 kinase complex of S. cerevisiae is regulated by proteolysis and phosphorylation. EMBO J. 11, 1773–1784 (1992).
Bergeron-Sandoval, L.P., Safaee, N. & Michnick, S.W. Mechanisms and consequences of macromolecular phase separation. Cell 165, 1067–1079 (2016).
Gemayel, R. et al. Variable glutamine-rich repeats modulate transcription factor activity. Mol. Cell 59, 615–627 (2015).
Fishbain, S. et al. Sequence composition of disordered regions fine-tunes protein half-life. Nat. Struct. Mol. Biol. 22, 214–221 (2015).
McDonald, M.J., Wang, W.C., Huang, H.D. & Leu, J.Y. Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 9, e1000622 (2011).
Lenz, C., Haerty, W. & Golding, G.B. Increased substitution rates surrounding low-complexity regions within primate proteins. Genome Biol. Evol. 6, 655–665 (2014).
Huntley, M.A. & Clark, A.G. Evolutionary analysis of amino acid repeats across the genomes of 12 Drosophila species. Mol. Biol. Evol. 24, 2598–2609 (2007).
McDonald, M.J. et al. Mutation at a distance caused by homopolymeric guanine repeats in Saccharomyces cerevisiae. Sci. Adv. 2, e1501033 (2016).
Dreze, M. et al. 'Edgetic' perturbation of a C. elegans BCL2 ortholog. Nat. Methods 6, 843–849 (2009).
Woerner, A.C. et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351, 173–176 (2016).
Panigrahi, G.B., Lau, R., Montgomery, S.E., Leonard, M.R. & Pearson, C.E. Slipped (CTG)*(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat. Struct. Mol. Biol. 12, 654–662 (2005).
Mar Albà, M., Santibáñez-Koref, M.F. & Hancock, J.M. Amino acid reiterations in yeast are overrepresented in particular classes of proteins and show evidence of a slippage-like mutational process. J. Mol. Evol. 49, 789–797 (1999).
Shah, K.A. & Mirkin, S.M. The hidden side of unstable DNA repeats: mutagenesis at a distance. DNA Repair (Amst.) 32, 106–112 (2015).
Shah, K.A. et al. Role of DNA polymerases in repeat-mediated genome instability. Cell Rep. 2, 1088–1095 (2012).
Zhang, J. & Yang, J.R. Determinants of the rate of protein sequence evolution. Nat. Rev. Genet. 16, 409–420 (2015).
Narayanaswamy, R. et al. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc. Natl. Acad. Sci. USA 106, 10147–10152 (2009).
Chakrabortee, S. et al. Intrinsically disordered proteins drive emergence and inheritance of biological traits. Cell 167, 369–381.e12 (2016).
Caudron, F. & Barral, Y. A super-assembly of Whi3 encodes memory of deceptive encounters by single cells during yeast courtship. Cell 155, 1244–1257 (2013).
Levy, E.D., Landry, C.R. & Michnick, S.W. How perfect can protein interactomes be? Sci. Signal. 2, pe11 (2009).
Hancock, J.M. & Simon, M. Simple sequence repeats in proteins and their significance for network evolution. Gene 345, 113–118 (2005).
Jarosz, D.F., Taipale, M. & Lindquist, S. Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu. Rev. Genet. 44, 189–216 (2010).
Ekman, D., Light, S., Björklund, A.K. & Elofsson, A. What properties characterize the hub proteins of the protein-protein interaction network of Saccharomyces cerevisiae? Genome Biol. 7, R45 (2006).
Dosztányi, Z., Mészáros, B. & Simon, I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics 25, 2745–2746 (2009).
Jorda, J. & Kajava, A.V. T-REKS: identification of Tandem REpeats in sequences with a K-meanS based algorithm. Bioinformatics 25, 2632–2638 (2009).
Ward, J.J., McGuffin, L.J., Bryson, K., Buxton, B.F. & Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinformatics 20, 2138–2139 (2004).
Simon, M. & Hancock, J.M. Tandem and cryptic amino acid repeats accumulate in disordered regions of proteins. Genome Biol. 10, R59 (2009).
Harrison, P.M. & Gerstein, M. A method to assess compositional bias in biological sequences and its application to prion-like glutamine/asparagine-rich domains in eukaryotic proteomes. Genome Biol. 4, R40 (2003).
Harbi, D., Kumar, M. & Harrison, P.M. LPS-annotate: complete annotation of compositionally biased regions in the protein knowledgebase. Database (Oxford) 2011, baq031 (2011).
Cherry, J.M. et al. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res. 40, D700–D705 (2012).
Altenhoff, A.M., Schneider, A., Gonnet, G.H. & Dessimoz, C. OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 39, D289–D294 (2011).
Huang, W., Sherman, B.T. & Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
McGraw, K.O. & Wong, S.P. A common language effect size statistic. Psychol. Bull. 111, 361–365 (1992).
Weatheritt, R.J., Gibson, T.J. & Babu, M.M. Asymmetric mRNA localization contributes to fidelity and sensitivity of spatially localized systems. Nat. Struct. Mol. Biol. 21, 833–839 (2014).
Grissom, R.J. & Kim, J.J. Effect Sizes for Research: Univariate and Multivariate Applications (Routledge, 2012).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Amberd, D.C., Burke, D. & Strathern, J.N. Methods in Yeast Genetics: a Cold Spring Harbor Laboratory Course Manual (Cold Spring Harbor Laboratory Press, 2005).
Rossmann, M.P. & Stillman, B. Immunoblotting histones from yeast whole-cell protein extracts. Cold Spring Harb. Protoc. 2013, 625–630 (2013).
Eng, J.K., McCormack, A.L. & Yates, J.R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 (1994).
Perkins, D.N., Pappin, D.J., Creasy, D.M. & Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 (1999).
Hu, Z., Killion, P.J. & Iyer, V.R. Genetic reconstruction of a functional transcriptional regulatory network. Nat. Genet. 39, 683–687 (2007).
Teste, M.A., Duquenne, M., François, J.M. & Parrou, J.L. Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae. BMC Mol. Biol. 10, 99 (2009).
Acknowledgements
We thank B. Lenhard, C. Semple, M. Torrent, A.J. Venkatakrishnan, B. Lang and members of our group for stimulating discussions and comments on this study. This work was supported by the Medical Research Council (MC_U105185859; S.C., N.S.L., S.B. and M.M.B.), the Human Frontier Science Program (RGY0073/2010; M.M.B.), a European Molecular Biology Organization Long term fellowship (S.C.), the Young Investigator Program (M.M.B. and K.J.V.), ERASysBio+ (GRAPPLE; S.C., S.B. and M.M.B.), Marie Curie actions (FP7-PEOPLE-2011-IEF-299105; N.S.d.G.) and Cancer Research UK (P.L.C.). M.M.B. is supported as a Lister Institute Prize Fellow. We thank C. Taylor and C. d'Santos from the CRUK-Cambridge Institute proteomics core facility.
Author information
Authors and Affiliations
Contributions
S.C. and M.M.B. conceived the project and wrote the manuscript, incorporating input from all authors. S.C., K.J.V., S.B. and M.M.B. designed the study; S.C., G.C., N.S.L., E.I.-S. and S.B. collected the data sets and performed computational investigations; S.C., P.L.C., N.S.d.G. and R.G. designed and performed the experiments. All authors participated in interpreting the results. S.C. led the study, and M.M.B. supervised the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 2 Control calculations for physiological importance and functional versatility of HRPs.
(a) Scaled gene to phenotype (scaled-G2P) network of yeast non-essential genes, reconstructed by merging phenotypes that had more than 49% genetic overlap (left panel). Boxplot of distribution of the number of phenotypes in the scaled G2P network among HRPs and NonHRPs (right panel). Statistical significance was assessed using Wilcoxon rank sum test, with effect sizes displayed as CLES.
Different features such as (i) protein length (Ekman, D. et al., Genome Biol 7, R45, 2006) and (ii) low complexity sequences, especially amino acid compositional bias (Coletta, A. et al. BMC Syst Biol 4, 43, 2010), can influence the propensity of proteins to interact with multiple partners and hence its functionality. To investigate the effect of protein length on the functionality of HRPs, we obtained length matched controls and compared their distributions. To investigate whether the observed trends for functionality are similar between proteins with homorepeats and NonHRPs containing non-repeat amino acid bias we first identified all yeast NonHRPs that contain compositional bias. Boxplots of distribution of the number of protein-protein interactions and link communities in PPI network among (b) length-matched HRPs and NonHRPs and (d) HRPs and NonHRPs with amino acid bias. Statistical significance was assessed using Wilcoxon rank sum test and FDR corrected for multiple testing within each class, with effect sizes displayed as CLES. HRPs tend to have more interactions and are involved in more processes than their length-matched counterparts. HRPs have more protein interactions and participate in more biological processes than proteins with non-repetitive amino acid bias. Collectively, these results suggest that HRPs are more functionally versatile than proteins with similar lengths and those, which contain amino acid bias.
(c) Proteins with homorepeats are functionally more versatile than NonHRPs with similar biological functions. Significance estimates are provided as –log P values (upper panel) and effect size estimates are provided as median differences (lower panel) for each one of the 100 randomizations comparing distributions of HRPs and randomly selected but functionally similar NonHRPs for (i) No. of protein-protein interactions, (ii) No. of link communities in PPI network, (iii) No. of genetic interactions and (iv) No. of link communities in genetic network. Statistical significance was assessed using Wilcoxon rank sum test. The solid black line in the upper panel represents a p-value of 0.05.
Barplots showing percentages of HRPs and NonHRPs with a high number of (e) protein-DNA and (f) protein-RNA interactions. Since the number of data points was limited, we classified transcription factors and RNA binding proteins into tertiles representing proteins with low, medium and high number of protein-DNA and protein-RNA interactions, respectively. Statistical significance was assessed using Fisher’s exact test, with effect size represented by OR.
Supplementary Figure 3 Influence of the type of amino acid of the homorepeat on different features analyzed in this study.
(a) Estimation of Likelihood Ratios (LRs) from conditional probabilities for different types of features (categorical or quantitative) analyzed in this study and its interpretation. Quantitative features were categorized into low or high classes based on tertile cut-offs. (b) The heatmap presents LRs for all HRPs and proteins with different amino acid repeat types, with the numbers in the parenthesis the corresponding to the number of proteins with a particular amino acid type. Categorical features are marked with asterisks. For quantitative features, the class analyzed (high or low) is presented in the parenthesis. LRs greater than 1 are marked in green, with the intensity of the color representing its magnitude.
Supplementary Figure 4 Influence of the lengths of different amino acid repeat types on different features analyzed in this study.
Each cell in the heatmap represents the Pearson correlation coefficient defining the extent and type of correlation (positive/negative) between the lengths of a specific amino acid repeat (given as column names) and the feature analyzed (row names). The numbers in the parentheses indicate the number of proteins with a particular amino acid repeat type. Correlation coefficients >0.70 were found to be statistically significant upon correcting for multiple testing.
Supplementary Figure 5 PolyQ in Snf5 influences cell division and mediates protein-protein interactions.
(a) Budding index of cells after re-inoculation of stationary phase cultures from WT or ∆HR in YPD or YPG after 1, 3 and 4h. Statistical significance was assessed using ANOVA. (b) Immunoblot analysis for HA-tagged WT and ∆HR Snf5 in the immunoprecipitate. (c) Number of Snf5 protein-protein interactions previously known, as retrieved from BioGRID (Chatr-Aryamontri, A. et al., Nucleic acids research 41, D816–23, 2013) and identified in this study. (d) Proteins interacting with Snf5 in a polyQ dependent manner (observed in WT but absent in ∆HR) in YPD and YPG connected to the diverse biological processes. The interactors are identified with a minimum of 3 unique peptide hits in at least 2 of the 3 experiments and classified according to the associated biological processes. The pie chart on top shows the number of polyQ-mediated interactions in each condition.
Supplementary Figure 6 Stringent regulation of HRP activity by coarse and fine-tuning.
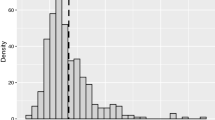
Previously we showed that intrinsically disordered proteins are tightly regulated (Gsponer, J., et al. Science 322, 1365–8, 2008). This raises the question of whether the stringent regulation of HRPs is confounded by protein disorder. To address this, we compared the regulatory features between HRPs and NonHRPs with low and high intrinsic disorder content. Boxplot of distributions of protein abundance, translational rate and protein half-life among HRPs and NonHRPs with (a) low and (b) high disorder content. Irrespective of the disorder content, HRPs tend to have lower translational rates and low protein half-life compared to NonHRPs, though the protein abundance is comparable. Thus, stringent protein regulation of HRPs is more pronounced compared to NonHRPs with similar disorder content. To investigate if HRPs and proteins with non-repetitive amino acid bias are regulated with similar stringency, we compared the regulatory features between HRPs and NonHRPs with amino acid bias. (c) Boxplots showing the distributions of protein abundance, translational rate and protein half-life among HRPs and NonHRPs with amino acid bias. HRPs tend to be less abundant with lower translational rates and protein half-lives. This suggests that HRPs are more stringently controlled than NonHRPs without amino acid bias. Statistical significance was determined using Wilcoxon rank sum test corrected for multiple testing within each class of comparisons. Effect sizes are provided as CLES. (d) Distribution (grey histogram) showing the random expectation of the over-expression toxicity HRPs, with the red arrow showing the observed number of over-expression toxicity HRPs. Enrichment of HRPs in over-expression toxicity genes was tested using permutation test, by performing 10,000 iterations. The Z-score indicates the distance of the actual observation to the mean of random expectation in terms of number of standard deviation. P-values were estimated as the ratio of randomly observed proteins greater than or equal to the number of actually observed HRPs to the total number of random samples (10,000).Over-representation of HRPs among over-expression toxicty genes implies that they are stringently regulated by coarse-tuning, i.e., by regulating their abundance. (e) Box plot showing distribution of density of PTMs among NonHRPs and HRPs. Density of PTMs for each protein was estimated by obtaining a ratio of the total number of PTMs identified in a protein to the total number of amino acids in that protein. The P-value was estimated using Wilcoxon rank sum test and effect size displayed as CLES. More PTMs in HRPs suggest a stringent fine-tuning of their activity. (f) Distribution of the density of PTMs within (shown as 0 in the X-axis) and 500 amino acids on either side of the HR.
Supplementary Figure 7 Pleiotropic effects, adaptability, functional versatility and proteostasis collectively differentiate HRPs from nonHRPs and highly disordered nonHRPs.
(a) The left panel depicts the work flow of a random forest (RF) model that was trained on eight features to distinguish (i) proteins with (HRPs) and without HRs (NonHRPs) and (ii) HRPs from highly disordered NonHRPs (HDP; intrinsic disorder >30%). The eight features considered for this machine learning approach pertained to pleiotropy, adaptability, functional versatility and proteostasis (right panel). The number of features for each class is provided in the parenthesis. (b) The RF model trained on the eight features is able to distinguish between HRPs and Non HRPs with an overall accuracy of 0.75. Test set precision, recall, f1 scores, and sample sizes (support) are provided for each class. Genes with three or more missing values among the eight features were disregarded. This resulted in 3819 genes with values for at least five features. For some of these genes, the missing values were imputed using multiple imputation by chained equations, using the `mice` R package (arguments to mice method: method=“pmm", m=5, maxit=5; van Buuren, S. et al. J Stat Softw 45, doi:10.18637/jss.v045.i03, 2011). The outcome class was balanced (data before balancing: 16% HRP, n=3819; data after balancing: 48%, n=1261) by down-sampling the majority class size to approximately the size of the minority class (down sampled majority n=700). The cleaned and balanced feature set was split into 70% training and 30% testing data. The training data was used as the input to a random forest model composed of 50 classification trees, with hyperparameters (purity criterion, maximum features nodes consider for splitting, and max tree depth) optimized using 10-fold cross-validation and grid search (yielding optima: criterion=“entropy”, max_features=“log2”, max_depth=None). (c) Importance of different features for distinguishing HRPs from NonHRPs relative to the most important feature, disorder fraction. The most predictive features are related to disorder and proteostasis, with the remaining features being of approximately equal importance, with relatively low contribution from essentiality. Since disorder fraction was an important feature for distinguishing HRPs from NonHRPs, we investigated if HRPs could be distinguished from highly disordered NonHRPs (HDPs; with disorder fraction >30%). (d) The RF model is able to distinguish HRPs from HDPs with an overall accuracy of 0.67. (e) Importance of different features for distinguishing HRPs from HDPs, relative to disorder fraction. Similar to our observations for NonHRPs, the most important features are related to proteostasis, with other features being of approximately equal importance and low contribution from essentiality.
Supplementary Figure 8 Evolutionary benefits associated with HRs.
(a) Box plot showing the distribution of sequence identities among similar NonHRP:NonHRP) and divergent (HRP:NonHRP) pairs of yeast paralogs. Statistical signficance was estimated using Wilcoxon rank-sum test, with effect size displayed as CLES. To test if our observations related to HR-associated amino acid substitutions (Fig. 8) are confounded by high number of protein- protein interactions or higher density of linear motifs or PTM sites associated with HRPs, we classified yeast proteins into three bins of low, medium and high using tertile cut-offs for each of the features (panel b). Linear motif residue density was estimated by obtaining the ratio of residues predicted to form putative linear motifs over the entire length of the protein. In each bin, we tested for differences in the proportion of NonHRPs and HRPs with and without amino acid substitutions in the functionally relevant sites. Barplots showing proportion of NonHRPs and HRPs with amino acid substitutions affecting (c) functionally relevant sites (putative linear motifs and/or PTM sites) among low, medium and high bins of proteins classified based on the number of protein-protein interactions, (d) putative linear motifs among different bins of proteins classified based on the density of linear motif residues and (e) PTM sites among different bins of proteins classified based on the density of PTM sites. Statistical significance within each bin for each attribute was tested using Fisher’s exact test and corrected for multiple testing (FDR). If a feature is a confounder, then there will not be any difference in the proportion of NonHRPs and HRPs with amino acid substitutions across different bins of that feature. Higher proportion of HRPs tend to harbor substitutions affecting functionally relevant sites compared to NonHRPs across all bins, with matched number of protein-protein interactions (panel c). Similarly, across all bins of linear motif residue density, higher proportion of HRPs harbor substitutions within putative linear motifs compared to the matched NonHRPs (panel d). HRPs show a significant difference for the proportion of genes with amino acid subsitutions affecting PTM sites especially in the ‘high’ bin of PTM site density (panel e). These findings suggest that the amino acid substitutions affecting functionally relevant sites in HRPs are independent of the number of protein interactions that a HRP participates in, or the density of linear motif residues or PTM sites. (f) Estimation of conditional probability for observing more amino acid substitutions than expected by chance in segments containing HRs and conditional probability for observing HRs in segments with more observed amino acid substitutions than expected by chance (denoted by P(O>E | HRs) and P(HRs | O>E), respectively). Each protein was divided into four equal segments and the expected number of amino acid substitutions per segment was calculated as shown. Subsequently, segments with more amino acid substitutions than expected and those that contained HRs were identified. Using a similar approach, segments with more amino acid substitutions in putative linear motifs were defined. Due to very few data points, substitutions at the PTM sites were not considered for estimating conditional probabilities. (g) Conditional probability values of finding more amino acid substitutions than expected by chance in HR segments and that of finding a HR given a segment contains more amino acid substitutions (first row). The second row provides the conditional probability values of finding more amino acid substitutions affecting putative linear motifs than expected by chance in HR segments and that of finding a HR given a segment contains more linear motif affecting amino acid substitutions.
Supplementary Figure 9 Essential HRPs constitute a part of the rapidly adaptable part of the proteome, facilitated by stringent proteostasis.
(a) Enrichment of Gene Ontology (GO) biological processes among essential HRPs and NonHRPs in yeast. Both essential HRPs and NonHRPs are over-represented in similar biological processes. Boxplot of distributions of the number of (b) protein-protein interactions, (c) link communities in protein-protein interaction network, (d) different GO terms a gene is associated with, reflecting its functional dviersity, (e) protein abundance, (f) translational rate and (g) protein half-life among essential HRPs and NonHRPs. Statistical significance was assessed using Wilcoxon rank sum test and P-values were corrected for multiple testing (FDR). (h) Sequence divergence among yeast essential HRPs and NonHRPs with their one-to-one orthologs in 73 fungal species belonging to 16 distinct fungal classes. Species names are abbreviated. Taxonomic and phylogenetic details of the fungal species are provided in Supplementary Notes. Median divergences of essential HRPs (red circle) and NonHRPs (grey circle) with their orthologs in each species are shown in the middle panel. The upper panel provides the statistical significance estimated by comparing the distribution of divergence of yeast essential HRPs and NonHRPs with their respective orthologs in each species. The black line shows a P-value cut-off of 0.01, corrected for multiple testing. The bottom panel shows the number of orthologs of yeast essential HRP (red) and NonHRP (grey) in each species. The average median difference between essential NonHRPs and HRPs is 6.3%, which corresponds to ~19 missense variant positions for a 300 amino acid long protein. Though both HRP and NonHRP essential genes show enrichment for similar processes, essential proteins that contain a HR are more functionally versatile, stringently regulated and undergo accelerated sequence divergence and thereby may constitute the rapidly adaptable part of the genome.
Supplementary Figure 10 Highly pleiotropic HRPs constitute a part of the rapidly adaptable part of the proteome, facilitated by stringent proteostasis.
We selected the top 33% of pleiotropic genes (defined by tertiles) in the G2P network and classified them into highly pleiotropic HRPs and NonHRPs. (a) Enrichment of Gene Ontology (GO) biological processes among yeast highly pleiotropic HRPs and NonHRPs. Boxplots of distributions of the number of (b) genetic interactions, (c) protein-protein interactions, (d) link communities in genetic and protein-protein interaction network, (e) different GO terms a gene is associated with, reflecting its functionality, (f) genes whose expression is altered upon deletion of regulators (from the gene-perturbation network), (g) protein abundance, (h) translational rate and (i) protein half-life among highly pleiotropic HRPs and NonHRPs. Statistical significance was assessed using Wilcoxon rank sum test, corrected for multiple testing. (j) Sequence divergence among yeast highly pleiotropic HRPs and NonHRPs with their one-to-one orthologs in 73 fungal species belonging to 16 distinct fungal classes. Species names are abbreviated. Taxonomic and phylogenetic details of the fungal species are provided in Supplementary Notes. Median divergences of highly pleiotropic HRPs (red circle) and NonHRPs (grey circle) with their orthologs in each species are shown in the middle panel. The upper panel provides the statistical significance estimated by comparing the distribution of divergence of yeast highly pleiotropic HRPs and NonHRPs with their respective orthologs in each species. The black line shows a P-value cut-off of 0.01, corrected for multiple testing. The bottom panel shows the number of orthologs of yeast highly pleiotropic HRP (red) and NonHRP (grey) in each species. The average median difference between highly pleiotropic NonHRPs and HRPs is 5.2% which corresponds to ~15 missense variant positions for a 300 amino acid long protein. These findings suggest that highly pleiotropic proteins that contain a HR affect diverse functions through multiple interactions, are stringently regulated, and undergo accelerated divergence and hence may constitute the rapidly adaptable part of the proteome. In contrast, hNonHRPs with housekeeping functions seem to constitute the relatively slowly evolving core of the proteome.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–10, Supplementary Tables 1 and 2, and Supplementary Notes 1–4 (PDF 6341 kb)
Supplementary Data Set 1
Protein interactors of Snf5 identified in this study (XLSX 126 kb)
Supplementary Data Set 2
Source data of computational and experimental studies (XLSX 7774 kb)
Rights and permissions
About this article

Cite this article
Chavali, S., Chavali, P., Chalancon, G. et al. Constraints and consequences of the emergence of amino acid repeats in eukaryotic proteins. Nat Struct Mol Biol 24, 765–777 (2017). https://doi.org/10.1038/nsmb.3441
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nsmb.3441
This article is cited by
-
Evolution of Transcript Abundance is Influenced by Indels in Protein Low Complexity Regions
Journal of Molecular Evolution (2024)
-
Lineage-specific protein repeat expansions and contractions reveal malleable regions of immune genes
Genes & Immunity (2022)
-
A framework for understanding the functions of biomolecular condensates across scales
Nature Reviews Molecular Cell Biology (2021)
-
Homopeptide and homocodon levels across fungi are coupled to GC/AT-bias and intrinsic disorder, with unique behaviours for some amino acids
Scientific Reports (2021)
-
Mutation–selection balance and compensatory mechanisms in tumour evolution
Nature Reviews Genetics (2021)
