
Abstract
Mesenchymal stem cells (MSCs) have multiple properties including anti-inflammatory and immunomodulatory effects in various disease models and clinical treatments. These beneficial effects, however, are sometimes inconsistent and unpredictable. For wider and proper application, scientists sought to improve MSC functions by engineering. We aimed to invent a novel method to produce synthetic biological drugs from engineered MSCs. We investigated the anti-arthritic effect of engineered MSCs in a collagen-induced arthritis (CIA) model. Biologics such as etanercept are the most successful drugs used in anti-cytokine therapy. Biologics are made of protein components, and thus can be theoretically produced from cells including MSCs. MSCs were transfected with recombinant minicircles encoding etanercept (trade name, Enbrel), which is a tumour necrosis factor α blocker currently used to treat rheumatoid arthritis. We confirmed minicircle expression in MSCs in vitro based on GFP. Etanercept production was verified from the conditioned media. We confirmed that self-reproduced etanercept was biologically active in vitro. Arthritis subsided more efficiently in CIA mice injected with mcTNFR2MSCs than in those injected with conventional MSCs or etanercept only. Although this novel strategy is in a very early conceptual stage, it seems to represent a potential alternative method for the delivery of biologics and engineering MSCs.
Similar content being viewed by others


Introduction
Tumour necrosis factor (TNF) α plays a critical role in the pathogenesis of rheumatoid arthritis (RA). Therefore, the development of a TNFα inhibitor has been vigorously pursued as an attractive treatment for RA1,2. Etanercept (Enbrel) is one of the most successful biological drugs against TNFα3,4. Etanercept is a synthetic protein drug that conjoins the p75 region of TNF receptor 2 (TNFR2) with the Fc portion of immunoglobulin5,6,7. Biological drugs are usually generated in vitro; however, they are some of the most expensive drugs in the market, which increases medical expenses for treatment8. The high cost is due to the recombination and purification processes required for human clinical applications9. Therefore, the search for an alternative treatment is ongoing.
Theoretically, biological drugs can also be produced in vivo because they are proteins formulated via translation and transcription of DNA sequences. In our previous studies, we attempted to elucidate a new method to deliver a biological drug into a living organism by delivering the DNA sequences encoding etanercept and abatacept9,10. Using minicircles encoding biological drugs, we confirmed the anti-inflammatory and immunosuppressive effects in vitro and in vivo. Minicircles are relatively small episomal vectors because the bacterial backbones are removed after gene amplification. The absence of the bacterial backbone and the non-integrating transfection system make minicircle vectors an attractive in vivo delivery tool for future clinical applications11. Minicircles are thought to be an ideal vehicle to transfer a gene of interest both in vitro and in vivo and have already been used in preclinical gene therapy research12,13. Minicircles were used in those attempts because their small size makes transfection more feasible and efficient without integration and the proteins they encode can be secreted in vivo with fewer safety issues11,12,14,15.
Mesenchymal stem cells (MSCs) are adult stem cells that have immunosuppressive effects against various autoimmune diseases including RA16,17,18. MSCs also have the ability to reconstruct damaged bone and cartilage19,20,21,22. MSCs may become a central material for regenerative medicine due to their immunomodulatory and anti-inflammatory effects and regenerative potency23,24. Although MSCs have several beneficial effects, some researchers claim that these effects are inconsistent and controversial25. Scientists are attempting to engineer MSCs in order to improve their function in various ways. It was reported that MSCs engineered with specific genes show improved curative effects26,27,28. To date, it remains difficult to transfer a foreign gene into MSCs24. Viruses are the main means by which genes are transferred into MSCs. However, the use of viruses can induce the integration of foreign genes, which is another hurdle for clinical applications. Therefore, an improved method to deliver a foreign gene into MSCs is required. Efficacy and safety are the main issues that need to be resolved for MSC engineering by gene transfer for clinical applications.
In this study, we attempted to generate engineered MSCs named mcTNFR2MSCs using a novel strategy. We generated a unique minicircle plasmid encoding etanercept (mcTNFR2). Using electroporation, we transfected MSCs with mcTNFR2 (mcTNFR2MSC). The generated mcTNFR2MSCs successfully produced etanercept as intended. mcTNFR2MSCs were superior at suppressing arthritis compared with conventional MSCs. We hope that the generation of mcTNFR2MSCs using minicircle vectors can make MSC-based treatment more applicable in the future.
Results
Scheme of the experimental concept and drug expression of the generated mcTNFR2
We designed and conducted our experiments following the concept showed in Fig. 1a. We generated mcTNFR2 by arabinose treatment. Then, the generated mcTNFR2 vector was transfected into MSCs to generate mcTNFR2MSCs. mcTNFR2MSCs were delivered to collagen-induced arthritis (CIA) mouse model by intraperitoneal injections to investigate the therapeutic effects on RA. First, minicircle production from parental plasmids was confirmed by gel electrophoresis (Fig. 1b). The generated mcTNFR2 had a size of 3 kb, almost half that of the parental plasmid. To confirm that the produced mcTNFR2 vector can synthesise and secrete soluble TNFR2-Fc protein (sTNFR2-Fc), human embryonic kidney 293 (HEK293T) cells were transfected with mcTNFR2 (mcTNFR2-293T). The isolated and concentrated sTNFR2-Fc was detected by immunoblotting (Fig. 1c). sTNFR2-Fc was strongly detected in the culture supernatant of mcTNFR2-transfected HEK 293 T cells(mcTNFR2-293T), but not in untreated cells or cells transfected with mock minicircle vectors (mcMock-293T). The amount of secreted drug was 1.8-fold higher than the known amount of commercial etanercept (250 ng/mL). We speculated that 231.25 ng/mL sTNFR2-Fc can be secreted per 5 × 106 cells. In summary, we generated mcTNFR2, and cells transfected with this vector synthesised the protein drug sTNFR2-Fc. The full-length gels and blots are included in supplements (Supplementary Fig. S1).

(a) Schematic diagram of experiments using mcTNFR2MSCs in a CIA mouse model. The concept is to produce synthetic biological drugs from mcTNFR2MSCs and to investigate the anti-arthritic effect of mcTNFR2MSCs in a CIA mouse model. (b) The representative gel image of parental and minicircle plasmids (mock and sTNFR2). Minicircles were produced by removing the bacterial backbone from the parental plasmid with arabinose. (c) Representative Western blot image and analysis to detect sTNFR2-Fc (n = 4). sTNFR2-Fc was detected in the conditioned media of mcTNFR2-transfected HEK293T cells. Etanercept-treated and untreated HEK293T cells were used as controls.
Generation of mcTNFR2MSCs by transfection with mcTNFR2
We verified that mcTNFR2 was functional in HEK293T cells; therefore, we attempted to transfect this vector into MSCs. To confirm the transfection efficacy of MSCs, mcMock and mcTNFR2 were transfected (Fig. 2a). GFP expression in MSCs transfected with mcTNFR2 (mcTNFR2MSCs) was similar to that in MSCs transfected with mcMock (mcMockMSCs). The transfection efficacy was higher for mcMockMSCs, but the overall transfection rate of mcTNFR2 was similar to that of mcMock (Fig. 2b). We examined if the transfected mcTNFR2MSCs could synthesise the protein drug in vitro (Fig. 2c). sTNFR2-Fc was highly detected in the culture supernatant of mcTNFR2MSCs; however, it was not detected in the culture supernatant of non-transfected MSCs or mcMockMSCs. After we confirmed the transfection of mcTNFR2 and expression of sTNFR2-Fc in mcTNFR2MSCs, we sought to confirm that transfection of a foreign gene or secretion of a protein drug did not disrupt the MSC phenotype. Expression of positive markers of MSCs (CD73 and CD105) in mcMockMSCs and mcTNFR2MSCs was similar to that in non-transfected MSCs (Fig. 2d). The levels of negative markers of MSCs (CD34 and CD45) were also similar to those in non-transfected MSCs. By analysing MSC marker expression, we confirmed that transfection and drug expression did not alter the MSC characteristics of mcTNFR2MSCs. Consequently, we confirmed that mcTNFR2MSCs were successfully generated and were able to maintain the characteristics of wild-type (WT) MSCs throughout the entire process. The generated mcTNFR2MSCs were also able to secrete the desired protein drug, sTNFR2-Fc, in vitro.

(a) Fluorescence images showing the expression of GFP at 48 h after transfection. Expression of GFP was similar in mcMockMSCs and mcTNFR2MSCs. Experiment was repeated 4–6 times and the representative image is shown. Scale bars: 500 μm. (b) The transfection efficacy of mcMock and mcTNFR2 was measured by counting GFP+ cells. (c) Concentration of sTNFR2-Fc in the conditioned media of MSCs transfected with minicircles encoding sTNFR2-Fc (mean ± SEM). The conditioned media were removed 48 h post-transfection and analysed by an ELISA. The experiment was repeated three times with each conditioned media. (d) Characterisation of mcTNFR2MSCs. The percentage of cells expressing each MSC marker was analysed. The analysed plots represent the fluorescence intensity and cell number per fluorescence channel. Negative control (isotype control) fluorescence is plotted on each panel as a black line. CD105-PE-Cy7 and CD73-FITC were used as positive markers. CD34-APC and CD45-PE were used as negative markers. Each histogram is a representative result of at least three MSC, mcMockMSC and mcTNFR2MSC samples (*P < 0.01; **P < 0.005; ***P < 0.001).
Functional analysis of the protein drug sTNFR2-Fc derived from mcTNFR2MSCs in vitro
RA joints are characterised by hyperplasia of fibroblast-like synoviocytes (FLSs), which is thought to be promoted by TNFα29. We verified the anti-migratory effect of sTNFR2-Fc secreted by mcTNFR2MSCs by performing scratch assay. Migration of RA-FLSs was observed upon TNFα stimulation. The migration of FLSs was significantly decreased by treatment with the culture media of mcTNFR2MSCs (Fig. 3a). The migration of FLSs treated with the culture media of non-engineered MSCs or commercial etanercept was also decreased, but not as much as that of FLSs treated with the culture media of mcTNFR2MSCs. The migration rate was measured (Fig. 3b). The number of migrating FLSs was highly increased in the negative control. The number of migrating FLSs was remarkably decreased upon treatment with the culture media of mcTNFR2MSCs. The invaded area in the scratch was measured. FLSs treated with the culture media of mcTNFR2MSCs failed to fill the scratched area compared with FLSs in the other conditions (Fig. 3c). In conclusion, we confirmed that mcTNFR2MSCs have superior suppressive effects on the migration of RA-FLSs compared with commercial etanercept and non-engineered MSCs.

(a) Representative microscopy image from scratch assay (n = 3). RA-FLS monolayers were scratched with a sterile pipette tip. Each well was treated with conditioned media of MSCs and HEK293T cells transfected with minicircles. Etanercept was used as a control. Phase-contrast microscopy images were acquired at 0 and 16 h after wounds were created. (b) Cell count of wound-invading cells. The number of invading RA-FLS cells was significantly higher in samples treated with the medium control and etanercept. (c) Percentage of invaded area. The tendency to migrate was calculated according to the following equation: percentage of invaded area = (1 − (wounded area at 16 h)/(wounded area at 0 h)) × 100. This statistical data represents the results of three individual experiments. (*P < 0.01; **P < 0.005; ***P < 0.001.)
In vivo injection of mcTNFR2MSCs inhibits the development of arthritis in CIA mice
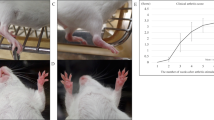
To investigate the effect of mcTNFR2MSCs in vivo, we injected CIA mice with mcTNFR2MSCs. After day 40, the arthritic severity score of CIA mice injected with mcTNFR2MSCs started to decrease and was continuously lower than in the other groups (Fig. 4a). The effect of mcTNFR2MSCs was histologically demonstrated by markedly decreased synovial hypertrophy in joints (Fig. 4b). Haematoxylin and eosin (H&E) staining showed diffuse cellular infiltration and pannus invasion in adjoining bone structures. CIA control mice showed massive damage in the tarsal bones. Joints in the mcTNFR2MSC-injected group showed less infiltration and invasion of the pannus compared with CIA control joints. Cartilage damage was confirmed by safranin O and toluidine blue staining. The level of cartilage damage was lower in the group injected with mcTNFR2MSCs than in the CIA control group. Joints in the MSC-injected group showed less cellular infiltration than those in the CIA control group; however, cartilage damage was similar in these two groups. The inflammation score was significantly lower in the mcTNFR2MSC-injected group than in the other groups (Fig. 4c). The joint destruction score was also significantly lower in the mcTNFR2MSC-injected group, while the joint destruction score in the MSC-injected group was about half of that in the CIA control group (Fig. 4d). These results confirmed that injection of mcTNFR2MSCs could reduce the symptoms of arthritis and that the therapeutic effect of mcTNFR2MSCs was better than that of non-engineered WT MSCs.

(a) Arthritis severity score of mice (n = 5 per group). MSCs and mcTNFR2MSCs were intraperitoneally injected three times. The arrows indicate the day of injection. (b) The representative histological images of cartilage and bone erosion. Joints were stained with H&E, safranin O and toluidine blue. Scale bars: 200 μm. (c) Inflammation score of histological data. The score was based on the extent of synovial hyperplasia and infiltration of leukocytes. (d) Joint destruction scores were based on cartilage loss estimated in samples stained with safranin O and toluidine blue as well as the extent of pannus formation. (*P < 0.01; **P < 0.005; ***P < 0.001).
In vivo detection of mcTNFR2MSCs
It has been reported that MSCs mostly migrate into the spleen by inflammatory cytokine-guided homing30. Therefore, we screened the spleen to validate the life-span of injected mcTNFR2MSCs in vivo. To examine the life-span of mcTNFR2MSCs, CIA mice were sacrificed at 3, 7, 15, and 30 days after the final mcTNFR2MSC infusion. GFP+ cells were frequently observed in the spleen from day 3, and expression of GFP started to increase on day 7 (Fig. 5a). GFP expression increased until day 15; however, it diminished and almost disappeared on day 30. The number of GFP+ cells suddenly increased between day 3 and day 7 (Fig. 5b). GFP+ cells slightly increased after day 7. On day 30, the number of GFP+ cells rapidly decreased to the level seen on day 3. The area of GFP positivity showed a similar pattern (Fig. 5c). Because the injected mcTNFR2MSCs originate from human, they were labelled using a specific antibody against human nuclei (h-Nuclei). h-Nuclei positivity in the spleen was observed only after 3 days (Fig. 5d). Expression was highest on day 7 and this was sustained until day 15. However, expression almost disappeared on day 30. We counted h-Nuclei+ cells and GFP+ cells to confirm the expression tendencies (Fig. 5e). The number of h-Nuclei+ cells started to increase earlier than the number of GFP+ cells (Fig. 5a and d). Expression of h-Nuclei was highest on day 7, which was about 1 week earlier than the peak in GFP positivity. Arthritic joints were also examined to determine if mcTNFR2MSCs migrated to the inflamed region (Fig. 5f). GFP+ cells were detected in joints even on day 3 after the final mcTNFR2MSC injection. Similar to the results for the spleen, GFP signals started to peak later than h-Nuclei signals. However, interestingly, co-localising signals of mcTNFR2MSCs were highly detected in joints on day 30. Based on our results, we conclude that the injected mcTNFR2MSCs populated the spleen after infusion. We confirmed that protein drug synthesis occurs after the migration of MSCs. Moreover, migration of mcTNFR2MSCs was detected in inflamed joints, which can explain the therapeutic effect in CIA mice.

(a) GFP expression of mcTNFR2MSCs in the spleen of CIA mice (n = 5 per time point) is shown by fluorescence. At 3, 7, 15, and 30 days after injection, the spleen was removed from CIA mice and analysed by immunofluorescence staining. Scale bars: 200 μm. (b) The number of GFP+ cells was counted in the spleen on each of these days after mcTNFR2MSC injection. (c) The percentage of the populated area was calculated by a computer program. (d) Time-course analysis of mcTNFR2MSCs in the spleen. At 3, 7, 15, and 30 days after mcTNFR2MSC injection, the spleen was removed from CIA mice and analysed by immunofluorescence staining with both anti-GFP and anti-h-Nuclei antibodies. Scale bars: 200 μm. (e) Analysis of numbers of GFP+ and h-Nuclei+ cells. (f) Time-course analysis of mcTNFR2MSCs in joints (n = 5 per time point). At 3, 7, 15, and 30 days after mcTNFR2MSC injection, joints were removed from CIA mice and analysed by immunofluorescence staining with anti-GFP and anti-h-Nuclei antibodies. Scale bars: 200 μm.
Anti-inflammatory and anti-osteoclastogenic effects of mcTNFR2MSCs
We confirmed that the injected mcTNFR2MSCs tended to migrate to the spleen. We hypothesised that they can influence the population of T lymphocytes in the spleen. Th17 is one of the major cell types that participate in the pathogenesis of RA; therefore, we investigated the population of Th17 cells. The Th17 cell population among splenocytes was analysed by fluorescence-activated cell sorting (Fig. 6a). The population of Th17 cells in the mcTNFR2MSC-injected group tended to decrease to almost half of that in the CIA control group and was similar to that in the WT group. We also confirmed that mcTNFR2MSCs were able to significantly decrease the Th17 cell population compared with WT MSCs (Fig. 6b). Next, we investigated the effect of mcTNFR2MSCs on osteoclastogenesis, which is involved in bone erosion and destruction in RA. Inflammatory cells, particularly the Th17 cell population, promote osteoclastogenesis in arthritis; therefore, we hypothesised that mcTNFR2MSCs would also suppress osteoclastogenesis. As expected, osteoclastogenesis was suppressed by conditioned media of mcTNFR2MSCs (Fig. 6c and d). These results suggest that mcTNFR2MSCs have a superior suppressive effect on the Th17 cell population and osteoclastogenesis compared with MSCs. In conclusion, we confirmed that mcTNFR2MSCs can alter the two major phenotypes involved in the pathogenesis of RA.

(a) Splenocytes of mice (n = 5 per group) were analysed by flow cytometry. The RORγt+ population of mcTNFR2MSC-treated mouse splenocytes was similar to that in WT mice. Flow cytometric data were analysed in triplicate. (b) Fewer RORγt+ cells were seen in mice administered mcTNFR2MSCs than in mice administered etanercept or MSCs. (c) Osteoclast differentiation was induced from WT bone marrow cells (n = 5). Cells were treated with conditioned media of MSCs and HEK293T cells transfected with the minicircle in vitro. Scale bars: 200 μm. (d) Osteoclasts were counted in three independent experiments. (*P < 0.01; **P < 0.005; ***P < 0.001).
Discussion
MSCs are reported to have immunosuppressive effects against various autoimmune diseases. However, there remain several controversies regarding the therapeutic and immunosuppressive effects of MSCs. Various studies investigated the anti-inflammatory role of MSCs in RA using CIA mice. Djouad et al. reported that MSCs do not have any effects in CIA mice31. However, various studies report beneficial effects of MSCs in CIA mice32,33,34,35,36,37. The use of various cell strains and differences in the infused cell number or the administration route make it difficult to compare and interpret the inconsistent results of previous studies38.
Several groups attempted to modify and improve the curative effects of MSCs because of these controversies. MSCs were transduced with viral vectors, including the genes encoding human proteins39 such as CTLA4Ig, BMP2 and IL-1028,39,40,41. Even though some viral vectors were confirmed to be safe for clinical use, an improved method that can entirely avoid the integration of viral vectors is required. In this study, we generated mcTNFR2MSCs using minicircle vectors containing the sequence of a human biological drug. The efficacy of minicircle vectors was previously confirmed by several researchers9,10,28,42,43,44. We previously confirmed that minicircle vectors encoding biological drugs (i.e., CTLA4Ig and tocilizumab) can repress the immune reaction in CIA mice9,10. Mun et al. reported that MSCs transfected with minicircles encoding CXCR4 are more likely to migrate to the injury site44. However, the effect of minicircle-transfected MSCs on CIA mice has not been reported. To our knowledge, this is the first study to confirm the therapeutic effect of minicircle-transfected MSCs in a RA animal model, namely, CIA.
A variety of cytokines act in the progression and pathogenesis of RA. Among several secreted cytokines, TNFα is thought to be a major player. It is also reported that the coexistence of TNFα can alter the action of MSCs31. MSCs are unable to suppress the proliferation of T lymphocytes in allogenic responses in the presence of TNFα. The concentration of an inflammatory cytokine (IL-6) is increased when MSCs are injected in the presence of TNFα. Therefore, MSCs that can block the action of TNFα can be useful for future applications.
Genes can be delivered into MSCs by non-viral methods, including electroporation and chemical transfection. MSCs transfected using Lipofectamine had a high viability; however, the transfection efficacy was lower than that achieved by electroporation. Considering both cell viability and transfection efficiency, we decided to deliver our vector by electroporation. In previous studies of gene delivery into MSCs, gene delivery was poor using chemical transfection, but was about 25–42% using electroporation, which is relatively high27. However, with the combination of minicircle vectors and electroporation, we were able to achieve a higher transfection efficacy of about 45–50%. The transfection efficacy was lower than in previous studies using viral vectors; however, it was still slightly higher than that in previous studies using electroporation24,45. It was recently reported that the microporation of minicircles can increase the transfection efficiency to 63–66%44. Mun et al. also reported that minicircles can be delivered under milder conditions because of their relatively small size and that cell viability is maintained at up to 96%. The transfection rate is still lower than that achieved using viral delivery methods; however, the integration of viral vectors is one reason why researchers are searching for an alternative tool for transfection.
After transfection, we confirmed that the original characteristics of MSCs were not lost due to the procedure or the soluble drug protein secreted by the minicircles. We also demonstrated that CD marker expression was similar in non-engineered MSCs and mcTNFR2MSCs. However, we cannot conclude that MSCs are exactly the same before and after engineering. We also could not confirm that TNFR2 and commercial etanercept were exactly the same, although the generated TNFR2 bound to an anti-etanercept antibody. This problem may be due to differences between batches of commercial etanercept. We think that the anti-arthritic effects of mcTNFR2MSCs in CIA mice provide indirect evidence of the existence of a synthetic protein similar to commercial etanercept.
Synovial hyperplasia is a major feature of RA. It is associated with pro-inflammatory cytokines including IL-1ß and TNFα29,46,47. Synoviocytes actively proliferate and migrate to adjacent structures. The migration of FLSs is an important pathophysiological phenotype, resulting in joint destruction and consequent dysfunction. Synoviocytes proliferate in a dose-dependent manner when treated with TNFα48. The Boyden chamber method was used to demonstrate that TNFα can induce the migration and invasion of FLSs in vitro49. We tested whether synthetic sTNFR2-Fc inhibited the migration of synoviocytes using an in vitro scratch assay. sTNFR2-Fc suppressed the migration of synoviocytes. There are many TNFα-mediated pathological activities other than cell migration. We extended these in vitro results to an in vivo animal model. The detection of mcTNFR2MSCs was highest between day 7 and day 15 in vivo. We also previously confirmed that minicircle expression can be maintained for 10 days. Based on these results, mcTNFR2MSCs were intraperitoneally injected with an interval of 10 days. Injected mcTNFR2MSCs had an anti-arthritic tendency, but there was no statistically significant difference in the arthritic score between mice injected with MSCs and those injected with mcTNFR2MSCs. However, tissue staining showed a remarkable difference between MSCs and mcTNFR2MSCs.
Cytokines and chemokines are associated with MSC migration. Previous studies attempted to show the migration of MSCs; however, divergent results were obtained due to the use of different MSC delivery methods in these studies. It was also reported that MSCs are detected in the spleen after intravenous and intraperitoneal delivery but not in joints using these protocols16,50,51,52. In contrast with these previous reports, we detected a small population of mcTNFR2MSCs in joints as well as in the spleen. Migrated human mcTNFR2MSCs expressing GFP were detectable for up to 30 days. The spleen and joints are full of vascular structures and are an important source of chemokines in CIA mice, which might explain why mcTNFR2MSCs tend to migrate into the spleen and joints. Interestingly, h-Nuclei positivity was detected slightly earlier than GFP positivity in the spleen. This can be explained by the initial migration of mcTNFR2MSCs and their subsequent production of TNFR2-Fc. Positive staining of h-Nuclei and GFP in the spleen and joints provides indirect evidence of the detection of mcTNFR2MSCs and sTNFR2-Fc in vivo.
Th17 cells and osteoclasts are two major cell types with roles in the pathogenesis of RA53,54,55,56. A large number of mcTNFR2MSCs migrated to the spleen; therefore, we investigated the effect of mcTNFR2MSCs on the Th17 cell population among splenocytes. We confirmed the population of RORγt+ cells among splenocytes isolated from mice in each group. Our results correlated with those of a study by Wang et al., which confirmed the inhibitory role of MSCs in the regulation of RORγ mRNA and protein expression57. Th17 cells stimulate bone resorption by inducing RANKL, which activates osteoclasts58. Osteoclasts are induced by treating monocyte-derived macrophages with RANKL in vitro. Macrophages are a major cell type that secretes TNFα59,60. When in vitro activated macrophages were treated with the culture supernatant of mcTNFR2MSCs, osteoclast formation was reduced. This inhibitory effect is thought to be due to the blockage of TNFα secretion by monocytes and macrophages, even though there was no additional TNFα treatment. However, the relatively high amount of sTNFR2-Fc secreted by HEK293T cells completely inhibited osteoclast formation. Several studies reported that TNFα synergises the effect of RANKL on osteoclastogenesis61,62,63. Lam et al. reported that TNFα can dramatically enhance osteoclast formation, even when RANKL alone cannot62. Kobayashi et al. also reported that TNFα stimulates gene expression of RANK in osteoclast precursor cells63. Therefore, it is thought that the complete inhibition of osteoclasts can be due to relatively increased blockade of TNFα.
MSCs are sometimes dependent on the environment. In an inflammatory environment, MSCs may act as an inflammation agonist and focus on surviving in the harsh conditions. The anti-inflammatory effects of MSCs are reduced or abrogated by their efforts to survive. If we can equip naïve MSCs with anti-inflammatory weapons, they can focus on eliciting anti-inflammatory effects and immune modulation.
In conclusion, etanercept-synthesising MSCs efficiently ameliorated arthritis in an experimental mouse model. Compared with non-engineered MSCs, mcTNFR2MSCs had a superior anti-arthritic activity due to their secretion of etanercept. Although this strategy is at the proof-of-concept stage, it represents a potential alternative method for the delivery of biologics through mcTNFR2MSCs and cell-based therapy.
Methods
Cell culture
A bone marrow-derived human MSC (hMSC) line (BM025SS13) was retrieved from The Catholic Institute of Cell Therapy, South Korea. hMSCs were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Gibco Life Technology, Gaithersburg, MD, USA) supplemented with 20% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco Life Technology). RA-FLSs were maintained in DMEM supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. HEK293T cells were maintained in DMEM supplemented with 7.5% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin. Cells were maintained at 37 °C in 5% CO2.
Production of minicircles
The minicircles (mcMock and mcTNFR2) were produced as described by Kay et al.12. Briefly, Escherichia coli ZYCY10P3S2T cells transformed with the parental plasmids were grown overnight at 37 °C in Terrific Broth containing 50 μg/mL kanamycin. A single colony was grown for 8 h in 2 mL of Luria-Bertani (LB) broth containing kanamycin. The cultures were combined with LB broth containing 0.02% l-(+)-arabinose and incubated at 30 °C for 5 h. Minicircle DNA was isolated using the Nucelobond Xtra Midi kit (Macherey-Nagel, Duren, Germany).
Minicircle transfection
HEK293T cells were transfected with the minicircle vectors using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. MSCs at passage 5 were transfected with minicircle vectors using an electroporation system (Neon Transfection System) according to the manufacturer’s instructions. MSCs (5 × 105 cells) were transfected with mcTNFR2 to generate sTNFR2-Fc-expressing cells. Expression of GFP in the cells was assessed by fluorescence microscopy at 48 h post-transfection. The pulse voltage and width conditions were based on the information provided by the Neon Transfection System regarding cell viability and transfection efficacy.
Detection of sTNFR2-Fc
The conditioned media of transfected HEK293T cells was collected 48 h after transfection. The media was incubated with protein A/G PLUS-agarose beads (Santa Cruz, CA, USA). The captured proteins were detached from the beads and detected by western blotting with an anti-etanercept antibody (Agrisera AB, Sweden). In total, 250ng of Enbrel (AMGEN, CA, USA) in PBS was also loaded onto the gel as a positive control.
Enzyme-linked immunosorbent assay (ELISA)
To analyse the amount of sTNFR2-Fc protein expressed by transfected cells, the culture media of MSCs transfected with the indicated minicircle vectors was analysed by an enzyme-linked immunosorbent assay (ELISA) at 48 h post-transfection. The levels of sTNFR2-Fc were quantified using a human sTNFR (80 kDa) Platinum ELISA (Affymetrix, San Diego, CA, USA), according to the manufacturer’s instructions. After applying stop solution, absorbance was measured at 450 nm.
Characterisation of MSC markers
The MSC markers reported by Choi et al.64 were examined. The immunophenotypes of non-transfected MSCs, mcMockMSCs and mcTNFR2MSCs at passage 5 were determined by flow cytometric analysis. The cells were incubated with an allophycocyanin (APC)-conjugated mouse anti-human CD34 antibody (Affymetrix, 17-0349), a phycoerythrin (PE)-conjugated mouse anti-human CD45 antibody (Affymetrix, 12-9459), a fluorescein isothiocyanate (FITC)-conjugated mouse anti-human CD73 antibody (Affymetrix, 11-0739) and a PE-cyanine 7 (Cy7)-conjugated mouse anti-human CD105 antibody (Affymetrix, 25-1057). The cells were permeabilised using a Foxp3 Transcription Factor Staining Buffer Set (Affymetrix, 00-5523). After cellular staining, cell populations were identified and estimated using an LSR Fortessa Cell Analyser (BD Biosciences, San Jose, CA, USA). The acquired data were analysed using FlowJo V10 single cell analysis software (TreeStar Inc., Ashland, OR, USA).
Scratch assay of cell migration
The conditioned media of MSCs and HEK293T cells transfected with or without minicircles was collected at 48 h post-transfection. RA-FLSs were seeded onto 6-well plates (Corning-Costar, Tokyo, Japan). The cell monolayer was scratched in a straight line with a P200 pipette tip. Cells were cultured in 2 mL of fresh DMEM supplemented with TNFα (20 ng/mL) or with DMEM alone as a negative control. To examine whether minicircle-derived drugs affect the migration of RA-FLSs, cells were treated with conditioned media of mcTNFR2MSCs for 16 h in the migration assay. In addition, the conditioned media of HEK293T cells transfected with mcTNFR2 and etanercept was used as a positive control and fresh MSC culture media was used as a negative control. Phase-contrast microscopy images were acquired at 0 and 16 h after wounds were created. Using ImageJ software, the invaded area was calculated as follows: percentage of invaded area = (1 − (wounded area at 16 h)/(wounded area at 0 h)) × 100.
Ethics
All procedures involving animals were in accordance with the Laboratory Animals Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the Guidelines and Policies for Rodent Experimentation provided by the Institutional Animal Care and Use Committee of the College of Medicine of The Catholic University of Korea. This study protocol was approved by the Institutional Review Board of The Catholic University of Korea (CUMC-2015-0084-01).
Induction of arthritis and evaluation of disease severity
Bovine type II collagen (CII; Chondrex, Redmond, WA, USA) was dissolved in 0.1 M acetic acid to a concentration of 2 mg/mL at 4 °C overnight. CII was emulsified at a ratio of 1:1 with complete Freund’s adjuvant (CFA; Chondrex) containing 2 mg/mL heat-killed Mycobacterium tuberculosis. Female DBA1/J mice (n = 5 per group, 6 weeks old; OrientBio, Seongnam, Korea) were intradermally injected at day 0 with 100 mL of the CII/CFA emulsion. The same concentration of CII emulsified with incomplete Freund’s adjuvant (Chondrex) was injected into mice as a booster immunisation 21 days after the first immunisation. Five mice were allocated to the WT group and five mice were each allocated to the CIA, etanercept-treated, MSC-treated and transfected MSC-treated (mcMock and mcTNFR2) groups. The severity of arthritis was monitored and scored as described previously65. The severity of inflamed paws was observed for 72 days after the first immunisation.
In vivo delivery of mcTNFR2MSCs
mcTNFR2MSCs (5 × 106 cells) were resuspended in 150 μL of PBS. Cells were delivered by intraperitoneal injection. Cells were injected three times at 20, 30, and 40 days after the first immunisation. Non-engineered WT MSCs and etanercept were used as controls. Mice were administered 25 μg of etanercept (Enbrel; Pfizer, New York, NY, USA) three times per week for 3 weeks after the first immunisation.
Histological evaluation of arthritis
The hind limbs of each mouse (n = 5 per group) were removed, fixed in 4% paraformaldehyde and decalcified in 10% (w/v) EDTA, and the samples were embedded in paraffin. Sections (4 μm thick) were stained with H&E, safranin O or toluidine blue. The inflammation and joint destruction scores were determined by three individual researchers using the procedure of Huckel et al.66. The inflammation score of each group was determined according to the severity of infiltration and pannus formation. The destruction score was determined based on cartilage and bone loss.
Immunofluorescence analysis
Sections of mouse spleen (n = 5 per time point) were fixed in pre-cooled acetone and incubated with 0.3% hydrogen peroxide prepared in 10% methanol for 15 min. After washing with PBS, the sections were blocked using an Avidin/Biotin Blocking Kit (Vector Laboratories, CA, USA). After incubation, blocking was performed following the manufacturer’s instructions of Mouse on Mouse Fluorescein Kit (Vector Laboratories). Sections were incubated with a mouse anti-h-Nuclei monoclonal antibody (Millipore, CA, USA) overnight. The next day, sections were incubated with Alexa Fluor 594-conjugated goat anti-mouse IgG (Invitrogen) for 1 h. After DAPI staining, sections were observed with a confocal microscope (Carl Zeiss LSM700, Prenzlauer, Berlin, Germany).
Th17 cell population
To detect Th17 cells in the mouse spleen, splenocytes were isolated from each group (n = 5 per group). The cells were incubated with an APC-conjugated rat anti-mouse CD4 antibody (BD Biosciences, 561091) and permeabilised using a Foxp3 Transcription Factor Staining Buffer Set (Affymetrix, 00-5523). The Th17-specific marker RORγt was detected using a PE-conjugated anti-human/mouse RORγt antibody (Affymetrix, 12-6988). After cellular staining, cell populations were identified and estimated using an LSR Fortessa Cell Analyser (BD Biosciences). The acquired data were analysed using FlowJo V10 single cell analysis software (TreeStar Inc.). Cells were first gated for the CD4+ proportion and then analysed for expression of RORγt.
In vitro osteoclast differentiation
Monocytes were isolated from bone marrow of WT mice (n = 5). Samples were treated with ACK buffer to eliminate red blood cells, washed with phosphate-buffered saline and filtered with a strainer. Cells were seeded and maintained in α-MEM supplemented with 10% FBS. The next day, cells were counted, and 3 × 105 cells were stimulated with 10 ng/mL macrophage colony-stimulating factor (M-CSF; Peprotech, Oak Park, CA, USA) for 48 h. Then, a second stimulation was provided by treatment with 30 ng/mL RANKL (Peprotech) and M-CSF. Cells were treated with conditioned media of transfected MSCs or HEK293T cells, adding the conditioned media of non-transfected MSCs as required. After 72 h, differentiated osteoclasts were stained for TRAP following the manufacturer’s guide. Stained cells were counted.
Statistical analysis
All experiments were repeated three or more times. The results are shown as mean and standard error of the mean. Error bars represent the standard error of the mean. Statistical analysis was performed and graphs were drawn using GraphPad Prism 5 (GraphPad). The T-test was applied to analyse non-parametric quantitative datasets, and the one-tailed p-value was calculated. *P < 0.01; **P < 0.005; and ***P < 0.001 indicated statistical significance.
Additional Information
How to cite this article: Park, N. et al. Etanercept-Synthesising Mesenchymal Stem Cells Efficiently Ameliorate Collagen-Induced Arthritis. Sci. Rep. 7, 39593; doi: 10.1038/srep39593 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Bluml, S., Scheinecker, C., Smolen, J. S. & Redlich, K. Targeting TNF receptors in rheumatoid arthritis. Int Immunol. 24, 275–281 (2012).
Faustman, D. & Davis, M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nar Rev Drug Discov. 9, 482–493 (2010).
Olsen, N. J. & Stein, C. M. New drugs for rheumatoid arthritis. N Engl J Med. 350, 2167–2179 (2004).
Van Vollenhoven, R. F. Unresolved issues in biologic therapy for rheumatoid arthritis. Nat Rev Rheumatol. 7, 205–215 (2011).
Moreland, L. W. Soluble tumor necrosis factor receptor (p75) fusion protein (ENBREL) as a therapy for rheumatoid arthritis. Rheum Dis Clin N Am. 24, 579–591 (1998).
Tsimberidou, A. et al. Recombinant human soluble tumor necrosis factor (TNF) receptor (p75) fusion protein Enbrel in patients with refractory hematologic malignancies. Cancer Chemoth Pherm. 50, 237–242 (2002).
Weinblatt, M. et al. A trial of etanercept, a recombinant tumor necrosis factor receptor: Fc fusion protein, in patients with rheumatoid arthritis receiving methotrexate. New Engl J Med. 340, 253–259 (1999).
Brennan, A. et al. Modelling the cost effectiveness of TNF-alpha antagonists in the management of rheumatoid arthritis: results from the British Society for Rheumatology Biologics Registry. Rheumatology. 46, 1345–1354 (2007).
Yi, H. et al. A new strategy to deliver synthetic protein drugs: self-reproducible biologics using minicircles. Scientific Reports. 4, 5961 (2014).
Rim, Y. A. et al. Self in vivo production of a synthetic biological drug CTLA4Ig using a minicircle vector. Scientific Reports. 4, 6935 (2014).
Chen, Z. Y., He, C. Y., Ehrhardt, A. & Kay, M. A. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo . Mol Ther. 8, 495–500 (2003).
Kay, M. A., He, C. Y. & Chen, Z. Y. A robust system for production of minicircle DNA vectors. Nat Biotechnol. 28, 1287–1289 (2010).
Gill, D. R., Pringle, I. A. & Hyde, S. C. Progress and prospects: the design and production of plasmid vectors. Gene Ther. 16, 165–171 (2009).
Chen, Z. Y., He, C. Y. & Kay, M. A. Improved production and purification of minicircle DNA vector free of plasmid bacterial sequences and capable of persistent transgene expression in vivo . Hum Gene Ther. 16, 126–131 (2005).
Gracey Maniar, L. E. et al. Minicircle DNA vectors achieve sustained expression reflected by active chromatin and transcriptional level. Mol Ther. 21, 131–138 (2013).
Mao, F. et al. Immunosuppressive effects of mesenchymal stem cells in collagen-induced mouse arthritis. Inflamm Res. 59, 219–225 (2010).
Augello, A., Tasso, R., Negrini, S. M., Cancedda, R. & Pennesi, G. Cell therapy using allogeneic bone marrow mesenchymal stem cells prevents tissue damage in collagen-induced arthritis. Arthritis Rheum. 56, 1175–1186 (2007).
Hoogduijn, M. J. et al. The immunomodulatory properties of mesenchymal stem cells and their use for immunotherapy. Int immunopharmacol. 10, 1496–1500 (2010).
Bruder, S. P. et al. Bone regeneration by implantation of purified, culture‐expanded human mesenchymal stem cells. J Orthop Res. 16, 155–162 (1998).
Gao, J. et al. Tissue-engineered fabrication of an osteochondral composite graft using rat bone marrow-derived mesenchymal stem cells. Tissue Eng. 7, 363–371 (2001).
Murphy, J. M., Fink, D. J., Hunziker, E. B. & Barry, F. P. Stem cell therapy in a caprine model of osteoarthritis. Arthritis Rheum. 48, 3464–3474 (2003).
Noel, D., Djouad, F. & Jorgense, C. Regenerative medicine through mesenchymal stem cells for bone and cartilage repair. Curr Opin Investig Drugs (London, England: 2000). 3, 1000–1004 (2002).
Aggarwal, S. & Pittenger, M. F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 105, 1815–1822 (2005).
Wang, W., Xu, X., Li, Z., Lendlein, A. & Ma, N. Genetic engineering of mesenchymal stem cells by non-viral gene delivery. Clin Hemorheol Micro. 58, 19–48 (2014).
Fehrer, C. & Lepperdinger, G. Mesenchymal stem cell aging. Exp Gerontol. 40, 926–930 (2005).
Dai, F. et al. hCTLA4-gene modified human bone marrow-derived mesenchymal stem cells as allogeneic seed cells in bone tissue engineering. Tissue Eng. 12, 2583–2590 (2006).
Jorgensen, C., Djouad, F., Apparailly, F. & Noel, D. Engineering mesenchymal stem cells for immunotherapy. Gene Ther. 10, 928–931 (2003).
Wu, S. et al. Ultrasound-targeted stromal cell-derived factor-1-loaded microbubble destruction promotes mesenchymal stem cell homing to kidneys in diabetic nephropathy rats. Int J NanoMed. 9, 5639–5651 (2014).
Hashizume, M., Hayakawa, N. & Mihara, M. IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology. 47, 1635–1640 (2008).
Sohni, A. & Verfaillie, C. M. Mesenchymal stem cells migration homing and tracking. Stem Cells Int. 2013, 130763 (2013).
Djouad, F. et al. Reversal of the immunosuppressive properties of mesenchymal stem cells by tumor necrosis factor alpha in collagen-induced arthritis. Arthritis Rheum. 52, 1595–1603 (2005).
Park, K. H. et al. Treatment of Collagen-Induced Arthritis Using Immune Modulatory Properties of Human Mesenchymal Stem Cells. Cell Transplant. 25, 1057–1072 (2016).
Wu, C. C., Liu, F. L., Sytwu, H. K., Tsai, C. Y. & Chang, D. M. CD146+ mesenchymal stem cells display greater therapeutic potential than CD146- cells for treating collagen-induced arthritis in mice. Stem Cell Res Ther. 7, 23 (2016).
Chen, M. et al. Adoptive transfer of human gingiva-derived mesenchymal stem cells ameliorates collagen-induced arthritis via suppression of Th1 and Th17 cells and enhancement of regulatory T cell differentiation. Arthritis Rheum. 65, 1181–1193 (2013).
Zhao, F. T., Yin, J. W., Liu, Q. F. & Xu, S. F. Effect of bone marrow mesenchymal stem cell transplant on synovial proliferation in rats with type II collagen-induced arthritis. Exp Clin Transplant. 11, 352–357 (2013).
Chen, B. et al. Flk-1+ mesenchymal stem cells aggravate collagen-induced arthritis by up-regulating interleukin-6. Clin Exp Immunol. 159, 292–302 (2010).
Augello, A., Tasso, R., Negrini, S. M., Cancedda, R. & Pennesi, G. Cell therapy using allogeneic bone marrow mesenchymal stem cells prevents tissue damage in collagen-induced arthritis. Arthritis Rheum. 56, 1175–1186 (2007).
Sullivan, C. et al. Allogeneic Murine Mesenchymal Stem Cells: Migration to Inflamed Joints in vivo and Amelioration of Collagen Induced Arthritis When Transduced to Express CTLA4Ig. Stem Cells and development. 22, 3203–3213 (2013).
Zhang, X. L. et al. Immunomodulatory and osteogenic differentiation effects of mesenchymal stem cells by adenovirus-mediated coexpression of CTLA4Ig and BMP2. J Orthop Res. 26, 314–321 (2008).
Dai, F. et al. hCTLA4-gene modified human bone marrow - Derived mesenchymal stem cells as allogeneic seed cells in bone tissue engineering. Tissue Eng. 12, 2583–2590 (2006).
Choi, J. J. et al. Mesenchymal stem cells overexpressing interleukin-10 attenuate collagen-induced arthritis in mice. Clin Exp Immunol. 153, 269–276 (2008).
Chen, Z. Y., He, C. Y., Ehrhardt, A. & Kay, M. A. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo . Mol Ther. 8, 495–500 (2003).
Huang, M. et al. Novel Minicircle Vector for Gene Therapy in Murine Myocardial Infarction. Circulation. 120, S230–S237 (2009).
Mun, J. Y., Shin, K. K., Kwon, O., Lim, Y. T. & Oh, D. B. Minicircle microporation-based non-viral gene delivery improved the targeting of mesenchymal stem cells to an injury site. Biomaterials. 101, 310–320 (2016).
Sprangers, A. J., Freeman, B. & Ogle, B. M. Electroporation can efficiently transfect hESC-derived mesenchymal stem cells without inducing differentiation. Open Stem Cell J. 3, 62–66 (2011).
Di Giovine, F. S., Nuki, G. & Duff, G. W. Tumour necrosis factor in synovial exudates. Ann Rheum Dis. 47, 768–772 (1988).
Saxne, T. et al. Detection of tumor necrosis factor alpha but not tumor necrosis factor beta in rheumatoid arthritis synovial fluid and serum. Arthritis Rheum. 31, 1041–1045 (1988).
Youn, J. et al. Regulation of TNF-alpha-mediated hyperplasia through TNF receptors, TRAFs, and NF-kappaB in synoviocytes obtained from patients with rheumatoid arthritis. Immunol Lett. 83, 85–93 (2002).
Liang, L. et al. Inhibitory effects of niclosamide on inflammation and migration of fibroblast-like synoviocytes from patients with rheumatoid arthritis. Inflamm Res. 64, 225–233 (2015).
MacDonald, G. I., Augello, A. & De Bari, C. Role of mesenchymal stem cells in reestablishing immunologic tolerance in autoimmune rheumatic diseases. Arthritis Rheum. 63, 2547–2557 (2011).
Choi, J. J. et al. Mesenchymal stem cells overexpressing interleukin-10 attenuate collagen-induced arthritis in mice. Clin Exp Immunol. 153, 269–276 (2008).
Gonzalez, M. A., Gonzalez-Rey, E., Rico, L., Buscher, D. & Delgado, M. Treatment of experimental arthritis by inducing immune tolerance with human adipose-derived mesenchymal stem cells. Arthritis Rheum. 60, 1006–1019 (2009).
Weaver, C. T., Hatton, R. D., Mangan, P. R. & Harrington, L. E. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 25, 821–852 (2007).
Bettelli, E., Korn, T. & Kuchroo, V. K. Th17: the third member of the effector T cell trilogy. Curr Opin Immunol. 19, 652–657 (2007).
Stockinger, B. & Veldhoen, M. Differentiation and function of Th17 T cells. Curr Opin Immunol. 19, 281–286 (2007).
Kato, H., Endres, J. & Fox, D. A. The roles of IFN-gamma versus IL-17 in pathogenic effects of human Th17 cells on synovial fibroblasts. Mod Rheumatol. 23, 1140–1150 (2013).
Wang, Q. et al. The allogeneic umbilical cord mesenchymal stem cells regulate the function of T helper 17 cells from patients with rheumatoid arthritis in an in vitro co-culture system. BMC Musculoskelet Disord. 13, 249 (2012).
Page, G. & Miossec, P. RANK and RANKL expression as markers of dendritic cell-T cell interactions in paired samples of rheumatoid synovium and lymph nodes. Arthritis Rheum. 52, 2307–2312 (2005).
Flynn, J. L. et al. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 2, 561–572 (1995).
Pfeffer, K. et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 73, 457–467 (1993).
Fuller, K., Murphy, C., Kirstein, B., Fox, S. W. & Chambers, T. J. TNFalpha potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology. 143, 1108–1118 (2002).
Lam, J. et al. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 106, 1481–1488 (2000).
Kobayashi, K. et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 191, 275–286 (2000).
Choi, E. et al. Transplantation of CTLA4Ig gene-transduced adipose tissue-derived mesenchymal stem cells reduces inflammatory immune response and improves Th1/Th2 balance in experimental autoimmune thyroiditis. J Gene Med. 13, 3–16 (2011).
Ju, J. et al. Oral administration of type-II collagen suppresses IL-17-associated RANKL expression of CD4+ T cells in collagen-induced arthritis. Immunol Lett. 117, 16–25 (2008).
Huckel, M. et al. Attenuation of murine antigen-induced arthritis by treatment with a decoy oligodeoxynucleotide inhibiting signal transducer and activator of transcription-1 (STAT-1). Arthritis Res Ther. 8, R17 (2006).
Acknowledgements
This work was supported by a grant of the Korea Healthcare Technology R&D project, Ministry for Health, Welfare & Family Affairs, Republic of Korea (HI16C2177).
Author information
Authors and Affiliations
Contributions
N.P. designed and performed the study, analysed the data and wrote the manuscript. Y.A.R. analysed the data and wrote the manuscript. J.K. performed the cell culture. H.J. obtained and stained the tissues. N.P., Y.A.R., H.Y., Y.K., Y.J., S.M.J., J.L., S.K.K. and S.H.P. analysed the data. J.H.J. designed and supervised the study, analysed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Park, N., Rim, Y., Jung, H. et al. Etanercept-Synthesising Mesenchymal Stem Cells Efficiently Ameliorate Collagen-Induced Arthritis. Sci Rep 7, 39593 (2017). https://doi.org/10.1038/srep39593
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep39593
This article is cited by
-
Effects of stepwise administration of osteoprotegerin and parathyroid hormone-related peptide DNA vectors on bone formation in ovariectomized rat model
Scientific Reports (2024)
-
Research progress of engineered mesenchymal stem cells and their derived exosomes and their application in autoimmune/inflammatory diseases
Stem Cell Research & Therapy (2023)
-
TNFα/TNFR2 signaling pathway: an active immune checkpoint for mesenchymal stem cell immunoregulatory function
Stem Cell Research & Therapy (2020)
-
Comparative study between human mesenchymal stem cells and etanercept as immunomodulatory agents in rat model of rheumatoid arthritis
Immunologic Research (2020)
-
Immunomodulatory plasticity of mesenchymal stem cells: a potential key to successful solid organ transplantation
Journal of Translational Medicine (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
