
Abstract
According to the nearly-neutral theory of evolution, the relative strengths of selection and drift shift in favour of drift at small population sizes. Numerous studies have analysed the effect of bottlenecks and small population sizes on genetic diversity in the MHC, which plays a central role in pathogen recognition and immune defense and is thus considered a model example for the study of adaptive evolution. However, to understand changes in genetic diversity at loci under selection, it is necessary to compare the genetic diversity of a population before and after the bottleneck. In this study, we analyse three fragments of the MHC DQA gene in woolly mammoth samples radiocarbon dated to before and after a well-documented bottleneck that took place about ten thousand years ago. Our results indicate a decrease in observed heterozygosity and number of alleles, suggesting that genetic drift had an impact on the variation on MHC. Based on coalescent simulations, we found no evidence of balancing selection maintaining MHC diversity during the Holocene. However, strong trans-species polymorphism among mammoths and elephants points to historical effects of balancing selection on the woolly mammoth lineage.
Similar content being viewed by others
Introduction
Small populations face an increased risk of extinction due to loss of genetic diversity, which is associated with inbreeding depression and loss of adaptive variation1. Inbreeding can lead to unmasking of deleterious alleles, while a decreased adaptive potential affects the population’s ability to adapt to environmental changes or to respond to pathogens2.
In vertebrates, the capability to resist pathogens is determined by the variability of genes of the major histocompatibility complex (MHC)3. The MHC encodes proteins that are responsible for antigen recognition and initiation of an immune response. In order to recognize and bind diverse pathogens, the expressed loci of MHC are highly polymorphic and it is thought that this diversity is maintained by a pathogen-mediated balancing selection4,5. Among other factors, selection pressure and investment in immune defense are associated with environmental and climatic conditions6,7. It has been shown that pathogen and parasite loads are lower in high latitude, cold and arid environments and under increased solar radiation8,9,10.
What effect a bottleneck has on MHC diversity has been a long-standing topic of discussion in research on natural selection11. There are two basic scenarios: selection either maintains diversity throughout the bottleneck, or the diversity is lost due to the effects of genetic drift12. From a conservation perspective, understanding this is important since low MHC diversity is associated with decreased ability to resist pathogens13,14. Thus, populations with few MHC alleles are more susceptible to infectious diseases due to decreased heterozygosity15,16 or absence of a resistant allele17,18.
A meta-analysis of studies on the effect of bottlenecks on MHC polymorphism12 supports the latter scenario and shows that in most of the studied populations MHC diversity decreased as a consequence of the bottlenecks. However, there are examples where, in contrast to neutral diversity, MHC polymorphism appears to have been maintained during bottlenecks19,20, including endangered species with small population sizes21,22.
In recent years, however, analyses of DNA from historical samples have been applied to directly study pre- and post-bottleneck MHC diversity in prairie-chickens23, peary caribou24 and New Zealand passerines25. These studies suggest that balancing selection is not only unable to maintain MHC diversity throughout a bottleneck, but in prairie-chickens and passerines the decrease of variability in MHC loci was more profound than in neutral markers (i.e. microsatellites).
To date, no studies have examined changes in MHC diversity leading up to a species’ extinction. In this study, we have used ancient DNA from the woolly mammoth (Mammuthus primigenius) to study the change in diversity in a MHC locus as the species went through a bottleneck and subsequently persisted at a small population size until its extinction ~4 cal ka (thousand calendar years ago). The woolly mammoth was a large herbivore with a widespread Holarctic distribution during the Pleistocene, but retreated to the northern edge of its range following the climatic changes at the Pleistocene-Holocene boundary26. Wrangel Island was the terminal refugium where the woolly mammoth survived an additional six thousand years after the mainland populations went extinct27,28, with an estimated mean effective population size of 326 individuals29. Several studies, including a whole-genome analysis of one of the last surviving Wrangel mammoths, have shown that isolation on Wrangel Island led to a decline in genetic diversity by 20% in autosomal heterozygosity and ~37% in allelic richness, likely due to a combination of inbreeding and genetic drift in the population prior to its extinction29,30. While previous studies have identified declines in neutral genetic diversity prior to a species’ extinction29,31,32, there is a lack of knowledge on whether adaptive genetic diversity also declined prior to these extinctions. However, based on the meta-analysis discussed above, as well as previous studies on historical DNA in other taxa, we hypothesize that balancing selection was unable to maintain MHC diversity in the Wrangel population. We thus expect to find a similar loss in MHC diversity as that previously observed in neutral markers.
Methods
DNA extraction and primer design
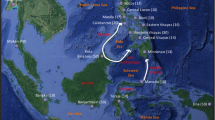
We analysed radiocarbon-dated samples (Table 1) representing i) the Pleistocene continental population in Chukotka and Wrangel while these areas were still a part of Beringia (>13 cal ka), and ii) the Wrangel Island population after rising sea levels led to its isolation (<10 cal ka; Fig. 1). We used a Dremel drill to obtain approximately 50 mg of powder from bones, teeth and tusks. DNA was extracted from the powder following a modified version33 of a silica-based ancient DNA extraction protocol34.

Wrangel Island and Pevek Region, Chukotka are highlighted. The light shaded area depicts the extent of dry land in the Late Pleistocene, before Wrangel’s isolation69. The map was drawn using Adobe® Photoshop® software (CS5, version 12).
We designed four new primer pairs (see Supplementary Table S1) targeting four short fragments of the MHC DQA gene that have been shown to be variable in both Asian and African savannah elephants35. Two of these fragments were located in exons and two in introns. We tested the primers by amplifying and sequencing four mammoth samples with high levels of DNA preservation. Amplifications were performed using 1× PCR Buffer, 0.2 μM of each primer, 0.2 mM of each dNTP, 2.5 mM MgCl2, 0.1 mg/mL BSA, 2U (0.4 μL) HotStar Taq DNA Polymerase (Qiagen), 2 μL of DNA extract and distilled water up to the total volume of 25 μL. Amplifications were carried out under following conditions: 10 min of initial denaturation at 95 °C; 55 cycles of denaturation of 30 sec at 94 °C, annealing of 30 sec at 50–54 °C (depending on the primer pair, see Supplementary Table S1) and extension of 30 sec at 72 °C; followed by a final extension 7 min at 72 °C. PCR products were cleaned using Exo-FAP (Fermentas) and sequenced on an ABI 3130xl (Applied Biosystems). All primer pairs yielded sequences that were almost identical to elephant MHC sequences when blasted against the NCBI database36.
454 pyrosequencing
We screened 82 mammoth samples by amplifying a 141-bp fragment using primers MPDQA-1F and MPDQA-1R. Thirty-six samples yielded a single clear band of expected size and 32 of these were selected for subsequent analyses. To create a pooled library for sequencing on the 454 pyrosequencing platform, each primer was equipped with 8 unique six-nucleotide tags designed to differ at at least two positions so that the risk of incorrect sorting due to sequencing errors in the tag sequences was minimized37. As both forward and reverse primers were tagged, we were able to pool 64 samples per locus (256 in total). This allowed us to include two PCR replicates for each of the 32 selected samples on the same 454 sequencing run.
Amplifications with tagged primers were performed with the same concentrations and PCR conditions as described above; except for using 0.2 μM of each primer and using 5 μL of DNA extract instead of 2 μL. We were able to successfully amplify 181 of 256 PCR products and these were pooled and cleaned using the Qiagen MinElute PCR Purification Kit with elution volume of 20 μL. Three loci showed consistent results and were in the pool represented by 51 (exon 2) or 52 (both intron 2a and exon 4) PCR products. Intron 2b worked only for 26 samples, likely due to the fact that this fragment was longer than the other fragments that were targeted.
To estimate the concentration of the cleaned pool, we ran 2 μL of the pool on an agarose gel and compared it to a dilution series of a GeneRuler 100 bp Plus DNA Ladder (Fermentas). Subsequently, we followed a modified version38 of the Rapid Library Preparation Method Manual and Sequencing Method Manual supplied with the GS Junior System (454 Life Sciences). The pool was sequenced in one GS Junior run.
Authenticity of results
We conducted all pre-PCR steps in a dedicated ancient DNA laboratory at the Swedish Museum of Natural History in Stockholm, which is physically separated from the modern DNA facilities. We used protective gloves, suits and face masks to prevent contamination. All surfaces were regularly cleaned with sodium hypochlorite and tools and reagents were cleaned by ultraviolet irradiation. We used negative controls with all extractions and PCRs (at least one per eight samples).
For samples that were scored as homozygotes, we added a third replicate to further reduce the risk that allelic dropout led to erroneous genotypes. These third replicates were sequenced by Sanger sequencing on the ABI 3130xl sequencer (Applied Biosystems) with primers MPDQA-1F, -1R and -3F being replaced by internal primers (see Supplementary Table S1) to secure a more efficient targeting of the mammoth MHC. We calculated the proportion of allelic dropout39 to verify that the probability of false negatives was lower than 0.05.
MHC is recognized as one of the hotspots of gene duplication in various taxonomic units40. However, there are three reasons why we are confident that our results represent variability at a single locus. First, a study of elephant MHC35 didn’t find any evidence of paralogues and showed that the DQA was successfully transcribed and expressed. Second, we never observed more than two alleles per locus in any of the mammoth individuals. Third, we mapped all alleles against the African elephant genome using the Burrows-Wheeler Aligner41 (BWA) and observed very high mapping quality scores (MP = 60), which strongly suggested that all alleles mapped only to a single position in the elephant genome.
Data processing
We used the 454 Sequencing System Software 2.9 to process the sequencing data generated in the GS Junior run. Reads that passed system filters were sorted according to 5′ and 3′ tags using the sfffile command and allowing for one mismatch. The sorted files in .sff format were converted into fasta and quality files using the sffinfo command. We used a perl script fastaQual2fastq.pl (https://github.com/josephhughes/Sequence-manipulation/blob/master/fastaQual2fastq.pl) to convert the fasta and quality files into fastq files. At this point, each fastq file represented (one replicate of) one individual and contained reads for all loci that were successfully amplified for that particular sample.
The reads were mapped to the reference Asian elephant sequence (GenBank no. GU369701) following steps 4–6.6 of a mapping pipeline for 454 sequencing data42. BWA 0.7.1041 was used to index the reference sequence and mapped the reads against the indexed reference sequence with the bwa mem algorithm. We used SAMtools 1.143 to convert the files containing mapped reads from SAM to BAM format, sort and index the BAM files. We performed the subsequent processing of BAM files in Geneious R744. Primer sequences were manually trimmed off the reads and BAM files containing all four loci were sorted into separate BAM files for each locus of each sample.
Since ancient DNA is generally of low quality, we used a rigorous step-by-step approach to score alleles for each sample: 1) We extracted one read for each cluster of identical reads that constituted at least 5% of the reads; 2) We aligned putative alleles from both replicates; 3) We discarded all singletons differing only by C/T and G/A changes, unless an identical sequence was found in any other of the samples or among the elephant MHC alleles.
Estimates of genetic diversity
For the diversity analyses we included three loci – exon 2, intron 2a and exon 4. Intron 2b was omitted because only half as many samples were successfully amplified and sequenced as for the other loci. We calculated genetic diversity of the mainland and Wrangel populations as observed and expected heterozygosity (HO and HE), the number of alleles (NA) and the number of unique alleles per population in Arlequin 3.5.2.145. We assessed the change in the frequency of heterozygous individuals between the mainland and Wrangel populations using a χ2 2 × 2 contingency table, where groups were defined as the mainland and Wrangel populations and categories were defined as the number of homozygotes and heterozygotes. Allelic richness was calculated with rarefaction as implemented in adze 1.046 using maximum standardized sample size of 24, equivalent to 12 diploid samples. To compare allelic richness between mammoths and African savannah elephants, we used the counts of individuals carrying an allele (see Supplementary Information). We did not include exon 4 because some of the elephant alleles were missing this part of the DQA sequence. We also created temporal statistical parsimony networks using the R-script TempNet 1.847 to illustrate the change in allelic composition between the two time periods.
Scans of selection
We scanned for signals of selection in MEGA 648, and calculated the ratios of non-synonymous and synonymous mutations (dN and dS) using the Nei-Gojobori/Jukes-Cantor method49. Values of dN/dS above 1 are considered as evidence of positive selection, while values below 1 indicate purifying selection. We also performed codon-based Z-tests of selection to estimate the probabilities of positive, neutral and purifying selection. We tested selection on entire fragments of exon 2 and exon 4. Moreover, since exon 2 contains antigen-binding regions (ABR) we also analysed exon 2 split into ABR and non-ABR codons. ABR were selected according to elephant ABR35.
Coalescent simulations of genetic drift
We used coalescent simulations to explore if the number of observed alleles in the Wrangel Island population could be explained under a null-hypothesis of genetic drift. To do this, we assumed that the Wrangel population can be modeled as continuous to an ancestral population having suffered a severe bottleneck ~12,000 years ago29. We performed coalescent simulations for a wide range of effective population sizes (NEB: 2–10,000 individuals) for a bottleneck of five generations of duration and a constant population size for the Wrangel population (NEW: 2–10,000 individuals) until extinction, both on an equally spaced log-scale. For each combination of parameters we performed 1,000 simulations for three DNA fragments of lengths 99, 89 and 92 base pairs representing the three MHC loci analyzed. In each simulation, we sampled individuals at the same time as the mean calibrated age of our samples (Table 1) and calculated the number of alleles in the ancestral population (samples > 12,000 years old) and the Wrangel Island population (<12,000 years old). The probability of observing at least 63% of the alleles of the ancestral population in the simulated Wrangel samples, corresponding to 12 out of the 19 alleles observed in our three loci, was reported. Generation time was fixed to 31 years50. Because new mutations in the 6,000 years between the bottleneck and the extinction of the Wrangel Island population are extremely unlikely, mutation rate and ancestral population sizes were adjusted to 1 × 10−7 and 100,000, respectively, in order to obtain between 13 and 25 alleles in the simulated ancestral samples. No transition bias was assumed. Simulations were performed using fastsimcoal251 and the number of alleles estimated with arlsumstats, both controlled by custom R scripts52.
Results
Loss of genetic diversity
In total, we observed seven alleles in exon 2 and intron 2a and five alleles in exon 4 (see Supplementary Figures S1–S4). In each of the three loci, we observed fewer alleles in the Wrangel Island population (<10 cal ka) than in the ancestral Pleistocene population (>13 cal ka; Table 2). In general, the allele frequencies were equally distributed with most of the values ranging from 0.021 to 0.292 (see Supplementary Table S2). The mean number of alleles in the Wrangel population was 37% lower than in the mainland population.
As illustrated in the temporal statistical parsimony networks, the less frequent alleles appear to have been lost after the isolation in intron 2a and exon 4 (Fig. 2). However, we did not observe this pattern in exon 2. In all three loci, the frequency of heterozygous individuals was lower in the more recent time period (<10 cal ka). This decrease was only statistically significant in exon 4 (χ2 = 12, P = 0.001) and marginally significant in intron 2a (χ2 = 3.556, P = 0.059), whereas it was non-significant in exon 2 (χ2 = 1.815, P = 0.178). Plots of the mean number of distinct alleles per locus against sample size (Fig. 3) showed that for all three loci the allelic richness is higher in the mainland population compared to the population on Wrangel Island. In the comparison of allelic richness between the woolly mammoth and African savannah elephant, elephants had lower allelic richness than mammoths, even compared to the bottlenecked Wrangel population (Fig. S5).

(a) exon 2, (b) intron 2a and (c) exon 4. Layers represent separate time periods before (>13 cal ka; in magenta) and after the Wrangel Island isolation (<10 cal ka; in turquoise). Numbers inside the circles indicate number of samples with the allele. Empty circles represent alleles absent in the time period.

(a) exon 2, (b) intron 2a and (c) exon 4. Coloured shapes represent the two populations: mainland >13 cal ka (magenta squares) and Wrangel <10 cal ka (turquoise triangles).
Positive and purifying selection
We estimated the ratio of non-synonymous to synonymous substitutions (dN/dS) in exons 2 and 4. Since exon 2 contains ABR, we performed analyses on ABR and non-ABR codons separately (Fig. 4). Although the dN/dS ratio for ABR in exon 2 was above 1, indicating positive selection, the Z-tests of selection showed no significant deviations from neutrality (Tables 3 and 4).

Highlighted are positions predicted as antigen-binding regions (ABR) in the elephant MHC35.
The non-ABR codons of exon 2 and the entire exon 4 had dN/dS ratios close to zero, suggesting that they are under purifying selection (Table 3). However, the Z-tests of selection were only statistically significant for exon 4 in mammoths >13 cal ka, which proved to be different from neutrality and under purifying selection. In mammoths from Wrangel Island after its isolation (<10 cal ka), none of the exons differed significantly from neutrality.
Simulations
Our coalescent simulations suggest that the null-hypothesis of genetic drift being responsible for a reduction of more than 37% of alleles in Wrangel Island samples with respect to the ones observed in the ancestral population could be rejected only for very small effective population sizes (roughly 2-155, Fig. 5). The size of the population at the bottleneck had almost no effect on this probability as long as at least 10 individuals composed the remaining population. For scenarios in which less than four individuals survived the bottleneck, observing at least 63% of the alleles in the Wrangel samples was highly unlikely, irrespective of the effective population size after the bottleneck. Coalescent simulations performed with one and ten generations of bottleneck duration were consistent with the results presented above (see Supplementary Information and Figs S6 and S7).

Two parameters were explored: effective population size during the bottleneck (NEB) and the size of the Wrangel population until extinction (NEW). The probability of observing at least the 63% of the alleles present in the ancestral population in the Wrangel Island samples is depicted in a yellow to red color scale. White to grey colors indicate combinations of parameters for which the simulations suggest a departure from the null-hypothesis (P < 0.05). The green68, blue29 and purple30 lines correspond to three estimates of the mean effective population size for Wrangel mammoths.
Discussion
Our results show that diversity in the MHC DQA gene decreased seemingly as a consequence of the decline in woolly mammoth population size that took place at the end of the last glaciation, when mammoths became isolated on Wrangel Island. Furthermore, the results from our coalescent simulations indicate that the observed loss of allelic diversity is consistent with what would be expected from genetic drift, and that there thus is no evidence for balancing selection having maintained diversity during the last 6,000 years of the mammoth’s existence. While it is important to remember that our analyses are based on only one gene in the MHC, our findings support the hypothesis that in small fragmented populations the stronger effect of genetic drift overshadows the power of balancing selection to maintain high MHC polymorphism. We observed a loss in allele numbers as well as heterozygosity, likely due to that the small population size on Wrangel Island led to genetic drift and inbreeding.
Although the difference in time spans of these populations should be considered, the mainland population covering ~40 ka and Wrangel population covering ~4 ka, it is unlikely that this could explain the difference in genetic diversity between these populations. Our findings of trans-species polymorphism among the woolly mammoth and extant elephants indicates that the variability in the woolly mammoth’s MHC is highly conserved. Trans-species polymorphism is a phenomenon typical for MHC when species share identical pairs of alleles as a result of strong long-term balancing selection operating on the locus53. There is evidence of alleles shared between mice and rats that diverged about 10 million years ago54 (Mya) and humans and chimpanzees with divergence dating back about 6 Mya55,56. The ancestors of the woolly mammoth split from the last common ancestor with African savannah elephant about 6.6–8.8 Mya and the origin of the clade of woolly mammoths and Asian elephant is estimated at 5.8–7.8 Mya57. All alleles that we found in the woolly mammoth MHC DQA are either identical to African savannah and Asian elephant alleles, or they differ in one or two positions. Thus, it is highly unlikely that mutations would have occurred during a time span of 50,000 years, and that comparing samples from a ~40 ka time period with those from a ~4 ka time period would have led to any biases in the diversity estimates.
Analyses of neutral microsatellite diversity in a previous study29 revealed a 30% decrease in observed heterozygosity from 0.67 ± 0.22 to 0.46 ± 0.20 comparing mammoths before and after the isolation on Wrangel Island. The mean observed heterozygosity in the MHC DQA locus dropped by 45% from 0.92 ± 0.08 to 0.50 ± 0.14. This might suggest that the positively selected MHC diversity decreased more than neutral diversity, which is consistent with empirical studies23,25 and simulation models58 that suggest a greater loss of variation in the MHC compared to neutral markers. However, analysis of longer or multiple loci of the MHC is needed to verify this finding.
Our comparison of genetic diversity in the woolly mammoth and African savannah elephant, measured as allelic richness, revealed that, when standardized over sample size, mammoths had more alleles on MHC DQA than elephants. At first glance, this might seem surprising given that diseases are generally more common in the tropics59,60. On the other hand, climatic and environmental fluctuations have likely been more severe in the Arctic compared to tropics, and this could potentially have led to variation in selection on MHC. We also note that the higher diversity in mainland mammoths compared to African savannah elephants could be a result of past introgression between Siberian woolly mammoths and other mammoth populations, for example during periods of gene flow from North America as identified in earlier studies61,62,63.
A “hyper-disease” introduced to native Pleistocene fauna by newly arrived human populations has also been suggested as an explanation of the megafauna extinction64,65. However, the lack of evidence for a major temporal shift in DQA allele frequencies, as well as the observation of higher DQA diversity in mammoths compared to African savannah elephants, do not provide any support for the hypothesis that a hyper-disease led to the extinction of the woolly mammoth and other megafaunal species.
MHC is generally considered to be a locus under balancing selection. Although we found no statistically significant support for balancing selection in the ratio of nonsynonymous to synonymous mutations, we observed other patterns that could be explained as the effects of balancing selection. We found intermediate allele frequencies close to the frequency equilibrium, as is expected when selection keeps many variants in a population, and we also found a strong trans-species polymorphism.
Hughes and Nei66 pointed out distinct patterns of selection on MHC in humans and mice. While they found ABR sites of MHC to be regulated by positive selection, other regions of the sequences were under purifying selection. This observation is consistent with the assumption that purifying selection maintains the general structure of the protein conserved among different mammalian taxa67. Accordingly, our analyses suggest that exon 4 in the woolly mammoth MHC DQA locus was under purifying selection in the mainland population before the isolation on Wrangel Island (>13 cal ka), corresponding to the pattern in human and mouse MHC. Purifying selection could also explain the generally lower number of alleles that we observed at exon 4 compared to the other two loci.
Whether the observed loss in MHC allelic diversity can be explained by genetic drift alone depends on the effective population size after mammoths became isolated on Wrangel Island. There are three previous estimates of this post-isolation effective population size. In the first68, the authors used the island’s size, the estimated mammoth body size, and population density of herbivores in arctic environments to estimate a mean effective size of 240 individuals in Wrangel Island, assuming a 1:1 sex ratio and a relationship of 0.5 between carrying capacity and effective population size. Second, a study based on coalescent simulations of microsatellite data indicated an effective Holocene population size of 500 individuals30. Recently, a third study29 based on a newly calibrated mutation rate obtained from two complete mammoth genomes estimated the mean effective population size on Wrangel Island to 328 individuals. All these estimates fall within the ranges for which our coalescent simulations indicate that the temporal change in the number of observed alleles on Wrangel Island can be explained by genetic drift alone. Therefore, we find no evidence that other evolutionary forces, such as balancing selection, are required to explain the maintenance of diversity at this MHC locus during the Holocene.
Conclusions
Analyses of three short fragments of the MHC DQA gene in the woolly mammoth before and after the isolation on Wrangel Island showed that genetic diversity decreased in the Wrangel population, possibly as a result of genetic drift caused by the bottleneck and ensuing small population size. While it is important to remember that our analyses are based on only three short parts of one gene in the MHC, our results suggest that genetic drift had an effect on adaptive genetic variation in the last surviving woolly mammoth population.
Although we found no evidence of balancing selection during the Holocene, the trans-species polymorphism observed between the woolly mammoth and African savannah and Asian elephants can be considered strong evidence of historical long-term balancing selection. Our results represent the first analysis of MHC variation in an extinct species, which can be a useful primer for the study of endangered populations since MHC diversity plays a major role in immune defense and thus population persistence.
Additional Information
Accession codes: KX090927-KX090945.
How to cite this article: Pečnerová, P. et al. Changes in variation at the MHC class II DQA locus during the final demise of the woolly mammoth. Sci. Rep. 6, 25274; doi: 10.1038/srep25274 (2016).
References
Reed, D. H. & Frankham, R. Correlation between fitness and genetic diversity. Conser. Biol 17, 230–237 (2003).
Vilas, C., San Miguel, E., Amaro, R. & Garcia, C. Relative contribution of inbreeding depression and eroded adaptive diversify to extinction risk in small populations of shore Campion. Conserv Biol 20, 229–238 (2006).
Piertney, S. B. & Oliver, M. K. The evolutionary ecology of the major histocompatibility complex. Heredity 96, 7–21 (2006).
Penn, D. J., Damjanovich, K. & Potts, W. K. MHC heterozygosity confers a selective advantage against multiple-strain infections. P Natl Acad Sci USA 99, 11260–11264 (2002).
Spurgin, L. G. & Richardson, D. S. How pathogens drive genetic diversity: MHC, mechanisms and misunderstandings. P R Soc B 277, 979–988 (2010).
Moller, A. P. Evidence of larger impact of parasites on hosts in the tropics: investment in immune function within and outside the tropics. Oikos 82, 265–270 (1998).
Horrocks, N. P. C. et al. Environmental proxies of antigen exposure explain variation in immune investment better than indices of pace of life. Oecologia 177, 281–290 (2015).
Tong, Y. Y. & Lighthart, B. Solar radiation has a lethal effect on natural populations of culturable outdoor atmospheric bacteria. Atmos Environ 31, 897–900 (1997).
Moyer, B. R., Drown, D. M. & Clayton, D. H. Low humidity reduces ectoparasite pressure: implications for host life history evolution. Oikos 97, 223–228 (2002).
Guernier, V., Hochberg, M. E. & Guegan, J. F. Ecology drives the worldwide distribution of human diseases. Plos Biol 2, 740–746 (2004).
Radwan, J., Biedrzycka, A. & Babik, W. Does reduced MHC diversity decrease viability of vertebrate populations? Biol Conserv 143, 537–544 (2010).
Sutton, J. T., Nakagawa, S., Robertson, B. C. & Jamieson, I. G. Disentangling the roles of natural selection and genetic drift in shaping variation at MHC immunity genes. Mol Ecol 20, 4408–4420 (2011).
Obrien, S. J. et al. Genetic-Basis for Species Vulnerability in the Cheetah. Science 227, 1428–1434 (1985).
Obrien, S. J. & Evermann, J. F. Interactive Influence of Infectious-Disease and Genetic Diversity in Natural-Populations. Trends Ecol Evol 3, 254–259 (1988).
Arkush, K. D. et al. Resistance to three pathogens in the endangered winter-run chinook salmon (Oncorhynchus tshawytscha): effects of inbreeding and major histocompatibility complex genotypes. Can J Fish Aquat Sci 59, 966–975 (2002).
Sauermann, U. et al. Increased reproductive success of MHC class II heterozygous males among free-ranging rhesus macaques. Hum Genet 108, 249–254 (2001).
Paterson, S., Wilson, K. & Pemberton, J. M. Major histocompatibility complex variation associated with juvenile survival and parasite resistance in a large unmanaged ungulate population (Ovis aries L.). P Natl Acad Sci USA 95, 3714–3719 (1998).
Langefors, K., Lohm, J. & von Schantz, T. Allelic polymorphism in MHC class IIB in four populations of Atlantic salmon (Salmo salar). Immunogenetics 53, 329–336 (2001).
Aguilar, A. et al. High MHC diversity maintained by balancing selection in an otherwise genetically monomorphic mammal. P Natl Acad Sci USA 101, 3490–3494 (2004).
Oliver, M. K. & Piertney, S. B. Selection Maintains MHC Diversity through a Natural Population Bottleneck. Mol Biol Evol 29, 1713–1720 (2012).
Xu, S. X. et al. Considerable MHC Diversity Suggests That the Functional Extinction of Baiji Is Not Related to Population Genetic Collapse. Plos One 7, e30423 (2012).
Stiebens, V. A., Merino, S. E., Chain, F. J. J. & Eizaguirre, C. Evolution of MHC class I genes in the endangered loggerhead sea turtle (Caretta caretta) revealed by 454 amplicon sequencing. Bmc Evol Biol 13, doi: 10.1186/1471-2148-13-95 (2013).
Eimes, J. A. et al. Rapid loss of MHC class II variation in a bottlenecked population is explained by drift and loss of copy number variation. J Evolution Biol 24, 1847–1856 (2011).
Taylor, S. S., Jenkins, D. A. & Arcese, P. Loss of Mhc and Neutral Variation in Peary Caribou: Genetic Drift Is Not Mitigated by Balancing Selection or Exacerbated by Mhc Allele Distributions. Plos One 7, e36748 (2012).
Sutton, J. T., Robertson, B. C. & Jamieson, I. G. MHC variation reflects the bottleneck histories of New Zealand passerines. Mol Ecol 24, 362–373 (2015).
Lister, A. M. & Stuart, A. J. The impact of climate change on large mammal distribution and extinction: Evidence from the last glacial/interglacial transition. Cr Geosci 340, 615–620 (2008).
Vartanyan, S. L., Arslanov, K. A., Tertychnaya, T. V. & Chernov, S. B. Radiocarbon Dating Evidence for Mammoths on Wrangel Island, Arctic-Ocean, until 2000-Bc. Radiocarbon 37, 1–6 (1995).
Stuart, A. J., Sulerzhitsky, L. D., Orlova, L. A., Kuzmin, Y. V. & Lister, A. M. The latest woolly mammoths (Mammuthus primigenius Blumenbach) in Europe and Asia: a review of the current evidence. Quaternary Sci Rev 21, 1559–1569 (2002).
Palkopoulou, E. et al. Complete Genomes Reveal Signatures of Demographic and Genetic Declines in the Woolly Mammoth. Curr Biol 25, 1395–1400 (2015).
Nyström, V. et al. Microsatellite genotyping reveals end-Pleistocene decline in mammoth autosomal genetic variation. Mol Ecol 21, 3391–3402 (2012).
Dalén, L. et al. Partial Genetic Turnover in Neandertals: Continuity in the East and Population Replacement in the West. Mol Biol Evol 29, 1893–1897 (2012).
Knapp, M. et al. First DNA sequences from Asian cave bear fossils reveal deep divergences and complex phylogeographic patterns. Mol Ecol 18, 1225–1238 (2009).
Brace, S. et al. Serial population extinctions in a small mammal indicate Late Pleistocene ecosystem instability. P Natl Acad Sci USA 109, 20532–20536 (2012).
Yang, D. Y., Eng, B., Waye, J. S., Dudar, J. C. & Saunders, S. R. Technical note: Improved DNA extraction from ancient bones using silica-based spin columns. Am J Phys Anthropol 105, 539–543 (1998).
Archie, E. A. et al. Major histocompatibility complex variation and evolution at a single, expressed DQA locus in two genera of elephants. Immunogenetics 62, 85–100 (2010).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic Local Alignment Search Tool. J Mol Biol 215, 403–410 (1990).
Binladen, J. et al. The Use of Coded PCR Primers Enables High-Throughput Sequencing of Multiple Homolog Amplification Products by 454 Parallel Sequencing. Plos One 2, e197 (2007).
Palkopoulou, E. et al. Synchronous genetic turnovers across Western Eurasia in Late Pleistocene collared lemmings. Glob Change Biol, doi: 10.1111/gcb.13214 (2016).
Gagneux, P., Boesch, C. & Woodruff, D. S. Microsatellite scoring errors associated with noninvasive genotyping based on nuclear DNA amplified from shed hair. Mol Ecol 6, 861–868 (1997).
Kulski, J. K., Shiina, T., Anzai, T., Kohara, S. & Inoko, H. Comparative genomic analysis of the MHC: the evolution of class I duplication blocks, diversity and complexity from shark to man. Immunol Rev 190, 95–122 (2002).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Clarke, A. C. et al. From cheek swabs to consensus sequences: An A to Z protocol for high-throughput DNA sequencing of complete human mitochondrial genomes. BMC Genomics 15, 68 (2014).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10, 564–567 (2010).
Szpiech, Z. A., Jakobsson, M. & Rosenberg, N. A. ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24, 2498–2504 (2008).
Prost, S. & Anderson, C. N. K. TempNet: a method to display statistical parsimony networks for heterochronous DNA sequence data. Methods Ecol Evol 2, 663–667 (2011).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol 30, 2725–2729 (2013).
Nei, M. & Gojobori, T. Simple Methods for Estimating the Numbers of Synonymous and Nonsynonymous Nucleotide Substitutions. Mol Biol Evol 3, 418–426 (1986).
Rohland, N. et al. Genomic DNA sequences from mastodon and woolly mammoth reveal deep speciation of forest and savanna elephants. Plos Biol 8, e1000564 (2010).
Excoffier, L. et al. Robust demographic inference from genomic and SNP data. Plos Genetics, 9(10), e1003905 (2013).
R Development Core Team (2013) R Foundation for Statistical Computing: Vienna, Austria http://www.R-project.org, ISBN 3-900051-00-3.
Takahata, N. & Nei, M. Allelic Genealogy under Overdominant and Frequency-Dependent Selection and Polymorphism of Major Histocompatibility Complex Loci. Genetics 124, 967–978 (1990).
Figueroa, F., Gunther, E. & Klein, J. Mhc Polymorphism Predating Speciation. Nature 335, 265–267 (1988).
Lawlor, D. A., Ward, F. E., Ennis, P. D., Jackson, A. P. & Parham, P. Hla-a and Hla-B Polymorphisms Predate the Divergence of Humans and Chimpanzees. Nature 335, 268–271 (1988).
Mayer, W. E. et al. Nucleotide-Sequences of Chimpanzee Mhc Class-I Alleles–Evidence for Trans-Species Mode of Evolution. Embo J 7, 2765–2774 (1988).
Rohland, N. et al. Proboscidean mitogenomics: Chronology and mode of elephant evolution using mastodon as outgroup. Plos Biol 5, e207, 1663–1671 (2007).
Ejsmond, M. J. & Radwan, J. MHC diversity in bottlenecked populations: a simulation model. Conserv Genet 12, 129–137 (2011).
Kutz, S. J. et al. The Arctic as a model for anticipating, preventing, and mitigating climate change impacts on host-parasite interactions. Vet Parasitol 163, 217–228 (2009).
Weber, D. S. et al. Low MHC variation in the polar bear: implications in the face of Arctic warming? Anim Conserv 16, 671–683 (2013).
Barnes, I. et al. Genetic structure and extinction of the woolly mammoth, Mammuthus primigenius. Curr Biol 17, 1072–1075 (2007).
Debruyne, R. et al. Out of America: Ancient DNA Evidence for a New World Origin of Late Quaternary Woolly Mammoths. Curr Biol 18, 1320–1326 (2008).
Palkopoulou, E. et al. Holarctic genetic structure and range dynamics in the woolly mammoth. Proc R Soc B 280, 20131910 (2013).
MacPhee, R. & Marx, P. In Natural Change and Human Impact in Madagascar (eds S. M. Goodman & B. D. Patterson ), 169–217 (Smithsonian Institution Press, 1997).
Lyons, S. K. et al. Was a ‘hyperdisease’ responsible for the late Pleistocene megafaunal extinction? Ecol Lett 7, 859–868 (2004).
Hughes, A. L. & Nei, M. Pattern of Nucleotide Substitution at Major Histocompatibility Complex Class-I Loci Reveals Overdominant Selection. Nature 335, 167–170 (1988).
Furlong, R. F. & Yang, Z. Diversifying and purifying selection in the peptide binding region of DRB in mammals. J Mol Evol 66, 384–394 (2008).
Nyström, V. et al. Temporal genetic change in the last remaining population of woolly mammoth. Proc R Soc B 277, 2331–2337 (2010).
Elias, S. A. & Crocker, B. The Bering Land Bridge: a moisture barrier to the dispersal of steppe-tundra biota? Quat Sci Rev 27, 2473–2483 (2008).
Acknowledgements
The authors are grateful to Veronica Nyström Edmark for assistance with sampling. We would like to thank Uppmax for providing computational infrastructure. The genetic analyses were funded through a grant from the Swedish Research Council (VR grant 2012-3869). We thank Estelle Proux-Wéra from the Science for Life Laboratory for advice on the bioinformatic aspects of the study and Eleftheria Palkopoulou for help with the general workflow of the project. We would also like to thank Christopher Wheat for insightful comments on the analytical part of the project.
Author information
Authors and Affiliations
Contributions
L.D. designed the study; S.V. provided samples; P.P. performed lab work; P.P. and D.D. performed data analysis; P.P., D.D. and L.D. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Pečnerová, P., Díez-del-Molino, D., Vartanyan, S. et al. Changes in variation at the MHC class II DQA locus during the final demise of the woolly mammoth. Sci Rep 6, 25274 (2016). https://doi.org/10.1038/srep25274
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep25274
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
