
Abstract
The recent discovery that DNA methylation survives in fossil material provides an opportunity for novel molecular approaches in palaeogenomics. Here, we apply to ancient DNA extracts the probe-independent Methylated Binding Domains (MBD)-based enrichment method, which targets DNA molecules containing methylated CpGs. Using remains of a Palaeo-Eskimo Saqqaq individual, woolly mammoths, polar bears and two equine species, we confirm that DNA methylation survives in a variety of tissues, environmental contexts and over a large temporal range (4,000 to over 45,000 years before present). MBD enrichment, however, appears principally biased towards the recovery of CpG-rich and long DNA templates and is limited by the fast post-mortem cytosine deamination rates of methylated epialleles. This method, thus, appears only appropriate for the analysis of ancient methylomes from very well preserved samples, where both DNA fragmentation and deamination have been limited. This work represents an essential step toward the characterization of ancient methylation signatures, which will help understanding the role of epigenetic changes in past environmental and cultural transitions.
Similar content being viewed by others


Introduction
The retrieval of DNA from archaeological and palaeontological material provides direct genetic information about the evolutionary past1,2. However, DNA fragmentation and chemical damage start shortly after an organism dies, making ancient DNA (aDNA) analyses particularly challenging. Many technologies have been developed to improve the characterization of aDNA since the early days of bacterial cloning3, which, together with the development of High-Throughput Sequencing (HTS) technologies, opened access to an increasing number of ancient genomes3,4. The extraordinary throughput of HTS technologies is, however, often hampered by the abundance of environmental microbial DNA in aDNA extracts5 and shotgun sequencing can only achieve high-coverage genomes from exceptionally well-preserved material where microbial colonization is minimal (e.g.6,7,8).
Several target enrichment methods have been developed to circumvent these limitations. Most are based on the hybridization of probes tiled on markers of interest to DNA library templates9,10. Within a single enrichment experiment, the number of loci targeted can vary from the mitochondrial genome and a few nuclear genes11 to entire bacterial genomes12, human chromosomes13 or complete mammalian genomes10,14. Hybridization-based target enrichment approaches are subject to a variety of experimental biases, including the possible over-representation of long inserts15, %GC-rich genomic regions16 and repetitive elements14. Additionally, as ancient DNA library inserts are generally short, the size of adapters can influence the enrichment efficiency17. The number of mismatches between probes and library molecules, which increases with the evolutionary distance and the amount of post-mortem DNA damage, can also reduce annealing efficiency, which can represent a problem in situations where no closely related species are available for designing probes18.
Probe-free enrichment methods have been developed to avoid introducing bias while annealing probes to targets. With the ‘uracil selection’ method, ancient DNA templates carrying damage (in the form of deaminated cytosines) can be separated from those showing no damage during library preparation10. Deep sequencing of the damage-enriched fraction has shown enrichment of 1.1–4.7-fold on a range of Neanderthal specimens and performance is expected to increase with damage levels10.
Methyl Binding Domains (MBD) based enrichment is another probe-free enrichment method but directly operates on DNA extracts, prior to library construction. It exploits the affinity of MBD for methylated CpG dinucleotides (mCpG) so as to separate complex metagenomic assemblages into two fractions19, the first of which binds to MBD and is mostly of vertebrate origin, while the second does not bind and is mostly microbial19. This is so because vertebrate genomes are globally methylated, with 70–80% of methylated cytosines being located in a CpG context in the human genome20, in contrast to bacterial genomes where methylation marks are often found in non-CpG contexts21. This method could provide the basis for new enrichment approach in aDNA research, where microbial DNA often represents the vast majority of the DNA templates extracted. Surprisingly, it has so far been used in only a single aDNA study, which focused on the analysis of 200–2,800 year-old barley seeds from a single archaeological site in southern Egypt22. The potential of the method, thus, still remains largely unexplored.
Here, we applied MBD enrichment to a selection of mammalian fossil specimens preserved across a wide geographical and temporal range. We identify experimental conditions that influence the overall efficiency of MBD enrichment and conclude that the method is only compatible with a cost-effective characterization of ancient methylomes when DNA fragmentation and deamination are limited.
Results
The MBD enrichment procedure tested in this study relies on the affinity of MBD2−Fc for mCpGs. The MBD2−Fc protein corresponds to the fusion of the human MBD2 domain and the Fc tail fragment of human IgG119. Following incubation, mCpG-rich DNA molecules bound to MDB2−Fc can be recovered using paramagnetic beads coated with the protein A, which bind to the Fc tail. The supernatant (MBD−) is expected to contain mostly DNA molecules showing low mCpG content in contrast to the enriched fraction (MBD+). We explored the performance of this MBD enrichment system using 20 aDNA extracts from soft and calcified tissues (Table 1; Supplementary Table S1), including specimens preserved in a variety of environments: seven from arctic regions (the Palaeo-Eskimo Saqqaq individual from Greenland that was previously genome sequenced6, two Siberian woolly mammoths, three polar bears and one and three equids from Canada and Russia, respectively) and four equids from a cave, karstic formations and a submerged terrestrial site. These specimens spanned a wide temporal range, extending from the Holocene (~4,000 years before present, BP) to the Late Pleistocene (>45,000 years BP). Following MBD enrichment, both MBD+ and MBD− fractions were used to construct DNA libraries and were deep-sequenced to generate a total of 409 million paired-end sequences on Illumina HiSeq 2500 and MiSeq platforms (Supplementary Table S3.7).
Endogenous contents
Since post-mortem deamination progressively transforms mCpGs into TpGs, we first evaluated whether sufficient amounts of mCpGs were preserved to enable successful MBD enrichment. We were able to build Illumina DNA libraries from both the MBD+ and MBD− fractions for all samples investigated. After Illumina sequencing, we used stringent alignment parameters and quantified the proportion of molecules of endogenous origin based on the number of both low and high-quality hits (hereafter referred to as MQ0 and MQ25 filters) that mapped uniquely to the reference genomes (for extinct species, genomes of phylogenetically-close species). The MQ0 filter allowed us to quantify the fraction of the sequences that aligned against repetitive elements while the MQ25 filter identified high-quality unique hits23. For almost all comparisons (90%), the proportion of unique endogenous reads in the MBD+ fraction was lower than that in the MBD− fraction (Fig. 1B). This represented only a 1.2–1.3-fold reduction for the three polar bear specimens, but the reduction reached about two orders of magnitude in some of the equine and mammoth specimens (90–138-fold for MQ0 and MQ25 filters, respectively; Supplementary Table S2.1). The reduction of the proportion of endogenous reads could be partly explained by an increase in the proportion of PCR duplicates in MBD+ libraries (relative to MBD− libraries), which was somewhat comparable to the reduction observed in endogenous contents (6 to 391-fold) (Fig. 1A, Supplementary Table S2.2).

Endogenous content, clonality and number of non-unique reads of DNA libraries constructed from MBD+ and MBD− fractions.
(A) Clonality estimates, expressed as the percentage of hits that were PCR duplicates both for those not filtered for mapping quality (MQ0, darker shade) and those filtered for mapping quality (MQ25, lighter shade). (B) Endogenous DNA content, expressed as the natural logarithm of the fractions of unique hits over the total number of reads retained both for those not filtered for mapping quality (MQ0, lighter shade) and those filtered for mapping quality (MQ25, darker shade). (C) Fraction of nuclear DNA reads non-uniquely mapped to the reference genome calculated on the full-length reads (minimum 2,000 per sample) not filtered for mapping quality (MQ0). MBD+ = MBD enriched fraction. MBD− = uncaptured fraction.
MBD enrichment efficiency
In contrast to the nuclear genome, the mitochondrial genome is globally hypo-methylated, if at all24. We exploited this to evaluate the efficiency of MBD enrichment. To avoid biasing our estimates, only specimens where both MBD+ and MBD− fractions had a sufficient number of mitochondrial unique hits were considered (MQ25 and ≥20 hits). As expected, the contribution of mitochondrial DNA (mtDNA) to the pool of endogenous sequences was found to decrease significantly in the MBD+ fractions relative to the MBD− fractions (Supplementary Table S2.3). This was observed across all tissues, but most dramatically for one polar bear bone (PB25) and two mammoth tissues (heart - MZh_1, muscle - Mzm_1), which showed a 15.6-fold to 23.6-fold reduction (Supplementary Table S2.3).
Similarly, using CpG island (CGI) annotations available for the human and horse reference genomes, we found that, in the MBD+ fraction, CGIs were on average 9.3-fold less covered than the rest of the genome (MQ25 filter; Supplementary Table S3.1). This is in line with the overall hypo-methylation state of CGIs25.
Since methylation is involved in the silencing of transposable elements (TEs), we also tested for differences in the representation of TEs between MBD+ and MBD− fractions. We found that sub-optimal hits (i.e., mapping to multiple genomic regions identified using the MQ0 filter) represented a larger proportion of endogenous reads in the MBD+ fraction than in the MBD− fraction (average = 2.4-fold increase, Supplementary Table S3.2). To confirm that this was driven by TEs, which are present in multiple copies within the genome, we stratified coverage differences for annotated TEs in MBD+ and MBD− fractions. We found a higher abundance of TEs in the MBD+ fraction, which were on average 2.1-fold more abundant than in the MBD− fraction (Supplementary Table S3.3).
To further assess the efficiency of MBD enrichment, we took advantage of the work by Pedersen and colleagues26, which quantified genome-wide methylation levels for the Palaeo-Eskimo Saqqaq individual. We used the sequence data presented by Rasmussen and colleagues6 and the regional methylation proxy (Ms) defined by Pedersen and colleagues26 to estimate methylation levels within 100 bp sliding windows. We then performed MBD enrichment on DNA extracts from the Palaeo-Eskimo Saqqaq individual and tested whether the MBD+ fraction covered genomic regions with higher Ms values than the MBD− fraction. MBD+ sequences were found to overlap regions with higher Ms values than MBD− sequences for the two minimal coverage thresholds considered (Fig. 2, Supplementary Table S3.5).

Measure of regional methylation level based on the average CpG→TpG mismatch frequencies observed at read starts (Ms), as defined by Pedersen and colleagues26.
Ms was calculated within 100 bp genomic windows and two minimal coverage thresholds were tested (50 and 60 per window, respectively).
Altogether, the under-representation of mtDNA and CGIs in the MBD+ fraction, coupled with the over-representation of TEs in the MBD+ fraction and the presence of DNA fragments showing higher methylation rates in the Palaeo-Eskimo Saqqaq sequence data, demonstrate the overall efficiency of MBD enrichment.
Size and base compositional bias
We next evaluated whether endogenous DNA fragments recovered in MBD+ and MBD− fractions exhibited similar molecular properties. Only samples showing at least 2,000 high-quality hits mapping uniquely against their respective reference genome (MQ25 filter) were considered so as to enable reliable estimates of base compositional bias. We implemented linear mixed models analyses to assess statistical differences within the two fractions for %GC content, CpG densities and the size of library inserts. We found that MBD+ fractions were significantly enriched in endogenous reads carrying higher %GC contents and CpG densities, with the exception of a single sample (MZh_1, Figs 1C and 3A; Supplementary Fig. S1; Supplementary Tables S4.1 and S4.2).

Molecular characteristics of DNA library inserts and post-mortem DNA degradation.
(A) Percentage of CpGs in the nuclear DNA, normalized by read length. (B) Length of DNA library inserts. (C) Cytosine deamination probability in double-stranded DNA context (δd). (D) Cytosine deamination probability in single-stranded DNA contexts (δs). DNA degradation parameters were calculated using mapDamage 227. A minimum threshold of 2,000 reads was applied to all data shown (mapping quality filtered, MQ25) so as to retrieve unbiased distributions. MBD+ = MBD enriched fraction. MBD− = uncaptured fraction.
In MDB+ fractions, CpG densities increased up to 3.1-fold compared to that observed for other dinucleotides (Supplementary Tables S5.1 and S5.2). Interestingly, we detected a decrease in the number of ApAs, ApTs, TpAs and TpTs in the MBD+ fraction, coupled with an increase in the number of CpCs, GpCs and GpGs. These differences were statistically significant when tested for the three species with a minimum number of three individuals (except for CpG and GpC in polar bears; Supplementary Table S5.2). We also found that endogenous DNA inserts from MBD+ fractions were, on average, longer than in MBD− fractions (Fig. 3B). This difference was statistically significant in the woolly mammoth (with the exception of MZh_1, Supplementary Table S4.2), where MBD+ DNA inserts were on average ~21-bp longer than those from the MBD− fraction. MBD+ DNA library inserts were on average ~35-bp longer than those present in the MBD− fraction amongst equids and ~56-bp longer amongst polar bears. These differences were statistically significant (Supplementary Tables S4.1 and S4.2).
We observed that the relative amount of endogenous inserts showing at least one CpG generally increased with the size of DNA inserts. This trend was only apparent in MBD+ fractions, however and the inverse was found in MBD− fractions (Supplementary Fig. S4), suggesting that MBD enrichment separated methylated and unmethylated fractions more efficiently on long DNA templates. This most likely reflects enhanced DNA binding to MBD2−Fc with increasing numbers of mCpG targets, as has been previously reported19.
The bias observed toward DNA templates that are longer and richer in mCpG is expected to contribute to the reduced molecular complexity of MBD+ fractions as the vast majority of endogenous DNA templates present in aDNA extracts consists of short DNA molecules3,4. To test this, we calculated the number of PCR duplicates found in the MBD+ fractions (relative to the MBD− fractions, MQ25 filter; Supplementary Table S2.2), which provides a measure of the loss of molecular complexity in the MBD+ fraction. We observed an inverse relationship with the median size of MBD+ endogenous inserts (Supplementary Fig. S3). No relationship was found with other DNA damage related parameters (data not shown), confirming that the level of DNA fragmentation is a fundamental limitation in respect to the efficiency of MBD enrichment.
DNA damage
Given that post-mortem cytosine deamination transforms methylated CpGs into TpGs and that the efficiency of MBD2−Fc binding increases for mCpG-rich DNA templates, we next evaluated whether MBD enrichment was biased against recovering fragments altered by post-mortem cytosine deamination, as described by Smith and colleagues22. We used mapDamage 227 to quantify a series of post-mortem DNA damage parameters for each DNA library, including cytosine deamination rates at overhangs (δs) and in double-stranded (δd) DNA contexts and a proxy for the average length of overhanging ends (λ). We applied a threshold of 2,000 high-quality hits (MQ25 filter) as described above to the estimation of damage parameters.
Although not statistically significant (Supplementary Tables S4.3, S4.4 and S4.5), we found that both δs and δd rates were generally lower in the MBD+ fraction than in the MBD− fraction of a given specimen (Fig. 3C,D). On average, we found a 1.8-fold reduction in the δd rates of MBD+ fractions (relative to MBD− fractions). This reduction was of lower magnitude for δs rates (1.2-fold). This probably reflects that mCpGs present in overhangs of aDNA templates have a limited contribution to DNA binding to MBD2−Fc, probably owing to their limited size (estimated here to be 0.9–2.8 bp-long on average; Supplementary Fig. S2). Accordingly, no particular shift was observed in the length of overhangs from MBD+ and MBD− fractions (Supplementary Table S4.5, Supplementary Fig. S2).
We next evaluated whether cytosine deamination occurs at faster rates when CpG dinucleotides are methylated, as expected from genetic assay based on reversion of mutations in single-stranded28 and double-stranded29 DNA contexts and as previously observed in archeological barley material from Qasr-Ibrim, Egypt22. For this, we used C→T mutation rates to calculate the relative proportion of cytosine deaminations detected at CpG sites and other CpN dinucleotide contexts (MQ25 filter; Fig. 4). We observed that this proportion was up to 8.3-fold higher in endogenous sequences from the MBD+ fractions relative to MBD− fractions (Supplementary Table S3.6), except for two samples showing minimal sequence data (MKm_2 and MZm_1). This adds to the body of empirical evidence supporting faster cytosine deamination rates at methylated epialleles22,29. This fast decay of mCpGs also participates to reducing the molecular complexity of MBD+ libraries.

Relative amount of CpGs to TpGs transitions over all other kind of possible C->T mutations in CpN contexts.
A minimum threshold of 2,000 reads was applied.
Depth-of-coverage
We next tested for differential patterns of depth-of-coverage between MBD+ and MBD− fractions (Fig. 5). These analyses were restricted to samples showing sufficient sequencing data (namely polar bear, the Palaeo-Eskimo Saqqaq and the Khroma mammoth bones and hair/skin samples) and were normalized by the overall depth-of-coverage for bases covered at least once and performed on both quality-filtered (MQ25) and not quality-filtered (MQ0) reads. MBD+ distributions were shifted towards higher coverage when compared to MBD− distributions (Fig. 5). This indicates that for a given sequencing effort, MBD enrichment can achieve the sequencing of target regions to a higher coverage relative to no enrichment.

Per-base coverage distributions.
The dominant class of uncovered bases is not shown and the distributions are restricted to bases covered at least once in the nuclear genome. A minimum threshold of 2,000 reads was applied.
Microbial contents
Although MBD enrichment is designed to target vertebrate methylated DNA, MBD+ libraries were found to include DNA inserts of microbial origin (Supplementary Table S6). To compare the bacterial diversity of MBD+ and MBD− fractions, we excluded samples for which less than 2 genera were identified in either of the two fractions. The microbial profiles obtained from MBD− fractions segregated according to the type of environment in which the samples were collected (permafrost for mammoths and polar bears vs. caves for equids; Supplementary Fig. S7). At the genus-level, MBD+ and MBD− fractions were more similar to soil microbial communities than to a broad repertoire of human microbiomes (Supplementary Fig. S6). This is in line with previous studies5 and rules out extensive DNA contamination from microbes of human origin during and after excavation.
For most samples, MBD+ and MBD− fractions showed similar Shannon diversity indices (Supplementary Table S7) and little segregation by hierarchical clustering (Supplementary Fig. S5). This confirms that MBD enrichment does not significantly bias microbial profiles towards a particular bacterial group, as previously shown19. The equine specimens, however, except for sample EQB, stand as an exception. In these samples, class-level profiles showed differences between the MBD+ and MBD− fractions (Supplementary Fig. S8) and Linear Discriminant Analyses (LDA)30 identified significant enrichment for Afipia, Rhodopseudomonas, unclassified Methylocystaceae and Acetobacteraceae in MBD− fractions and for Burkholderia and Pseudomonas in MBD+ fractions (Supplementary Fig. S9). This suggests that, in these cases, MBD enrichment shows some level of selectivity with regard to the enrichment of certain microbial DNA present in the DNA extracts.
Discussion
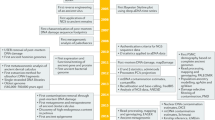
The recent discovery that epigenetic marks can be tracked through evolutionary time has opened new avenues for aDNA research22,31,32,33. In this study, we explore the efficiency and potential biases that may result from MBD enrichment of aDNA extracts (Fig. 6). Not unexpectedly, the method appears to be principally biased towards recovering CpG-rich templates. As a result, long molecules, which show a higher density of CpGs and therefore higher binding potential to MBD2−Fc, are preferentially enriched. The specificity of the method toward mCpGs is also apparent in the Palaeo-Eskimo Saqqaq sequence data, where the MBD+ fraction shows higher regional methylation scores than the MBD− fraction, in the lower coverage of mtDNA and CGI and in the over-representation of TEs in MBD+ fractions. In addition, DNA templates for which fewer cytosines have been deaminated (and therefore for which fewer mCpGs were converted to TpGs, which represent inappropriate targets for MBD enrichment), are over-represented in enriched fractions.

Properties of MBD enrichment for ancient DNA.
The MBD+ fraction favors DNA templates with a longer size and a higher number of mCpGs. This represents a limited fraction of the molecules present in aDNA extracts. mtDNA is under-represented in the MBD+ fraction while higher coverage is observed for hyper-methylated regions of the nuclear genome, including TEs.
These features have several important consequences for the analysis of aDNA extracts. First, aDNA molecules are extensively fragmented after an organism dies. Real-time PCR experiments have shown a 10 to 100-fold decrease in the number of amplifiable templates for every doubling of the amplicon sizes34. NGS data from a range of environmental conditions have confirmed that endogenous aDNA molecules become exponentially rare with increasing fragment length. Although nucleosomes can sometimes protect DNA from fragmentation and introduce periodicity patterns in depth-of-coverage and size distributions26, DNA inserts rarely exceed 100 bp. The observed bias towards recovering long DNA fragments, thus, unavoidably results in a decrease in the total number of molecules that are available for sequencing in the MBD+ fractions post-enrichment, which translates into reduced molecular complexity (and increased clonality) of DNA libraries. This bias is enhanced due to the faster cytosine deamination rates observed for methylated epialleles, since the relatively few number of methylated sites present in short templates will be relatively rapidly converted into inappropriate TpG targets. Additionally, from the billions of nucleotides that comprise mammalian genomes, only ~30 million CpGs are found20,35, of which only a subset are methylated in a tissue-specific manner. This means that the pool of endogenous molecules available for MBD enrichment shows lower complexity than the overall genome. We also found that microbial and organellar DNA templates represent an important proportion within MBD+ fractions and with the experimental conditions presented here, the population of endogenous sequences that lack CpGs but are still present in MBD+ fractions represents 1.0–3.2% of the data for polar bears, 2.9–30.9% for equids, 13.4–48.8% for woolly mammoths and 46.28% for the Palaeo-Eskimo Saqqaq (Supplementary Tables S3.4). Altogether, the relatively limited efficiency and drop in complexity associated with MBD enrichment as well as the substantial off-target carry-over observed suggest that MBD enrichment is probably not adequate for analyzing the vast majority of aDNA extracts, which generally show extensive DNA fragmentation, deamination and microbial contamination.
However, despite the biases described above, MBD enrichment provided access to a fraction of the methylome from a diversity of remains originating from both permafrost and non-permafrost environments. This demonstrates that a significant proportion of mCpGs are still intact in a variety of aDNA extracts. Importantly, for the three polar bear samples, the total fraction of the genome that is covered after sequencing MBD+ fractions, as calculated from the cumulative length of unique high-quality hits observed for every million sequences generated, was found to be, on average, higher in MBD+ than in MBD− fractions (0.041-fold vs. 0.027-fold; Supplementary Table S2.4). It follows that for these samples, the reduction in complexity and endogenous content of DNA libraries constructed from MBD+ fractions is compensated for by an increase in the DNA library insert size. Common to these samples is a high endogenous content (23.3–46.7%; Supplementary Table S2.1) and relatively moderate fragmentation levels (endogenous DNA fragments in MBD+ and MBD− fractions are on average 62.3 and 100.0 bp long, which is 48.1–99.4 bp longer than that of the Palaeo-Eskimo Saqqaq; Fig. 3B, Supplementary Table S4.1).
We therefore suggest that for similar samples, MBD enrichment holds the potential to be used to obtain complete genome sequences while measuring ancient methylation levels at the same time. This most certainly restricts the applicability of the method to the most recent and/or most preserved specimens, which represents only a minority of specimens present in the paleontological and archeological record. We, thus, caution against the method for DNA extracts showing significant levels of deamination and fragmentation (e.g. median sizes ≤80 bp).
Ideally, both MBD+ and MBD− fractions should be sequenced to help identify regions differentially covered in the two fractions. This might provide in the future an alternative to methods currently available for recovering ancient epigenetic information based on bisulphite conversion31 and differential patterns of CpG→TpG mutations at methylated and unmethylated sites26,32. Given the decrease in endogenous DNA content and the increase in clonality observed for MBD+ libraries, we recommend performing MBD enrichment on large volumes of aDNA extracts whenever possible. This will increase the pool of DNA templates available for enrichment and the molecular complexity of DNA libraries constructed on MBD+ fractions. Alternatively, DNA extracts that have been concentrated following additional purification could be used for MBD enrichment (although this will also lead to a substantial loss of DNA molecules during purification). In recognition of the observed bias towards CpG-rich DNA fragments, we recommend the use of specific DNA polymerases, such as Accuprime Pfx, and, more generally, PCR conditions that limit the over-amplification of %GC-rich DNA templates36. Other modifications to the procedure described here, for example involving different washing and elution conditions following DNA binding to MBD2−Fc37, might also contribute toward releasing the whole diversity of methylated DNA molecules present in the extract and limiting the carry-over of organellar and other unmethylated templates into the MBD+ fraction.
We note that the observed preference for mCpG-rich fragments introduces a bias in the quantification of post-mortem DNA degradation. Specifically, DNA templates where mCpG sites have been less affected by cytosine deamination – the most common aDNA damage – are more likely to be enriched. Epialleles also show higher deamination rates while methylated than not. The resultant pool of DNA fragments that make up the MBD+ fraction is, thus, unlikely to reflect the true level of post-mortem DNA damage. This observation was first noted by Smith and colleagues22, who described deamination reactions at unmethylated and methylated cytosines as two complementary forces, both contributing to the global estimate of post-mortem DNA deamination rates.
Although DNA degradation reactions are known to take place at faster rates with increasing temperature at depositional sites38, we observed striking differences in the molecular preservation of the different woolly mammoth tissues (Table 1; Fig. 3). For Khroma, bone endogenous DNA content was 1.6 to 140-fold higher than in soft tissues (Supplementary Table S2.1) and cytosine deamination occurred up to 2.4-fold slower (Fig. 3, Supplementary Table S4.4). This suggests that DNA degradation and post-mortem microbial colonization are greater in soft tissues, most likely due to the rapid collapse of cellular membranes and disruption of the tissue organization, are greater in soft tissues, as previously suggested39.
Our data also indicate that cytosine deamination levels cannot provide a direct estimate of the time since a fossil was buried, since we observed a large variation in cytosine deamination levels at overhangs across the tissues from the same mammoth specimen (Khroma; Fig. 3D, Supplementary Table S4.4). This contrasts with the linear relationship between time and C→T nucleotide misincorporation rates at sequence starts observed by Sawyer and colleagues40 in a diversity of preservation environments, but is in line with other studies inside41 and outside17 permafrost regions, where high variability of deamination rates where observed across DNA libraries prepared from the same bone samples42. This is also in agreement with previous reports showing different deamination rates in the DNA molecules isolated from pathogenic microbes and those from their human host43.
In this study, we have identified DNA fragmentation and degradation as the most important limiting factor for the efficiency of MBD enrichment. We have suggested experimental conditions and procedures to help characterize ancient methylation marks through MBD enrichment. Pending technologies capable of accurately identifying base modifications while sequencing, MBD enrichment provides a method complementary to those currently available for characterizing ancient methylomes but appears only appropriate for a limited fraction of ancient specimens affected by minimal DNA fragmentation and deamination. Together, these approaches are essential steps towards the evaluation of the role of epigenetic reprogramming during major evolutionary transitions33, such as the Industrial Revolution, the transition from hunting-gathering to farming, the domestication of plants and animals and the climatic warming that followed the Last Glacial Maximum.
Methods
Samples and DNA extraction
We extracted aDNA from 20 ancient and palaeontological specimens (human, mammoth, polar bear and several equine species) using previously published methods from6,41,44 (Table 1 and Supplementary Table S1). DNA extraction, MBD enrichment and DNA library building were performed in state-of-the-art aDNA facilities at the Centre for GeoGenetics, Copenhagen, Denmark. Specimens were selected to represent a range of time periods (Holocene and Late Pleistocene) and environmental preservation conditions (permafrost and caves).
Since methylomes are tissue-specific, we investigated the robustness of the MBD enrichment approach to tissue types using both calcified and soft tissues, taking advantage of two exceptional woolly mammoth specimens known as Khroma and Zhenya. Khroma was excavated in October 2008 in the Khroma River, Yana-Indigirka lowland, Yakutia, Eastern Siberia; the specimen’s radiocarbon age was >45,000 years45. We extracted 0.137 to 1.350 grams of fresh samples from skeletal muscle, a piece of skin, a piece of skin covered with hair and two different bones. For Zhenya, which was radiocarbon dated to 44,750 (+950/−700) year BP and excavated in September 2012 on the right bank of the Yenisei River Gulf, Taymyr Peninsula, Western Siberia46, 0.195–1.447 grams of fresh samples of liver, skeletal muscle and cardiac muscles were extracted for DNA. All aDNA extracts were stored at −20 °C before being subjected to MBD enrichment.
MBD enrichment
MBD enrichment of the aDNA extracts was performed using either the EpiMark® Methylated DNA Enrichment Kit (catalogue number E2600, New England BioLabs) or the NEBNext® Microbiome DNA Enrichment Kit (E2612), with slight modifications (Supplementary Table S1). For each enrichment reaction, we coupled 10 μL of human MBD2 protein fused to the Fc tail of human IgG1 (MBD2−Fc) to 1 μL of paramagnetic protein A beads (MBD2−Fc/Protein A Magnetic Bead) by incubating with 11 μL of 1X Bind/Wash Buffer for 15 min at Room Temperature (RT) with agitation. MBD2−Fc/Protein A Magnetic Beads were then concentrated using a magnetic rack and washed twice using 1 mL 1X Bind/Wash Buffer before being re-suspended in 11 μL 1X Bind/Wash Buffer. A volume of 10 μL of washed beads was mixed with 21.25–50 μL of aDNA extract, 20 μL of 5X Bind/Wash Buffer and supplemented with sterile water up to 100 μL, before being incubated for 20 minutes at RT with agitation. The fraction of DNA molecules that could bind to MBD2−Fc/Protein A Magnetic Beads was concentrated using a magnetic rack. The supernatant was saved as the fraction of the extract that could not bind to the MBD2−Fc/Protein A Magnetic Beads (hereafter, MBD−). MBD2−Fc/Protein A Magnetic Beads were washed three times with 1 mL of 1X Bind/Wash Buffer (or 1 mL of 0.4 M KCl for the Palaeo-Eskimo Saqqaq sample), before the fraction enriched for methylated DNA (hereafter, MBD+) was eluted in 150 μL TE with 15 μL proteinase K after 30 min incubation at 37 °C (except for the Palaeo-Eskimo Saqqaq MBD+, which was eluted in 100 μl of 1 M KCl, after 1 h incubation at 37 °C). Both MBD+ and MBD− fractions were purified using MinElute columns (QIAGEN) and 21.25 μL of EB buffer following the manufacturer’s instructions, except that the final elution was performed following a 15 min incubation step at 37 °C. For one polar bear sample (PB44), this MinElute purification step was replaced by ethanol precipitation and resuspension in 21.25 μL EB and 15 min incubation at 37 °C (Supplementary Table S1).
DNA library preparation and sequencing
For each sample, we constructed Illumina DNA libraries using both MBD+ and MBD− fractions and the procedure from Meyer and Kicher (2010)47, with the slight modifications from Seguin-Orlando and colleagues48. Half the volume of each library was amplified in a 50 μL PCR reaction using 5 units AmpliTaq Gold (Life Technologies). PCR cycling conditions consisted of an initial denaturation step for 10 min at 92 °C, followed by 8 to 15 cycles of 30 sec denaturation at 92 °C, 30 sec annealing at 60 °C and 40 sec elongation at 72 °C. The final elongation step lasted for 7 min at 72 °C. When flat DNA profiles were observed using the 2100 Bioanalyzer (Agilent) High-Sensitivity DNA Assay, a second round of PCR amplification for 6–12 cycles was performed in four parallel reactions, as described by Seguin-Orlando and colleagues48. Final products were purified on a MinElute column (QIAGEN) and eluted in 20 μL EB after 15 min incubation at 37 °C. Amplified libraries were quantified using the 2100 Bioanalyzer (Agilent) High-Sensitivity DNA Assay, pooled with other indexed libraries and sequenced on Illumina HiSeq 2500 and MiSeq platforms at the Danish National High-Throughput DNA Sequencing Centre. For all mammoth samples, the other half of each DNA library was subjected to two more independent rounds of PCR amplification and shipped to the Kurtchatov Institute, Moscow, Russia for sequencing on the GAIIx platform. For the Palaeo-Eskimo Saqqaq sample, both halves of the DNA libraries were amplified independently and sequenced in Denmark. The first position of both pair-ended reads generated in Russia was trimmed due to base calling issues at that position, after checking for DNA damage signature profiles with mapDamage 227. All sequence information generated in this study is available for download at the Short Read Archive (Accession Nb. PRJNA260386).
Sequence alignment
Sequencing reads were aligned against reference genomes using the PALEOMIX pipeline49. Seeding was disabled while mapping and two different alignment files were obtained per sample, one without applying a mapping quality threshold (MQ0) and one filtering for a mapping quality above 25 (MQ25 filter). For equine samples, sequencing reads were aligned against the nuclear chromosomes from the horse reference genome EquCab250 and against the horse mitochondrial reference genome (Accession Nb. NC_001640.1)51. The sequence data from the Palaeo-Eskimo Saqqaq individual was aligned to the GRCh37/hg19 assembly of the human genome and to the rCRS sequence (Accession Nb. NC_012920) for the mtDNA52,53. The scaffolds from the African elephant genome loxAfr3 (Accession Nb. GCA_000001905.1) and the Mammuthus primigenius mitochondrial sequence from Rogaev and colleagues54 (Accession Nb. DQ316067) were used for aligning mammoth reads. For mammoth and equine specimens, we used standard mapping parameters, except that the -n option from BWA was relaxed to 0.03 to compensate for the evolutionary distance to the genomes used as reference. The polar bear nuclear scaffolds from Miller and colleagues55 (SRA054912) and the mitochondrial reference from Lindqvist and colleagues56 (Accession Nb. GU573488.1) were used to align sequencing reads generated from polar bear DNA libraries.
All the analyses described below were performed on samples that showed a minimum number of 2,000 reads for the nuDNA and 20 reads for the mtDNA.
MBD+ vs. MBD− comparisons
DNA damage
The package mapDamage 227 was used to characterize DNA fragment length distributions and nucleotide misincorporation patterns. We also estimated the parameters for the DNA damage model, namely deamination rates in double strands (δd) and single strands (δs) and the probability of reads not terminating in overhangs (λ). The analyses were run with BAM alignments, after removing duplicates and filtering for a minimum mapping quality of 25, against reference genomes as input files, with default settings. We also evaluated whether MBD+ and MBD− fractions differed in their amount of DNA damage using the two-sample t-test on the posterior means of the damage parameters δd, δs and λ. All statistical tests were implemented in R version 3.0.157. Tests were carried out as three independent statistical comparisons between MBD+ and MBD− fractions on samples carrying at least 2,000 reads and on species with a minimum of three samples: Homo sapiens (HSSR), Mammuthus primigenius (Khroma, Zhenya), Ursus maritimus (PB9, PB25, PB44) and Equus ferus (EQ26, EQ34, EQ36).
Read size, GC content and CpG densities
We performed a linear mixed effect analysis to compare MBD+ and MBD− fractions for their size distribution, %CG and CpG densities, all calculated using seqtk comp (available at https://github.com/lh3/seqtk) on collapsed reads. We used a linear mixed effect model, with the overall mean μ and the fraction type covariate τi as fixed effects:


The within-sample dependence is modeled as the random factor νj; furthermore the error εijk and the νj terms are independent and identically distributed, respectively. Mixed effects model fitting was achieved by applying the lmer function (option REML = FALSE) in the R package lme4 (http://cran.r-project.org/package=achievedlme4). The asymptotic chi-square test was used to assess the statistical significance of the likelihood difference.
Linear mixed models were restricted to samples carrying a minimum of 2,000 collapsed reads in each MBD+ and MBD− fractions (one Homo sapiens - the Palaeo-Eskimo Saqqaq individual, twelve samples of Mammuthus primigenius, three samples of Ursus maritimus, three samples of Equus ferus and one sample of Equus lambei). The availability of different tissues for the mammoth specimens allowed us to implement one linear model per library, enabling each tissue type to be modelled separately.
Dimer composition
The relative representation of CpGs and all other dinucleotides in the two fractions (MBD+ and MDB−) was calculated as the total number of each possible dinucleotide normalized over the total number of dinucleotides observed. The calculation was restricted to samples carrying at least 2,000 collapsed reads. Differences between MBD+ and MBD− fractions were statistically assessed by two-sample t-tests performed on mean distributions. We carried out three independent statistical comparisons between MBD+ and MBD− fractions with respect to the three species with a minimum of three samples: Mammuthus primigenius (Khroma, Zhenya), Ursus maritimus (PB9, PB25, PB44) and Equus ferus (EQ26, EQ34, EQ,36).
Genome coverage
We investigated possible differences in patterns of genome coverage between samples, tissues and MBD+/MBD− fractions, focusing on the Palaeo-Eskimo Saqqaq individual, polar bear samples and the mammoth bones, hair and skin tissues, which all showed enough sequencing data to implement such analyses (above 0.0005X mean genome coverage). We used samtools depth58 to retrieve the distribution of coverage for positions covered at least once. Spearman’s rank-correlation was carried out on a random subset of two million reads per sample to statistically assess the relationships between MBD+ and MBD− fractions for differential coverage per mapping position in the nuclear genome.
Distribution of transposable elements
We compared MBD+ and MBD− fractions for their distribution of TEs using two complementary approaches. First, we used the proportion of reads with one or more sub-optimal reads (non-null X1:i tag and null X0:1 tag) as a proxy for TEs, since TEs represent regions found at multiple copies in the genome. Secondly, we directly estimated the relative amount of repetitive elements in MBD+ and MBD− fractions, using the human, equine and elephantid sequence data, as BED annotations for repetitive elements were available from the UCSC Genome Browser and for the polar bear we used the annotation kindly made available by the authors from Liu and colleagues59. Coverage within and outside annotated repetitive elements was calculated using samtools depth58.
Distribution of CpG islands
We compared the MBD+ and MBD− fractions for their representation in CpG islands (CGIs), which are globally hypo-methylated25. This analysis was restricted to the human and equine data, since BED annotations of those genomes were available for CGIs. Samples showing less than 2,000 collapsed reads mapping to such regions were disregarded to enable reliable coverage estimates. Coverage within and outside the CpG islands was calculated using the coverage tool from PALEOMIX49.
Read length and presence of CpG
We tested if the length shift toward longer reads in the MBD+ fraction was related to the presence of CpGs by calculating the relative abundance of reads showing the presence of at least one CpG in MBD+ and MBD− within different size categories (<30 bp, 30–40 bp, 40–50 bp, 50–60 bp, 60–70 bp, 70–80 bp, 80–90 bp, 90–100 bp, 100–200 bp, 200–300 bp, >300 bp). The number of reads with at least one CpG within a category was normalized over the total number of reads in that category and the relative importance of that specific category in the total number of reads considered.
MBD+ and MBD− microbial profiling
We compared MBD+ and MBD− fractions for their distribution of microbial taxonomic groups and their respective relative abundances. Microbial profiles were obtained using MetaPhlAn (version 1.7.760) as implemented within the metagenomic module of PALEOMIX49 and as described in Der Sarkissian and colleagues5 and Schubert and colleagues49. To limit the impact of false positive identification in downstream analyses, low-abundance taxa (less than 1%) were disregarded.
The Shannon diversity index of each microbial profile was computed using genus relative abundance data and the function diversity of the vegan package in R (http://cran.r-project.org/package=vegan). Principal Coordinate Analysis (PCoA) of Bray-Curtis distances among profiles was performed at the genus level using the R function pcoa. Manhattan distance-based relationships amongst profiles were assessed at the genus level by hierarchical clustering using the R package pvclust61. The support for each cluster was estimated through p-values (Approximately Unbiased and Bootstrap Probabilities) estimated from 10,000 bootstrap pseudo-replicates. Further statistical comparisons between the microbial profiles of MBD+ and MBD− fractions were carried out using the program LEfSe30. Consistent differences in taxon abundances were tested using the non-parametric Kruskal-Wallis sum-rank test and the unpaired Wilcoxon test. A linear discriminant analysis also estimated the effect size of taxonomical covariates at the origin of the fraction differences (threshold on logarithmic linear discriminant analysis score for discriminative features = 2.0). Microbial profiles were also compared with publicly available MetaPhlAn profiles of soil5,62 and human samples63 by PCoA based on Bray-Curtis distances.
Additional Information
Accession codes: All sequences generated in this study are available for download at the Short Read Archive (Accession Nb. PRJNA260386).
How to cite this article: Seguin-Orlando, A. et al. Pros and cons of methylation-based enrichment methods for ancient DNA. Sci. Rep. 5, 11826; doi: 10.1038/srep11826 (2015).
References
Orlando, L. & Cooper, A. Using Ancient DNA to Understand Evolutionary and Ecological Processes. Annu. Rev. Ecol. Evol. Syst. 45, 573–598 (2014).
Ermini, L., Der Sarkissian, C., Willerslev, E. & Orlando, L. Major transitions in human evolution revisited: A tribute to ancient DNA. J. Hum. Evol. 79, 4–20 (2014).
Der Sarkissian, C. et al. Ancient genomics. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20130387 (2015).
Shapiro, B. & Hofreiter, M. A Paleogenomic Perspective on Evolution and Gene Function: New Insights from Ancient DNA. Science 343, 1236573 (2014).
Der Sarkissian, C. et al. Shotgun microbial profiling of fossil remains. Mol. Ecol. 23, 1780–1798 (2014).
Rasmussen, M. et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature 463, 757–762 (2010).
Meyer, M. et al. A High-Coverage Genome Sequence from an Archaic Denisovan Individual. Science 338, 222–226 (2012).
Schubert, M. et al. Prehistoric genomes reveal the genetic foundation and cost of horse domestication. Proc. Natl. Acad. Sci. USA 111, E5661–9 (2014).
Ávila-Arcos, M. C. et al. Application and comparison of large-scale solution-based DNA capture-enrichment methods on ancient DNA. Sci. Rep. 1, 74 (2011).
Gansauge, M.-T. & Meyer, M. Selective enrichment of damaged DNA molecules for ancient genome sequencing. Genome Res. 24, 1543–1549 (2014).
Fabre, P.-H. et al. Rodents of the Caribbean: origin and diversification of hutias unravelled by next-generation museomics. Biol. Lett. 10, 20140266 (2014).
Schuenemann, V. J. et al. Targeted enrichment of ancient pathogens yielding the pPCP1 plasmid of Yersinia pestis from victims of the Black Death. Proc. Natl. Acad. Sci. USA 108, E746–752 (2011).
Fu, Q. et al. DNA analysis of an early modern human from Tianyuan Cave, China. Proc. Natl. Acad. Sci. USA 110, 2223–2227 (2013).
Carpenter, M. L. et al. Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries. Am. J. Hum. Genet. 93, 852–864 (2013).
Enk, J. M. et al. Ancient whole genome enrichment using baits built from modern DNA. Mol. Biol. Evol. 31, 1292–1294 (2014).
Briggs, A. W. et al. Targeted Retrieval and Analysis of Five Neandertal mtDNA Genomes. Science 325, 318–321 (2009).
Rohland, N., Harney, E., Mallick, S., Nordenfelt, S. & Reich, D. Partial uracil-DNA-glycosylase treatment for screening of ancient DNA. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20130624 (2015).
Mason, V. C., Li, G., Helgen, K. M. & Murphy, W. J. Efficient cross-species capture hybridization and next-generation sequencing of mitochondrial genomes from noninvasively sampled museum specimens. Genome Res. 21, 1695–1704 (2011).
Feehery, G. R. et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS One 8, e76096 (2013).
Ziller, M. J. et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–481 (2013).
Murray, I. A. et al. The methylomes of six bacteria. Nucleic Acids Res. 40, 11450–11462 (2012).
Smith, O. et al. Genomic methylation patterns in archaeological barley show de-methylation as a time-dependent diagenetic process. Sci. Rep. 4, 5559 (2014).
Schubert, M. et al. Improving ancient DNA read mapping against modern reference genomes. BMC Genomics 13, 178 (2012).
Rebelo, A. P., Williams, S. L. & Moraes, C. T. In vivo methylation of mtDNA reveals the dynamics of protein-mtDNA interactions. Nucleic Acids Res. 37, 6701–6715 (2009).
Deaton, A. M. & Bird, A. CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022 (2011).
Pedersen, J. S. et al. Genome-wide nucleosome map and cytosine methylation levels of an ancient human genome. Genome Res. 24, 454–466 (2014).
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. F. & Orlando, L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013).
Frederico, L. A., Kunkel, T. A. & Shaw, B. R. A sensitive genetic assay for the detection of cytosine deamination: determination of rate constants and the activation energy. Biochemistry 29, 2532–2537 (1990).
Shen, J. C., Rideout, W. M., 3rd & Jones, P. A. The rate of hydrolytic deamination of 5-methylcytosine in double-stranded DNA. Nucleic Acids Res. 22, 972–976 (1994).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011).
Llamas, B. et al. High-resolution analysis of cytosine methylation in ancient DNA. PLoS One 7, e30226 (2012).
Gokhman, D. et al. Reconstructing the DNA Methylation Maps of the Neandertal and the Denisovan. Science 344, 523–527 (2014).
Orlando, L. & Willerslev, E. Evolution. An epigenetic window into the past? Science 345, 511–512 (2014).
Schwarz, C. et al. New insights from old bones: DNA preservation and degradation in permafrost preserved mammoth remains. Nucleic Acids Res. 37, 3215–3229 (2009).
Sved, J. & Bird, A. The expected equilibrium of the CpG dinucleotide in vertebrate genomes under a mutation model. Proc. Natl. Acad. Sci. USA 87, 4692–4696 (1990).
Dabney, J. & Meyer, M. Length and GC-biases during sequencing library amplification: a comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. Biotechniques 52, 87–94 (2012).
Rauch, T. & Pfeifer, G. P. Methylated-CpG island recovery assay: a new technique for the rapid detection of methylated-CpG islands in cancer. Lab. Invest. 85, 1172–1180 (2005).
Smith, C. I., Chamberlain, A. T., Riley, M. S., Stringer, C. & Collins, M. J. The thermal history of human fossils and the likelihood of successful DNA amplification. J. Hum. Evol. 45, 203–217 (2003).
Forbes, S. L. Decomposition chemistry in a burial environment. Soil Analysis in Forensic Taphonomy. New York: CRC Press, Taylor & Francis 203–223 (2008).
Sawyer, S., Krause, J., Guschanski, K., Savolainen, V. & Pääbo, S. Temporal Patterns of Nucleotide Misincorporations and DNA Fragmentation in Ancient DNA. PLoS One 7, e34131 (2012).
Orlando, L. et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 499, 74–78 (2013).
Meyer, M. et al. A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature 505, 403–406 (2013).
Schuenemann, V. J. et al. Genome-wide comparison of medieval and modern Mycobacterium leprae. Science 341, 179–183 (2013).
Yang, D. Y., Eng, B., Waye, J. S., Dudar, J. C. & Saunders, S. R. Technical note: improved DNA extraction from ancient bones using silica-based spin columns. Am. J. Phys. Anthropol. 105, 539–543 (1998).
Fisher, D. C. et al. X-ray computed tomography of two mammoth calf mummies. J. Paleontol. 88, 664–675 (2014).
Mashchenko, E. et al. The partial carcass of the mammoth “Zhenya” (Mammuthus primigenius) from western Taymyr Peninsula, Russia: preliminary analysis and results. in VIth International Conference on Mammoths and their Relatives, Grevena - Siatista (ed. Scientific Annals, School of Geology, Aristotle University of Thessaloniki, Greece) 102, 121–122 (2014).
Meyer, M. & Kircher, M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb. Protoc. 2010, db.prot5448 (2010).
Seguin-Orlando, A. et al. Ligation Bias in Illumina Next-Generation DNA Libraries: Implications for Sequencing Ancient Genomes. PLoS One 8, e78575 (2013).
Schubert, M. et al. Characterization of ancient and modern genomes by SNP detection and phylogenomic and metagenomic analysis using PALEOMIX. Nat. Protoc. 9, 1056–1082 (2014).
Wade, C. M. et al. Genome sequence, comparative analysis and population genetics of the domestic horse. Science 326, 865–867 (2009).
Xu, X. & Arnason, U. The complete mitochondrial DNA sequence of the horse, Equus caballus: extensive heteroplasmy of the control region. Gene 148, 357–362 (1994).
Andrews, R. M. et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat. Genet. 23, 147 (1999).
Anderson, S. et al. Sequence and organization of the human mitochondrial genome. Nature 290, 457–465 (1981).
Rogaev, E. I. et al. Complete mitochondrial genome and phylogeny of Pleistocene mammoth Mammuthus primigenius. PLoS Biol. 4, e73 (2006).
Miller, W. et al. Polar and brown bear genomes reveal ancient admixture and demographic footprints of past climate change. Proc. Natl. Acad. Sci. USA 109, E2382–90 (2012).
Lindqvist, C. et al. Complete mitochondrial genome of a Pleistocene jawbone unveils the origin of polar bear. Proc. Natl. Acad. Sci. USA 107, 5053–5057 (2010).
Core Team, R. R.: A Language and Environment for Statistical Computing. (2014).
Li, H. et al. The Sequence Alignment/Map (SAM) Format and SAMtools. Bioinformatics btp352 25, 2078–2079 (2009).
Liu, S. et al. Population genomics reveal recent speciation and rapid evolutionary adaptation in polar bears. Cell 157, 785–794 (2014).
Segata, N. et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 9, 811–814 (2012).
Suzuki, R. & Shimodaira, H. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542 (2006).
Fierer, N. et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc. Natl. Acad. Sci. USA 109, 21390–21395 (2012).
The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012).
Acknowledgements
This work was supported by the Danish Council for Independent Research, Natural Sciences (FNU); the Danish National Research Foundation (DNRF94); “Chaires d’Attractivité 2014” IDEX, University of Toulouse; a Marie-Curie Career Integration grant (FP7 CIG-293845); the International Research Group Program (IRG14-08), Deanship of Scientific Research (King Saud University, Saudi Arabia). CG and LE were supported by Marie-Curie Intra-European Fellowships (FP7-IEF-328024 and FP7 IEF-302617). BS was supported by the Gordon and Betty Moore Foundation. Equid samples were obtained from the GNL Quintero project and the Natural and Cultural History Museum of the Atacama Desert (Museo de Historia Natural y Cultural del Desierto de Atacama). We thank the Smithsonian Institution for allowing us to sample Equus lambei and use its data for this study; S. Liu for providing access to the annotations on the polar bear genome; T. Brand, P. Selmer Olsen and the staff of the Danish National High-throughput DNA Sequencing Centre for technical assistance.
Author information
Authors and Affiliations
Contributions
L.O. designed and coordinated the study. A.S.O., E.B. and L.O. performed molecular experiments. C.G., C.D., L.E., G.L., A.S. and L.O. analyzed data. A.N., E.L., P.L., G.M.D., E.S., A.T., T.S., B.S., E.W., E.P., K.A.S.A. and L.O. provided samples, material and reagents. C.G., C.D., L.E. and L.O. wrote the article, with significant input from A.S.O., E.L., T.S., A.H.A., S.A.A., K.A.S.A. and B.S. and all other coauthors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Seguin-Orlando, A., Gamba, C., Sarkissian, C. et al. Pros and cons of methylation-based enrichment methods for ancient DNA. Sci Rep 5, 11826 (2015). https://doi.org/10.1038/srep11826
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11826
This article is cited by
-
Ancient DNA analysis
Nature Reviews Methods Primers (2021)
-
A whole mitochondria analysis of the Tyrolean Iceman’s leather provides insights into the animal sources of Copper Age clothing
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
