
Abstract
Several strategies have been used to clone large DNA fragments directly from bacterial genome. Most of these approaches are based on different site-specific recombination systems consisting of a specialized recombinase and its target sites. In this study, a novel strategy based on phage ϕBT1 integrase-mediated site-specific recombination was developed and used for simultaneous Streptomyces genome engineering and cloning of antibiotic gene clusters. This method has been proved successful for the cloning of actinorhodin gene cluster from Streptomyces coelicolor M145, napsamycin gene cluster and daptomycin gene cluster from Streptomyces roseosporus NRRL 15998 at a frequency higher than 80%. Furthermore, the system could be used to increase the titer of antibiotics as we demonstrated with actinorhodin and daptomycin and it will be broadly applicable in many Streptomyces.
Similar content being viewed by others


Introduction
Streptomyces are high-GC Gram-positive bacteria well known for their ability to produce a wide variety of medically and agriculturally useful antibiotics and related compounds1. Genes responsible for the biosynthesis of a specific secondary metabolite are usually arranged in clusters that vary in size from a few to over 100 kb2. To gain insight into the biosynthesis and regulation of antibiotics in Streptomyces, it is of great importance to clone their gene clusters. Recently, various approaches have been developed to clone gene clusters directly from bacterial genomic DNA. These methods include RecET-mediated linear-plus-linear homologous recombination (LLHR)3, oriT-directed capture system4 and transformation-associated recombination (TAR)5. The RecET-mediated LLHR was successful in cloning gene clusters (10 to 52 kb in length) from the genome of Photorhabdus luminescens into expression vectors in Escherichia coli3. The oriT-directed capture system has been used to clone regions up to 140 kb from the genome of Burkholderia pseudomallei6 and 200 kb from megaplasmid of Sinorhizobium meliloti4. However, the use of this system was limited to Gram-negative bacteria that can be established as conjugation donors6. Taking advantage of the natural in vivo homologous recombination of Saccharomyces cerevisiae, TAR cloning strategy was used to capture a 21.3 kb enterocin gene cluster from Salinispora pacifica CNT-1507 and a 67 kb taromycin A biosynthetic gene cluster from Saccharomonospora sp. CNQ-4908.
The ability to delete large genomic fragments within Streptomyces genome is of great interest for genetic manipulations of Streptomyces. Several strategies have been developed for a number of bacteria. Some methods are based on the meganuclease I-SceI system which involves the meganuclease I-SceI of Saccharomyces cerevisiae and its 18 bp recognition sequence9,10. Many of them are based on site-specific recombination systems consisting of a specialized recombinase and its target sites. Nearly all site-specific recombinases fall into two families, the tyrosine recombinases and the serine recombinases11. The recombination systems of the tyrosine recombinase family include Cre/loxP from the P1 phage12, Dre/rox from the P1-like transducing phage D613 and the Flp/FRT from yeast14. The Cre, Dre and Flp proteins are the tyrosine recombinases which catalyze reciprocal site-specific recombination of DNA at loxP, rox and FRT sites, respectively. Integrases (Int) from Streptomyces temperate phage ϕC31 and ϕBT1 belong to serine recombinase family. They catalyze site-specific recombination of the phage attachment site (attP) with the bacterial attachment site (attB), resulting in the formation of two hybrid sites (attL and attR)15,16. Both ϕC31 and ϕBT1 attP-int loci have been used to construct versatile vectors which can integrate into different attB sites in Streptomyces15,16. To increase the diversity of attP-attB pair of ϕBT1, 15 mutated attP-attB pairs (attP01-attB01 → attP15-attB15) were generated by PCR mutagenesis of the central dinucleotide sequence of attB and attP17. The Cre/loxP system was successfully used for the deletion of large fragments in Magnetospirillum gryphiswaldense and several Streptomyces species18,19,20. However, the use of ϕC31 and ϕBT1 integrase in this aspect has not been exploited.
We devised a novel strategy for Streptomyces genome engineering and cloning of antibiotic gene clusters. This method is based on phage ϕBT1 attP-attB-int system and requires two single crossovers for targeted integration of mutated attB and attP into the recipient chromosome. Using the system, we easily cloned 25 kb fragment containing actinorhodin (act) gene cluster from S. coelicolor M145, 45 kb fragment containing napsamycin (nap) gene cluster and 157 kb fragment containing daptomycin (dap) gene cluster from S. roseosporus NRRL 15998. In addition, this method could be used to improve the titer of antibiotics by increasing copy numbers of antibiotic gene clusters.
Results
Construction of pUC119- and pKC1139-based plasmids
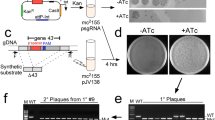
Our strategy used in this study requires both homologous and site-specific recombinations. The homologous recombinations were used for targeted integration of the mutated attB and attP into Streptomyces chromosome, while the ϕBT1 integrase-mediated site-specific recombination was employed to excise targeted region of interest from the chromosome (Fig. 1). The mutated attB and attP sites were chosen to avoid site specific recombination with the endogenous attB site in Streptomyces genome and consequently undesirable DNA rearrangements. Sites of attB6 and attP6 were randomly chosen from the 15 mutated attP-attB pairs. For the integration of attB6 into Streptomyces chromosome, pUC119-based suicide plasmids (pSV::attB6-act, pSV::attB6-nap and pSV::attB6-dap) were constructed. These plasmids are derivatives of pUC119 containing the kanamycin-resistance gene (neo), the origin of transfer (oriT) from plasmid RK2 (for the intergeneric conjugation between E. coli and Streptomyces), attB6 and a 2.0 kb homologous region flanking 5′ end of the targeted regions (Fig. S1a). We also constructed pKC1139-based plasmids (pKC1139::attP6-act, pKC1139::attP6-nap and pKC1139::attP6-dap) for the integration of attP6 into Streptomyces chromosome. These plasmids are derivatives of pKC1139 containing attP6 and a 2.0 kb homologous region flanking 3′ end of the targeted regions (Fig. S1b).

Schematic diagram of antibiotic gene cluster cloning from Streptomyces chromosome.
Initially, a pUC119-based suicide plasmid (pSV::attB6Up) carrying attB6 and a region homologous to 5′ end of the cluster is introduced into the chromosome by a single crossover. A second plasmid pKC1139::attP6Dn is based on pKC1139 carrying attP6 and a region homologous to 3′ end of the cluster. When the incubation temperature is higher than 34°C, pKC1139::attP6Dn turns into a non-replicating plasmid and then is integrated into the chromosome by a single crossover. Expression of ϕBT1 integrase (encoded in the plasmid pIJ10500) leads to excision of the pKC1139 backbone with gene cluster of interest, leaving behind the suicide vector pUC119::neo and 42 bp attL6 site. aac(3)IV: apramycin resistance gene; neo: kanamycin resistance gene; ori: temperature-sensitive origin of replication from pSG5; rep: rep encoding a replication initiator protein from pSG5.
Cloning of act gene cluster from S. coelicolor M145
To test this strategy, we first chose to clone the well-studied act gene cluster (SCO5070-SCO5092) from S. coelicolor M145. For this purpose, pSV::attB6-act and pKC1139::attP6-act were introduced into the recipient chromosomes via single-crossover homologous recombination to obtain double-cointegrate strain Sco-actB6P6 (Fig. 1). Further introduction of pIJ10500 (an integrative plasmid containing the ϕBT1 integrase gene) into Sco-actB6P6 allowed subsequent excision of 23 kb act gene cluster from S. coelicolor M145, leaving behind the suicide vector pUC119::neo, a scar of 42 bp attL site and pIJ10500 integrated within SCO4848. Excision of the gene cluster was confirmed by PCR analysis using both genomic and plasmid DNA as templates (Fig. 2). For 9 out of 10 exconjugates tested, the ϕBT1 integrase-mediated excision of act gene cluster occurred at a frequency of 90%. Furthermore, the presence of attL and attR in the amplified fragments was confirmed by DNA sequencing (Fig. 3). To recover the plasmid containing the entire act gene cluster (pKC1139::act) from Streptomyces, the DNA extract containing pKC1139::act from M145-MCact was used to transform E.coli Top10. Plasmid DNA from four apramycin resistant E. coli colonies was confirmed by BamHI and NotI digestion, respectively. The restriction fragments showed correct band patterns (Fig. S2a).

Confirmation of the excision events by PCR amplifications.
(A) The schematic diagram showing the position of primers in the chromosome of double-cointegrate strains. (B) Agarose gel electrophoresis showing PCR amplified fragments. PCR templates in the upper panels are genomic DNAs from S. coelicolor M145 or S. roseosporus NRRL 15998 (G) and ten randomly selected double-cointegrate strains with pIJ10500 (M145-MCact, Sro-MCnap or Sro-MCdap), while PCR templates in the lower panels are plasmid DNAs including pKC1139 (P) and ten different clones of pKC1139::act, pKC1139::nap and pKC1139::dap. The primers used and the expected size of amplification fragments were indicated.

Representative excision of the act gene cluster from S. coelicolor M145.
(A) Nucleotide sequence of attB6, attP6, attL6 and attR6. The mutated core dinucleotide (CT) at which the crossover occurs is in bold. (B) Verification of attL6 and attR6 by DNA sequencing. Sequences of attL6 and attR6 from DNA sequencing are underlined.
To verify the cluster is complete, the recombinant plasmid pKC1139::act was introduced into S. coelicolor M1146 (M1146) to obtain M1146-MCact. Unlike M1146 and M1146-pKC1139 (S. coelicolor M1146 containing empty vector pKC1139), M1146-MCact regained the ability to produce the blue pigment actinorhodin (Fig. 4a). These results showed that the cloned act gene cluster was complete and functional.

Comparison of actinorhodin production in S. coelicolor M145 and its derivatives.
(A) Comparison of actinorhodin production (blue pigment) of S. coelicolor M145 and its derivatives. Photograph was taken from the bottom of the plate after grown on R5MS agar medium for 4 days at 28°C. Representative image of three independent experiments with similar results was shown. (B) Actinorhodin titers of S. coelicolor M145 and its derivatives grown in 50 ml of R5MS at 28°C. Error bars show standard deviations.
Deletion of act gene cluster from S. coelicolor M145
To delete the act gene cluster from S. coelicolor M145, a single colony of M145-MCact was randomly chosen for the removal of pKC1139::act. After three rounds of nonselective growth at 28°C and subsequent cultivation at 40°C, approximately 5% of M145-MCact colonies lost pKC1139::act. Strains lacking the act gene cluster (M145-Dact) were first confirmed by PCR (data not shown) and then patched on R5MS solid agar plate for visual comparison of actinorhodin production. Unlike S. coelicolor M145 that could produce both blue pigment actinorhodin and red pigment undecylprodigiosins, M145-Dact could only produce the red pigment undecylprodigiosins (Fig. 4a). This was further validated by no actinorhodin production of M145-Dact in R5MS liquid culture (Fig. 4b).
Cloning and deletion of nap and dap gene cluster from S. roseosporus NRRL 15998
To clone gene cluster of medium and large sizes, we used the same strategy to clone nap and dap gene cluster from S. roseosporus NRRL 15998. Excision of nap gene cluster from S. roseosporus NRRL 15998 occurred in 9 out of 10 exconjugates and excision of dap gene cluster from S. roseosporus NRRL 15998 occurred in 8 out of 10 exconjugates (Fig. 2). Like pKC1139::act, plasmid containing nap gene cluster (pKC1139::nap) was passed through E.coli Top10 and isolated plasmid DNA was confirmed by BglII and EcoRI digestion, respectively (Fig. S2b). The cloned fragment covers a contiguous DNA region of 45 kb from SSGG02973 to SSGG03009. For pKC1139::dap, the plasmid was isolated directly from Streptomyces and confirmed with restriction digestion (Fig. S2c). The 157 kb fragment covering SSGG00215-SSGG00287 contains the complete dap gene cluster. Similar to that of act gene cluster, the removal of pKC1139::nap and pKC1139::dap from Sro-MCnap and Sro-MCdap generated strains lacking nap and dap gene clusters (Sro-Dnap and Sro-Ddap).
Improvement of antibiotic titers
The pKC1139 contains a temperature-sensitive origin of replication from pSG5, which is a medium copy plasmid with an approximate 20–50 copy numbers per chromosome21. When cultured at 28°C, pKC1139 exists as autonomous plasmid in Streptomyces. In S. coelicolor M145, there is only one copy of act gene cluster in the chromosome. After the ϕBT1 integrase-mediated excision, the act gene cluster was transferred into pKC1139. An increase in copy number of act gene cluster will improve actinorhodin production. This was confirmed both on R5MS agar plate (Fig. 4a) and in R5MS liquid culture (Fig. 4b). It should be noted that the titer of actinorhodin in M1146-MCact was even higher than that of M145-MCact. Similarly, daptomycin titer could also increase after the excision of dap gene cluster from its chromosome location in S. roseosporus NRRL 15998. Cultures of Sro-MCdap and S. roseosporus NRRL 15998 were subjected to bioassay against S. aureus, the results showed that Sro-MCdap exhibited bigger inhibition zones against S. aureus than S. roseosporus NRRL 15998 at time intervals from 2–5 days (Fig. 5a). This was further verified by comparison of daptomycin from fermentation broth of S. roseosporus NRRL 15998 and Sro-MCdap by high-performance liquid chromatography (HPLC) analysis (Fig. 5b). In addition, we noticed that existence of extra copy numbers of antibiotic gene clusters caused a slowdown in growth of Streptomyces. When cultured on AS-1 agar medium, growth of Sro-MCnap and Sro-MCdap are severely impaired, especially at earlier stages of cultivation (Fig. S3). This phenotype was most likely attributed to the metabolic burden of extra copy numbers of antibiotic gene clusters. This assumption is based on the observation that growth of Sro-Dnap (devoid of nap gene cluster) and Sro-Ddap (devoid of dap gene cluster) are converted back to that of S. roseosporus NRRL 15998 (Fig. S3).

Analysis of daptomycin production in S. roseosporus NRRL 15998 (WT) and Sro-MCdap.
(A) Bioassay of daptomycin against S. aureus. After grown on AS-1 agar for 2–5 days at 28°C, the patches of WT and Sro-MCdap were overlaid with cultures of S. aureus and the zone of inhibition was assessed after overnight incubation at 37°C. Representative images of three independent experiments with similar results are shown. (B) HPLC analysis of fermentation filtrates from WT and Sro-MCdap after incubation for 4 days. Components of daptomycin were indicated by comparison with standards.
To examine the stability of multiple copy plasmids in Streptomyces, two randomly chosen strains of Sro-MCdap were passed consecutively for five or ten times on AS-1 plates supplemented with or without apramycin. Biological activities of these stains (G5 and G10) were compared with that of the original Sro-MCdap (G0). All Sro-MCdap strains exhibited similar inhibitory activity against S. aureus (Fig. S4), suggesting that pKC1139-derived large plasmids are stable in the engineered Streptomyces in the presence or absence of selective pressure.
Discussion
We have established an efficient method for genome engineering and direct cloning of gene clusters in Streptomyces. The strategy is based on phage ϕBT1 attP-attB-int system and provides several advantages over similar methods. First, it can be used for the deletion of large fragment (up to 157 kb) from Streptomyces genome. In the meantime, the large fragment containing gene cluster of interest was cloned into pKC1139 simultaneously. Another advantage of our strategy is that it could clone gene cluster in size up to 157 kb. This is the largest size ever reported in Gram positive bacteria and should be good enough for most antibiotic gene clusters. Last, our strategy can be used to improve the titer of industrial important antibiotics by creating strains with extra copy numbers of antibiotic biosynthetic gene clusters.
The ϕBT1 attP-attB-int system is helpful for genetic modifications of Streptomyces genome at multiple sites. In addition to the intact attP-attB pair, there are 15 mutated attP-attB pairs which can be recognized by ϕBT1 integrase17. Multiple rounds of large fragment deletion can be achieved with the following modifications. (1) Relocation of the attB6 sequence (or any other mutated attB) to the downstream of the 2.0 kb homologous fragment in pSV::attB6Up. This change will allow the excision of the pUC119::neo backbone from Streptomyces genome together with pKC1139. (2) Construction of an autonomous helper plasmid containing a temperature-sensitive origin of replication from pSG5, the origin of transfer (oriT) from plasmid RK2 and ϕBT1 integrase gene. This plasmid can ensure the high efficient excision of large fragment from Streptomyces genome and subsequent removal of ϕBT1 integrase. With these modifications, there is only 42 bp attL site left in the chromosome of Streptomyces.
Genome analysis suggested that S. roseosporus NRRL 15998 has potential capacity to produce napsamycins22,23. However, the production of napsamycins in S. roseosporus NRRL 15998 has not been reported. With this strategy, we cloned nap gene cluster in pKC1139 to generate pKC1139::nap. It can be manipulated extensively in E. coli. These manipulations include replacement of vector backbone with integrative plasmid and deletion or constitutive expression of regulatory gene by PCR targeting24. The modified gene cluster can be transferred into heterologous hosts for expression. It can also be transferred back into the mutant devoid of nap gene cluster after removal of pKC1139::nap. Detection of napsamycin in these strains will shed light on the activation of cryptic gene clusters in Streptomyces.
In some industrial overproducing strains generated by traditional mutagenesis, amplification of biosynthetic gene cluster has been observed25,26,27. Based on these observations, controlled amplification of gene cluster was used to increase the productivity of commercially important antibiotics. Integration of an additional copy of gene cluster for nikkomycin and gougerotin biosynthesis led to an increased production of nikkomycin and gougerotin by Streptomyces ansochromogene28 and Streptomyces graminearus29, respectively. The zouA-mediated gene amplification of act gene cluster in S. coelicolor M145 led to a 20-fold increase in actinorhodin production30. The zouA encodes a site-specific relaxase similar to TraA protein which catalyzes RecA-independent site-specific recombination. The recombination sites of ZouA are oriT-like RsA and RsB30. In this study, we reported the amplification of gene clusters mediated by phage ϕBT1 integrase and improved antibiotic titers in the engineered Streptomyces strains. We believe that the system described here could be used readily to increase antibiotic titers in many Streptomyces and possible other actinomycetes.
Methods
Bacterial strains, plasmids, primers and growth conditions
Bacterial strains and plasmids used in this study are listed in Table 1 and primers are listed in Table S1. S. coelicolor M145 and S. roseosporus NRRL 15998 were used for cloning of act, nap and dap gene clusters. S. coelicolor M1146 is an engineered derivative of S. coelicolor M145 that lacks gene clusters for actinorhodin (ACT), undecylprodigiosins (RED), cryptic polyketide (CPK) and calcium-dependent antibiotic (CDA) biosynthesis31. Staphylococcus aureus was used as an indicator strain for daptomycin bioassay. E. coli Top10 was used as a general host for propagating plasmids. E. coli ET12567 (pUZ8002) was used as a host for transferring DNA from E. coli to Streptomyces by intergeneric conjugation32.
For general purpose, S. coelicolor M145 and its derivatives were grown on mannitol soya flour medium (MS) agar or in yeast extract-malt extract (YEME) liquid medium32. For actinorhodin production, S. coelicolor M145 and its derivatives were grown on R5MS agar or in R5MS liquid medium33. S. roseosporus NRRL 15998 was cultured on AS-1 agar medium or in tryptic soy broth (TSB) liquid medium34. All Streptomyces stains were maintained at 28°C unless specified otherwise. General approaches for E. coli or Streptomyces manipulations were performed according to standard protocols32,35. When necessary, the final antibiotic concentrations used for selection of E. coli transformants were as follows: ampicillin, 100 μg ml−1; apramycin, 100 μg ml−1; kanamycin, 100 μg ml−1; chloramphenicol, 12.5 μg ml−1. For selection of Streptomyces transformants, the final antibiotic concentrations were, kanamycin, 50 μg ml−1 in MS for S. coelicolor and 20 μg ml−1 in AS-1 for S. roseosporus; apramycin, 50 μg ml−1 in MS for S. coelicolor and 10 μg ml−1 in AS-1 for S. roseosporus; hygromycin, 50 μg ml−1 in MS or AS-1 for Streptomyces; nalidixic acid, 25 μg ml−1 in MS or AS-1 for Streptomyces.
Construction of plasmids
Of the 15 mutated attP-attB pairs17, attP6-attB6 was randomly chosen for this experiment. The sequences of attB6 and attP6 were obtained by overlapping PCR. For construction of pSV::attB6-act, a 2.0 kb fragment flanking 5′ end of the act gene cluster was amplified from genomic DNA of S. coelicolor M145 with primer pair act-Up F/act-Up R. The amplicon was diluted 1:100 and used as templates for the second round of PCR with primer pair attB6-in F/act-Up R. The product from the second amplification reaction was diluted again and underwent a third run with primer pair attB6-out F/act-Up R. The final product was digested with HindIII/BamHI and then inserted into the corresponding sites of pUC119::neo to generate pUC119::neo-attB6-act. The origin of transfer (oriT) from plasmid RK2 was amplified from pKC1139 with primer pair oriT F and oriT R, subsequently digested with EcoRI and inserted into the EcoRI site of pUC119::neo-attB6-act to generate pSV::attB6-act (Fig. S1a). For construction of pKC1139::attP6-act, a 2.0 kb fragment flanking 3′ end of the act gene cluster was amplified from genomic DNA of S. coelicolor M145 with primer pair act-Dn F/act-Dn R. The amplicon with 1:100 dilution served as templates for the second round of PCR with primer pair attP6 F/act-Dn R. The final product was digested with HindIII/EcoRI and then inserted into the corresponding sites of pKC1139 to generate pKC1139::attP6-act (Fig. S1b).
For construction of pSV::attB6-nap and pSV::attB6-dap, a 2.0 kb fragment flanking 5′ end of the nap and dap gene cluster was amplified from genomic DNA of S. roseosporus NRRL 15998 with primer pairs nap-Up F/nap-Up R and dap-UpF/dap-Up R, respectively. The product was digested with XbaI/BamHI and used to replace the 2.0 kb fragment upstream of the act gene cluster in pSV::attB6-act. For construction of pKC1139::attP6-nap and pKC1139::attP6-dap, a 2.0 kb fragment flanking 3′ end of the nap and dap gene cluster was amplified from genomic DNA of S. roseosporus NRRL 15998 using primer pairs nap-Dn F/nap-Dn R and dap-Dn F/dap-Dn R, respectively. The product was digested with BamHI/EcoRI and used to replace the 2.0 kb fragment downstream of the act gene cluster in pKC1139::attP6-act. To ensure the authenticity of DNA sequences, all PCR products were verified by sequencing.
Construction of double-cointegrate strains
To insert attB6 and attP6 at sites flanking the act gene cluster of S. coelicolor, pSV::attB6-act and pKC1139::attP6-act were conjugated into S. coelicolor M145. The pSV::attB6-act is unable to replicate alone in Streptomyces and selection with kanamycin allows to select exconjugants in which pSV::attB6-act is inserted into the S. coelicolor genome. The pKC1139::attP6-act is a derivative of the E. coli–Streptomyces shuttle vector pKC1139 that contains a Streptomyces temperature-sensitive origin of replication from pSG515. When the incubation temperature is higher than 34°C, pKC1139::attP6-act turns into non-replicating plasmid and attP6 was then inserted into S. coelicolor genome with selection of apramycin to obtain double-cointegrate strain Sco-actB6P6. Similar strategy was used for the construction of double-cointegrate strains Sro-napB6P6 and Sro-dapB6P6.
Excision of targeted regions
The integrative plasmid pIJ1050036 is a derivative of pMS82 which contains the phage ϕBT1 integrase gene and integrates intragenically into SCO4848 encoding a putative integral membrane protein16. It was conjugated into a randomly selected strain Sco-actB6P6, Sro-napB6P6 and Sro-dapB6P6, respectively. The exconjugants were initially selected with hygromycin and ten randomly chosen exconjugants were passed twice on MS or AS-1 plates supplemented with kanamycin and apramycin and subject to genomic and plasmid extraction. Excision of targeted region from Streptomyce genome was analyzed by PCR amplifications using genomic DNA templates and primer pairs B6-VF/actDn-VR, B6-VF/napDn-VR and B6-VF/dapDn-VR. In the meantime, PCR amplifications were performed with plasmid DNA template by using primer pairs P6-VF/actUp-VR, B6-VF/napUp-VR and B6-VF/dapUp-VR. Strains with excision of the targeted regions were designated as M145-MCact, Sro-MCnap and Sro-MCdap, respectively.
Deletion of the antibiotic gene clusters from Streptomyces
A single colony of M145-MCact was randomly chosen and passed three times on nonselective MS plates at 28 °C. Spores were harvested, serially diluted and then spread on MS agar. After growing for 4 days at 40°C, colonies were replicated on MS agar plates containing kanamycin or apramycin. Strains lacking the act gene cluster (M145-Dact) are apramycin sensitive (Aprs) and kanamycin resistant (Kanr). Aprs and Kanr strains were further verified by PCR. In a similar way, strains lacking the nap and dap gene clusters (Sro-Dnap and Sro-Ddap) were obtained by the removal of pKC1139::nap and pKC1139::dap from Sro-MCnap and Sro-MCdap.
Actinorhodin quantification
To quantitate actinorhodin production, S. coelicolor M145 and its derivatives were grown in 50 ml of R5MS at 28°C. 1 ml culture was harvested in a time-course and treated with KOH (1 N final concentration) and titer was calculated by measuring the absorbance at 640 nm37.
Production and analysis of daptomycin
Small-scale fermentation of daptomycin was carried out by following the procedures described previously38,39 with minor modifications. In brief, starter culture was grown in TSB for 48 h, 1 ml of starter culture was transferred to A355 (1% [wt/vol] glucose, 1.5% [vol/vol] glycerol, 1.5% [wt/vol] soya peptone, 0.3% [wt/vol] NaCl, 0.5% [wt/vol] malt extract, 0.5% [wt/vol] yeast extract, 0.1% [vol/vol] Tween 80 and 2% [wt/vol] MOPS, pH 7.0) and grown for 36 h as seed culture and 1 ml of seed culture was transferred into a shake flask containing 50 ml A346 (1% [wt/vol] glucose, 2% [wt/vol] soluble starch, 0.5% [wt/vol] yeast extract, 0.5% [wt/vol] casein and 4.6% [wt/vol] MOPS, pH 7.0). The cultures were incubated for different time points at 28°C before fermentation broths were collected by centrifugation.
For daptomycin analysis, culture broths were centrifuged at 13,000 × g for 10 min to remove the mycelia. The supernatants were filtered through a Millipore membrane (pore diameter, 0.22 μm) and 50 μl of sample was used for HPLC analysis. Separation of daptomycin was achieved with an Agilent 1100 HPLC system and a ZORBAX SB-Aq column (5 μm pore size, 4.6 by 250 mm). HPLC conditions were described as follows: gradient elution with buffer A (0.01% [vol/vol] trifluoroacetic acid in acetonitrile) and buffer B (0.01% [vol/vol] trifluoroacetic acid in ddH2O), flow rate at 1.0 ml/min, ultraviolet detection at wavelength of 224 nm. The elution profile was a linear gradient of 10%–100% buffer A over 22 min, a hold at 100% buffer A over 3 min, a linear gradient of 100%–10% buffer A over 2 min and a final hold at 10% buffer A over 3 min.
Bioassay against S. aureus was performed as previously described with modifications38. In brief, S. roseosporus and its derivatives were patched on AS-1 agar. After incubation for 2–5 days at 28°C, agar plugs were prepared from the patches, placed on the surface of an empty Petri dish and overlaid with culture of indicator strain in soft nutrient agar containing 5 mM CaCl2. The zone of inhibition was assessed after overnight incubation at 37°C.
References
Niu, G. & Tan, H. Biosynthesis and regulation of secondary metabolites in microorganisms. Sci. China Life Sci. 56, 581–583 (2013).
Bibb, M. J. Regulation of secondary metabolism in streptomycetes. Curr. Opin. Microbiol. 8, 208–215 (2005).
Fu, J. et al. Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol. 30, 440–446 (2012).
Chain, P. S., Hernandez-Lucas, I., Golding, B. & Finan, T. M. oriT-directed cloning of defined large regions from bacterial genomes: identification of the Sinorhizobium meliloti pExo megaplasmid replicator region. J. Bacteriol. 182, 5486–5494 (2000).
Shao, Z., Luo, Y. & Zhao, H. Rapid characterization and engineering of natural product biosynthetic pathways via DNA assembler. Mol. Biosyst. 7, 1056–1059 (2011).
Kvitko, B. H., McMillan, I. A. & Schweizer, H. P. An improved method for oriT-directed cloning and functionalization of large bacterial genomic regions. Appl. Environ. Microbiol. 79, 4869–4878 (2013).
Bonet, B., Teufel, R., Crusemann, M., Ziemert, N. & Moore, B. S. Direct capture and heterologous expression of Salinispora natural product genes for the biosynthesis of enterocin. J. Nat. Prod. 10.1021/np500664q (2014).
Yamanaka, K. et al. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc. Natl. Acad. Sci. U.S.A. 111, 1957–1962 (2014).
Fernandez-Martinez, L. T. & Bibb, M. J. Use of the meganuclease I-SceI of Saccharomyces cerevisiae to select for gene deletions in actinomycetes. Sci. Rep. 4, 7100, 10.1038/srep07100 (2014).
Lu, Z., Xie, P. & Qin, Z. Promotion of markerless deletion of the actinorhodin biosynthetic gene cluster in Streptomyces coelicolor. Acta Biochim. Biophys. Sin. (Shanghai) 42, 717–721 (2010).
Grindley, N. D., Whiteson, K. L. & Rice, P. A. Mechanisms of site-specific recombination. Annu. Rev. Biochem. 75, 567–605 (2006).
Sternberg, N. & Hamilton, D. Bacteriophage P1 site-specific recombination. I. Recombination between loxP sites. J. Mol. Biol. 150, 467–486 (1981).
Sauer, B. & McDermott, J. DNA recombination with a heterospecific Cre homolog identified from comparison of the pac-c1 regions of P1-related phages. Nucleic Acids Res. 32, 6086–6095 (2004).
Schweizer, H. P. Applications of the Saccharomyces cerevisiae Flp-FRT system in bacterial genetics. J. Mol. Microbiol. Biotechnol. 5, 67–77 (2003).
Bierman, M. et al. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116, 43–49 (1992).
Gregory, M. A., Till, R. & Smith, M. C. Integration site for Streptomyces phage ϕBT1 and development of site-specific integrating vectors. J. Bacteriol. 185, 5320–5323 (2003).
Zhang, L. et al. DNA cleavage is independent of synapsis during Streptomyces phage ϕBT1 integrase-mediated site-specific recombination. J. Mol. Cell. Biol. 2, 264–275 (2010).
Ullrich, S. & Schuler, D. Cre-lox-based method for generation of large deletions within the genomic magnetosome island of Magnetospirillum gryphiswaldense. Appl. Environ. Microbiol. 76, 2439–2444 (2010).
Komatsu, M., Uchiyama, T., Omura, S., Cane, D. E. & Ikeda, H. Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc. Natl. Acad. Sci. U.S.A. 107, 2646–2651 (2010).
Herrmann, S. et al. Site-specific recombination strategies for engineering actinomycete genomes. Appl. Environ. Microbiol. 78, 1804–1812 (2012).
Muth, G., Wohlleben, W. & Puhler, A. The minimal replicon of the Streptomyces ghanaensis plasmid pSG5 identified by subcloning and Tn5 mutagenesis. Mol. Gen. Genet. 211, 424–429 (1988).
Kaysser, L. et al. Identification of a napsamycin biosynthesis gene cluster by genome mining. Chembiochem 12, 477–487 (2011).
Niu, G. & Tan, H. Nucleoside antibiotics: biosynthesis, regulation and biotechnology. Trends Microbiol. 23, 110–119 (2015).
Gust, B., Challis, G. L., Fowler, K., Kieser, T. & Chater, K. F. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U.S.A. 100, 1541–1546 (2003).
Peschke, U., Schmidt, H., Zhang, H. Z. & Piepersberg, W. Molecular characterization of the lincomycin-production gene cluster of Streptomyces lincolnensis 78-11. Mol. Microbiol. 16, 1137–1156 (1995).
Fierro, F. et al. The penicillin gene cluster is amplified in tandem repeats linked by conserved hexanucleotide sequences. Proc. Natl. Acad. Sci. U.S.A. 92, 6200–6204 (1995).
Yanai, K., Murakami, T. & Bibb, M. Amplification of the entire kanamycin biosynthetic gene cluster during empirical strain improvement of Streptomyces kanamyceticus. Proc. Natl. Acad. Sci. U.S.A. 103, 9661–9666 (2006).
Liao, G. et al. Cloning, reassembling and integration of the entire nikkomycin biosynthetic gene cluster into Streptomyces ansochromogenes lead to an improved nikkomycin production. Microb. Cell Fact. 9, 6 (2010).
Jiang, L., Wei, J., Li, L., Niu, G. & Tan, H. Combined gene cluster engineering and precursor feeding to improve gougerotin production in Streptomyces graminearus. Appl. Microbiol. Biotechnol. 97, 10469–10477 (2013).
Murakami, T., Burian, J., Yanai, K., Bibb, M. J. & Thompson, C. J. A system for the targeted amplification of bacterial gene clusters multiplies antibiotic yield in Streptomyces coelicolor. Proc. Natl. Acad. Sci. U.S.A. 108, 16020–16025 (2011).
Gomez-Escribano, J. P. & Bibb, M. J. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 4, 207–215 (2011).
Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F. & Hopwood, D. A. Practical Streptomyces genetics. (John Innes Foundation, Norwich, U.K., 2000).
Okamoto, S., Taguchi, T., Ochi, K. & Ichinose, K. Biosynthesis of actinorhodin and related antibiotics: discovery of alternative routes for quinone formation encoded in the act gene cluster. Chem. Biol. 16, 226–236 (2009).
Nguyen, K. T. et al. A glutamic acid 3-methyltransferase encoded by an accessory gene locus important for daptomycin biosynthesis in Streptomyces roseosporus. Mol. Microbiol. 61, 1294–1307 (2006).
Sambrook, J. & Russell, D. W. Molecular cloning: a laboratory manual, 3rd ed. (Cold Spring Harbor Laboratory Press, NY, 2001).
Pullan, S. T., Chandra, G., Bibb, M. J. & Merrick, M. Genome-wide analysis of the role of GlnR in Streptomyces venezuelae provides new insights into global nitrogen regulation in actinomycetes. BMC Genomics 12, 175 (2011).
Bystrykh, L. V. et al. Production of actinorhodin-related “blue pigments” by Streptomyces coelicolor A3(2). J. Bacteriol. 178, 2238–2244 (1996).
Miao, V. et al. Genetic engineering in Streptomyces roseosporus to produce hybrid lipopeptide antibiotics. Chem. Biol. 13, 269–276 (2006).
Miao, V. et al. Daptomycin biosynthesis in Streptomyces roseosporus: cloning and analysis of the gene cluster and revision of peptide stereochemistry. Microbiology 151, 1507–1523 (2005).
MacNeil, D. J. et al. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111, 61–68 (1992).
Yang, H. et al. Autoregulation of hpdR and its effect on CDA biosynthesis in Streptomyces coelicolor. Microbiology 156, 2641–2648 (2010).
Paget, M. S., Chamberlin, L., Atrih, A., Foster, S. J. & Buttner, M. J. Evidence that the extracytoplasmic function sigma factor σE is required for normal cell wall structure in Streptomyces coelicolor A3(2). J. Bacteriol. 181, 204–211 (1999).
Li, R. et al. polR, a pathway-specific transcriptional regulatory gene, positively controls polyoxin biosynthesis in Streptomyces cacaoi subsp. asoensis. Microbiology 155, 1819–1831 (2009).
Acknowledgements
This work was supported by grants from the Ministry of Science and Technology of China (grant nos. 2012CB721103 and 2013CB734001) and the National Natural Science Foundation of China (grant nos. 31270110 and 31171202). We would like to thank Professor Mervyn Bibb and Dr. Chris D. Den Hengst (John Innes Centre, Norwich, UK) for providing S. coelicolor M1146 and pIJ10500, respectively. We also thank Dr. Guojian Liao (Southwest University, Chongqing, China) for the gift of daptomycin standard.
Author information
Authors and Affiliations
Contributions
D.D. and W.L. performed the experiments. T.Y. assisted with design of the project. L.H. assisted with the primary data analysis. N.G. conceived and designed the project and wrote the manuscript. T.H. supervised the project and revised the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplemental Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Du, D., Wang, L., Tian, Y. et al. Genome engineering and direct cloning of antibiotic gene clusters via phage ϕBT1 integrase-mediated site-specific recombination in Streptomyces. Sci Rep 5, 8740 (2015). https://doi.org/10.1038/srep08740
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08740
This article is cited by
-
An efficient method for targeted cloning of large DNA fragments from Streptomyces
Applied Microbiology and Biotechnology (2023)
-
Multiple copies of the oxytetracycline gene cluster in selected Streptomyces rimosus strains can provide significantly increased titers
Microbial Cell Factories (2021)
-
Streptomyces: host for refactoring of diverse bioactive secondary metabolites
3 Biotech (2021)
-
Molecular mechanism of mureidomycin biosynthesis activated by introduction of an exogenous regulatory gene ssaA into Streptomyces roseosporus
Science China Life Sciences (2021)
-
Evaluation of vector systems and promoters for overexpression of the acarbose biosynthesis gene acbC in Actinoplanes sp. SE50/110
Microbial Cell Factories (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
