
Abstract
The integration of different components into a hybrid nanosystem for the utilization of the synergistic effects is an effective way to design the electrocatalysts. Herein, we demonstrate a hybrid strategy to enhance the electrocatalytic property of hollow structured Pt nanoparticles for methanol oxidation reaction. This strategy begins with the preparation of bimetallic Ag-Pt nanoparticles with a core-shell construction. Element sulfur is then added to transform the core-shell Ag-Pt nanostructures into hybrid nanodimers consisting of Ag2S nanocrystals and remaining Pt domains with intact hollow interiors (Ag2S-hPt). Finally, Au is deposited at the surface of the Ag2S domain in each hetero-dimer, resulting in the formation of ternary Ag2S-Au-hPt nanocomposites with solid-state interfaces. The ternary nanocomposites exhibit enhanced electrocatalytic property toward methanol oxidation due to the strong electronic coupling between Pt and other domains in the hybrid particles. The concept might be used toward the design and synthesis of other hetero-nanostructures with technological importance.
Similar content being viewed by others


Introduction
Platinum (Pt) nanoparticles are catalytically active for the anodic reaction (methanol oxidation reaction, MOR) of the direct methanol fuel cell (DMFC)1,2,3. Hollowing platinum (Pt) nanoparticles with galvanic replacement or scarificial templates offers a promising approach to meet the high performance goals in electrocatalysis4,5,6,7,8,9. For intance, Fan and co-workers developed a photocatalytic approach using densely packed optically active porpyrins to template the synthesis of well-defined hollow Pt nanostructures which were excellent catalyst for the methanol oxidation reaction (MOR)7. The hollow interior increases the utilization of precious Pt metal by diminishing the number of its buried nonfunctional atoms and the increase in activity could be attributed mainly to the larger surface area of the hollow structure, where the porous or open shell allows the internal surface of the catalyst to be accessible to the reactants. However, at room and moderate temperatures, Pt nanomaterials are susceptible to the poisoning induced by carbon monoxide (CO), an intermediate product of methanol oxidation10,11,12,13, which could not be overcome by simply hollowing the interior of the electrocatalysts.
After a careful review of the recent literature, we found that the integration of materials with vastly different physical and chemical properties into a hybrid nanosystem for the utilization of the synergistic effects between different components is an effective way to design the electrocatalysts for improved activity and resistance to deactivation14,15,16,17. Analogous to the bi-functional theory based classical approaches of increasing the Pt catalytic performance for MOR through alloying with oxophilic metals (e.g. Ru)18,19,20,21, the design of hybrid electrocatalysts should be rational and based on a sufficiently good understanding of the reaction mechanism. For example, the importance of the Pt–CO bond in MOR has been well documented22,23. The chemisorption of CO on Pt involves the donation of lone pair electrons from the filled carbon σ orbital of CO to the empty 5d-orbital of Pt, which is compensated by the back donation of electrons from the Pt dπ to the π* orbitals of CO. A high local density of electrons around Pt is associated with weak chemisorption. With this understanding, Ag2S-Pt composite nanocatalysts have been developed to ameliorate CO deactivation24. The electron transfer from Ag2S to Pt in Ag2S-Pt nanocomposites due to the alignment of energy levels increases the electron density around the Pt sites, causing the weakening of CO chemisorption and hence the increase in MOR activity.
In this work, we will demonstrate a hybrid strategy to enhance the electrocatalytic property of hollow structured Pt nanoparticles toward MOR. In this strategy, bimetallic Ag-Pt nanoparticles with a core-shell construction are first prepared using seed-mediated growth method, which are dispersed in an organic medium and used as starting templates. Element sulfur is then used to transform the core-shell Ag-Pt nanostructures into hybrid nanodimers composed of Ag2S and Pt nanoparticles with hollow interiors (Ag2S-hPt). Finally, Au is deposited at one site on the surface of each Ag2S-hPt hetero-dimer, resulting in the formation of Ag2S-Au-hPt ternary nanocomposites with solid-state interfaces, which are important for the coupling occurrence among different domains in the nanocomposites. We will also demonstrate that the final ternary Ag2S-Au-hPt nanocomposites are more effective for MOR in comparison with their core-shell Ag-Pt and dimeric Ag2S-hPt ancestors due to the electronic coupling effect among the different domains in nanocomposites. This study offers a vivid example to exhibit the enhancement of the material properties by means of a structural tailoring. The concept might be used toward the design and synthesis of other hetero-nanostructures for catalytic applications other than methanol oxidation.
Results and discussion
Fig. 1 is a schematic illustration for the synthesis of ternary Ag2S-Au-hPt nanocomposites. Fig. S1a and c in Supplementary Information (SI) show the transmission electron microscopy (TEM) image and histogram of the Ag seeds, respectively, which were used for the subsequent preparation of core-shell Ag@Pt nanoparticles. As displayed, these Ag seeds are multiply twinned decahedral nanoparticles and have an average size of 9.3 nm with a standard deviation of 1.0 nm8,25. The high-resolution TEM (HRTEM) image (SI Fig. S1b) illustrated the lattice planes in these nanoparticles, showing an interplanar spacing of ~0.24 nm, which corresponded to the {111} planes of face-centered cubic (fcc) Ag (JCPDS Card File 893722).

Schematic illustration.
Schematic for the synthesis of ternary Ag2S-Au-hPt nanocomposites using core-shell Ag@Pt nanoparticles as starting templates.
In the strategy developed in this work, the preparation of core-shell Ag@Pt nanoparticles is an important step preceding the synthesis of Ag2S-hPt hetero-dimers and Ag2S-Au-hPt ternary nanocomposites. The core-shell Ag@Pt nanoparticles were synthesized using a seed-mediated growth method at elevated temperature. Fig. 2a and 2c show the TEM and HRTEM images of core-shell Ag@Pt nanoparticles prepared by successive reduction of AgNO3 and K2PtCl4 in oleylamine, where the core-shell structure has been confirmed by energy-dispersive X-ray (EDX) analysis of an arbitrarily chosen single particle in the high-angle annular dark-field scanning TEM mode8. The core-shell Ag@Pt nanoparticles were uniform in size and had an overall average size of 12.6 nm. The X-ray diffraction (XRD) pattern shows two distinct metal phases, which could be indexed to the cubic Ag and Pt, respectively (SI Fig. S2a).

Core-shell Ag@Pt nanoparticles and Ag2S-hPt heterodimers.
TEM images (a,b), HRTEM images (c,d) and EDX analyses (e,f) of core-shell Ag@Pt nanoparticles (a,c,e) and Ag2S-hPt hetero-dimers (b,d,f).
Subsequently, a previous protocol with slight modification was employed to synthesize the bimetallic Ag-hPt heterodimers26. The twinned structure of the Ag seeds provides an expeditious means to modify the internal structure of core-shell Ag@Pt nanoparticles. Multiply-twinned Ag nanoparticles are inherently unstable; known to be slowly etched by dissolved O2 and Cl− dissociated from the Pt precursor (K2PtCl4) and influence strongly on the continuity and compactness of the overlaid Pt shell8,27. The Ag+ released from the O2/Cl− etching of twinned Ag seeds diffuses out through the discontinuous Pt shell due to the prevailing Ag+ concentration gradient between the core of the Ag@Pt nanoparticles and the surrounding solution. This diffusion has been developed into a universal protocol for the production of noble metal nanoparticles with a hollow or cage-bell structure8. A variation of this protocol was used in this work to completely remove the Ag core from core-shell Ag@Pt nanoparticles, resulting in formation of hetero-dimers composed of Ag2S and remaining Pt nanoparticles with intact hollow interiors.
After mixing the core-shell Ag@Pt nanoparticles with element sulfur in toluene for 5 hours at 80°C, Ag diffused out from the interior of the core-shell Ag@Pt nanoparticles and was converted into Ag2S on the surface of Pt shells, forming Ag2S-hPt hetero-dimers (Ag2S and Pt nanoparticles with a hollow interior). The TEM image (Fig. 2b) and HRTEM image (Fig. 2d) after treatment with element sulfur show that only hybrid particles with strong imaging contrast at different domains are observed in the resulting products, which are very different from the starting core-shell nanoparticles (Fig. 2a and 2c). Since metals usually have strong imaging contrast owing to their high electron density28, the hybrid dimers formed by Ag2S and hollow Pt could be easily identified – the increase in the image contrast between the core and shell regions is an indication of metal depletion caused by the removal of the Ag cores, while the semiconducting Ag2S appears as lighter domain having a solid-state interface with the metal section. The conversion of Ag into Ag2S was supported by EDX analysis of the dimeric product. In comparison with that of core-shell Ag@Pt nanoparticles (Fig. 2e), as shown in Fig. 2f, besides Ag and the Pt metals, sulfur is also detected in the core-shell nanoparticles after the sulfur treatment. For the core-shell Ag@Pt nanoparticles, the broad absorption in the visible light region due to the surface plasmon resonance of Ag nanoparticles with structural defects also disappears after the sulfur treatment, as shown in SI Fig. S3, substantiating the conversion of Ag diffused from the core region of core-shell Ag@Pt nanoparticles into Ag2S in the hetero-dimers. A typical powder XRD pattern of the Ag2S-hPt hetero-dimers was shown in SI Fig. S2b and indicates the simultaneous presence of monoclinic Ag2S and face-centered cubic (fcc) Pt phases. TEM and HRTEM images in Fig. 2 show that the size and morphology of the remaining Pt domains in the hetero-dimers are virtually unchanged after treatment with element sulfur, suggesting that the diffusion of the Ag from the core of the core-shell nanoparticles did not cause the collapse of the particle geometry.
Ternary Ag2S-Au-hPt nanocomposites are found as the dominant product after aging the mixture of Ag2S-hPt hetero-dimers and HAuCl4 in toluene for 2 h, as indicated by the TEM and HRTEM images in Fig. 3. No additional reducing agent is needed. Dodecylamine (DDA) could reduce the Au3+ ions sufficiently in the presence of Ag2S-hPt hetero-dimers. Isolated Au nanoparticles are not observed, indicating that Au nucleates preferentially on the existing Ag2S-hPt heterodimers under the experimental conditions. In most cases, Au is deposited only at a single site on the Ag2S domain in each Ag2S-hPt hetero-dimers. The average diameter of the deposited gold patches is ca. 8.3 nm, which could be discernible by the strong brightness contrast in TEM and HRTEM images.

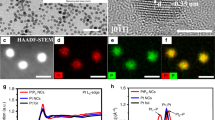
Ternary Ag2S-Au-hPt nanocomposites.
TEM image (a), HRTEM image (b), line-scan analysis (d) and elemental mapping (e ~ i) of a single particle (c) of the ternary Ag2S-Au-hPt nanocomposites as-prepared by depositing Au on the Ag2S-hPt heterodimers in toluene at room temperature.
As indicated by SI Fig. S4, the EDX analysis confirmed the presence of Ag, Au, Pt and S after aging the mixture of Ag2S-hPt hetero-dimers and Au3+ ions in toluene for 2 h at room temperature. The XRD pattern shown in SI Fig. S2c demonstrates a mixed phase of Au, Pt and Ag2S in the ternary Ag2S-Au-hPt nanocomposites. In addition, the appearance of a surface plasmon resonance at ca. 520 nm clearly indicates the formation of Au nanoparticles, as displayed in SI Fig. S3 (blue line). The broad feature of the surface plasmon resonance may indicate the presence of structural defects in the Au domains. The more direct evidences for the formation of ternary Ag2S-Au-hPt nanocomposites were provided by the line scanning analysis and elemental mapping of an arbitrary single composite nanoparticle (Fig. 3c) in the high-angle annular dark-field STEM mode. As shown in Fig. 3d for the line-scanning analysis, the Au and Pt signals are present at left- and right-hand side, respectively, whereas the Ag and S signals are only concentrated at the core region. The line-scanning analysis is well in accord with the nanoscale mapping results (Fig. 3e ~ 3i), which also manifest that the Au and Pt components are respectively distributed on the two sides of the ternary nanocomposites. Further, HRTEM image revealed that the crystal planes of Au were not parallel to those of Ag2S in each heterodimer nanoparticle (Fig. 3b), indicating that the growth of Au on the surface of Ag2S domain takes place in different orientations.
The core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers and ternary Ag2S-Au-hPt nanocomposites were loaded on Vulcan carbon and tested for their electrocatalytic property for the methanol oxidation reaction (MOR) at room temperature. As shown by the representative TEM images in SI Fig. S5, the core-shell particles, hetero-dimers and nanocomposites could be dispersed very well on the carbon support by conventional means and their structures are intact.
The electrochemically active surface areas (ECSAs) of core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers and ternary Ag2S-Au-hPt nanocomposites are determined using cyclic voltammetry (Fig. 4a). The specific ECSAs, based on the unit weight of Pt and calculated by integrating the charge associated with the hydrogen adsorption/desorption potential region after double-layer correction, are 41.6 m2 g−1 for core-shell Ag@Pt, 35.3 m2 g−1 for Ag2S-hPt hetero-dimers and 37.1 m2 g−1 for Ag2S-Au-hPt nanocomposites, respectively. The hollowing of core-shell Ag@Pt nanoparticles by element sulfur may lead to the increase of ECSAs by releasing the inner surface of Pt shell, whereas the growth of Ag2S domains on the outer surface of Pt shell would result in the decrease of ECSAs due to the solid-state interfaces between hollow Pt and Ag2S in the hetero-dimers, which may induce some blockage of the surface area of the Pt shells. These two effects might have offset each other, such that the ECSAs of Ag2S-hPt hetero-dimers are lower than that of core-shell Ag@Pt nanoparticles. Specifically, the ECSAs of Pt in ternary Ag2S-Au-hPt nanocomposites are similar to that in Ag2S-hPt hetero-dimers since the Au is only deposited at the surface of Ag2S domains in the hetero-dimers and has negligible influence on the ECSAs of the Pt domains.

Electrochemical measurements.
Cyclic voltammograms of the core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers and Ag2S-Au-hPt nanocomposites in argon-purged HClO4 (0.1 M) at room temperature at scan rate of 50 mV s−1 (a); 4f XPS spectra of Pt in the core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers and Ag2S-Au-hPt nanocomposites (b); energy level diagram for ternary Ag2S-Au-hPt nanocomposites showing the intraparticle charge transfer among different domains (c); room-temperature CO stripping from the core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers and Ag2S-Au-hPt nanocomposites in 0.1 M HClO4 (d); cyclic voltammograms of the core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers and Ag2S-Au-hPt nanocomposites in argon-purged HClO4 (0.1 M) with methanol (1 M) at scan rate of 20 mV s−1 (e); chronoamperograms of the core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers and Ag2S-Au-hPt nanocomposites at 0.45 V vs Ag/AgCl at room temperature in argon-purged HClO4 (0.1 M) with 1 M methanol (f).
An important feature in hybrid materials is the electronic coupling between the metal and semiconductor domains24,29,30,31. The Pt 4f X-ray photoelectron spectroscopy (XPS) spectra of the core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers and ternary Ag2S-Au-hPt nanocomposites were analyzed. As displayed in Fig. 4b, in comparison with the Pt 4f7/2 and 4f5/2 binding energies of core-shell Ag@Pt nanoparticles, an appreciable shift to lower values was observed in the Ag2S-hPt hetero-dimers and ternary Ag2S-Au-hPt nanocomposites, suggesting that electrons were transferred to Pt from other domains of the hetero-dimers or nanocomposites. The comparison of the Pt 4f XPS spectra between Ag2S-hPt hetero-dimers and ternary Ag2S-Au-hPt nanocomposites further reveals that the presence of the Au domain could promote this electron-donating effect. The decrease in the Pt 4f binding energies was ca. 0.7 eV in ternary Ag2S-Au-hPt nanocomposites and only ca. 0.3 eV in Ag2S-hPt hetero-dimers. This electron-donating effect to hPt domains could be explained by intra-particle charge transfer (Fig. 4c for the energy level diagram). The comparison between the electronic affinity of bulk Ag2S (3.63 eV) and the work function of Pt (5.65 eV) predicted that the alignment of energy levels in Ag2S and Pt would be favorable for electron transfer from Ag2S to hPt. Analogous charge transfer has been observed in the core-shell Au@PbS system, whereby the electrons transfer from PbS shell to the inner Au core results in the n-type to p-type change in hydrazine-treated PbS32. The electron transfer from Ag2S to hPt could also be described with the generation of a hole in the Ag2S domain. In the presence of Au domain (work function = 5.1 eV), the alignment of energy levels in Au and Ag2S would be favorable for electron transfer from Au to Ag2S to fill the hole generated by the electron transfer to hPt domains, further promoting the electron transfer from Ag2S to hPt in order for the Fermi levels to match at the interface. The electron-donating effect from Au to Ag2S has also been supported by the XPS analysis of Au 4f region (SI Fig. S6), whereby an appreciable shift in the 4f binding energies of Au in the ternary nanocomposites to higher values was found as compared to the Au 4f binding energies of monometallic Au particles33. Analogous to the electron donation from Sn to Pt in the Pt-Sn system34, the charge transfer from Ag2S to Pt in the Ag2S-hPt hetero-dimers or ternary Ag2S-Au-hPt nanocomposites leads to a substantial increase in the electron density around the Pt domains, resulting in the weaker chemisorption of CO, an intermediate product of methanol oxidation on the surface of hPt and hence promoting the MOR.
Fig. 4d shows the CO stripping voltammograms of core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers and ternary Ag2S-Au-hPt nanocomposites after the working electrode has been held at −0.15 V for 30 min in CO saturated 0.1 M HClO4. The CO stripping peaks of the Ag2S-hPt hetero-dimers and ternary Ag2S-Au-hPt nanocomposites shifted to a more negative potential as compared to the core-shell Ag@Pt nanoparticles, indicating a more facile CO removal and hence, an improved CO tolerance in practice. In addition, the CO stripping peak of ternary Ag2S-Au-hPt nanocomposites was located at a more negative potential than that of Ag2S-hPt hetero-dimers, suggesting a more facile CO removal from the hPt surfaces in the Ag2S-Au-hPt composite system. The ease of CO removal over Ag2S-hPt and Ag2S-Au-hPt relative to the core-shell Ag@Pt nanoparticles reflects the effectiveness of electron coupling among the different domains in hybrid particles.
Voltammograms of methanol oxidation were obtained in the potential window of 0.2–1 V at a swept rate of 20 mV s−1 (Fig. 4e). The current densities in the voltammograms were normalized by the ECSA of Pt. As expected, the comparison in current densities indicates that the Ag2S-hPt hetero-dimers and ternary Ag2S-Au-hPt nanocomposites show greater specific activities than those of the core-shell Ag@Pt nanoparticles. In particular, the ternary Ag2S-Au-hPt nanocomposites display the highest catalytic activities for methanol oxidation. The enhanced catalytic activity of Ag2S-hPt heterodimers and ternary Ag2S-Au-hPt nanocomposites could be attributed to the electronic coupling between Pt and the other domains in the hybrid particles. The long-term performance of core-shell Ag@Pt nanoparticles, Ag2S-hPt heterodimers and ternary Ag2S-Au-hPt nanocomposites in methanol oxidation was illustrated by the chronoamperograms in Fig. 4f. The slower rate of decay for the Ag2S-hPt heterodimers and ternary Ag2S-Au-hPt nanocomposites indicates their superior CO tolerance to the core-shell Ag@Pt nanoparticles.
In summary, a hybrid strategy has been demonstrated to enhance the electrocatalytic property of hollow structured Pt nanoparticles for methanol oxidation reaction. This strategy begun with the preparation of bimetallic Ag-Pt nanoparticles with a core-shell construction using the seed-mediated growth method. Element sulfur was then added to transform the core-shell Ag-Pt nanostructures into Ag2S-hPt hybrid nanodimers. Subsequently, Au was deposited at the surface of the Ag2S domain in each Ag2S-hPt hetero-dimer, resulting in the formation of ternary Ag2S-Au-hPt nanocomposites. In comparison with their core-shell parents, the Ag2S-hPt hetero-dimers and ternary Ag2S-Au-hPt nanocomposites were more active in catalyzing methanol oxidation reaction. The electronic coupling between hPt and other domains in hetero-dimers and ternary nanocomposites was the major contributor to the enhancement of MOR activity. Although it is difficult to apply these hybrid nanomaterials as practical electrocatalysts due to the low ECSA of Pt in the hybrid structure and the lack of strategies to maintain their complex structures after long-term use, this study offers a vivid example to demonstrate the tuning of the material properties by means of a hybrid approach and the concept might be used toward the design and synthesis of other hetero-nanostructures with technological importance.
Methods
General materials
Potassium tetrachloroplatinate(II) (K2PtCl4, 98%), silver nitrate (AgNO3, 99%), gold(III) chloride trihydrate (HAuCl4·3H2O, ACS reagent, >49.0% Au basis), sulfur powder (S, chemical grade) and dodecylamine (DDA, 98%) from Aldrich, oleylamine (95.4%, primary amine) from J&K Scientific, aqueous HClO4 solution (70%, ACS reagent) and Nafion 117 solution (5% in a mixture of lower aliphatic alcohols and water) from Aladdin Reagents, ethanol (99%), methanol (99%) and toluene (99.5%) from Beijing Chemical Works and Vulcan XC-72 carbon powders (XC-72C, BET surface area = 250 m2 g−1 and average particle size = 40 ~ 50 nm) from Cabot Corporation, were used as received. All glassware and Teflon-coated magnetic stir bars were cleaned with aqua regia, followed by copious washing with de-ionized water before drying in an oven.
Synthesis of core-shell Ag@Pt nanoparticles
The seed-mediated growth method was used for the synthesis of core–shell nanoparticles with a Ag core and a monometallic Pt shell. Typically, 51 mg of AgNO3 was added to 30 mL of oleylamine in a three-necked flask equipped with a condenser and a stir bar. The solution was heated to 150°C and kept at this temperature under flowing N2 for 1 h for the reduction of Ag+ ions by oleylamine. Then 62 mg of K2PtCl4 was added swiftly, followed by heating the mixture to 160°C and keeping there for 3 h under flowing N2 for the reduction of the Pt shell metal precursor. After the reactions, the core-shell Ag@Pt nanoparticles were recovered by precipitation with methanol, centrifugation and washing with methanol and re-dispersed in 30 mL of toluene.
Synthesis of Ag2S-hPt heterogeneous nanodimerts
For the synthesis of heterogeneous nanodimers consisting of Ag2S and Pt nanoparticles with hollow interiors, 32 mg of element sulfur was added to 20 mL of core-shell Ag@Pt nanoparticle solution in toluene. The molar ratio of S/Ag in the mixture was calculated to be ca. 5/1. The mixture was heated to 80°C for 5 h to extract Ag component from the core–shell nanoparticles and to convert the Ag into Ag2S on the surface of the leaving hollow structured Pt nanoparticles. The Ag2S-hPt hetero-dimers prepared as such were recovered by precipitation with methanol, centrifugation and washing with methanol and re-dispersed in 20 mL of toluene.
Phase transfer of Au ions from aqueous phase to toluene
The transfer of HAuCl4 (aqueous solution, 1 mM) from aqueous phase to toluene followed an ethanol-mediated protocol, which we reported previously6. Briefly, 50 mL of 1 mM aqueous HAuCl4 solution was mixed with 50 mL of ethanol containing 1 mL of DDA. After 3 min of stirring, 50 mL of toluene was added and stirring continued for another 1 min. Phase transfer of Au3+ ions would occur quickly and completely, as illustrated by the complete bleaching of the color in the aqueous phase. Upon the complete transfer of the Au3+ ions from water, the Au3+ ion concentration in toluene was 1 mM. The Au3+ ions in toluene were separated from the aqueous phase and kept for further experiments.
Synthesis of ternary Ag2S-Au-hPt nanocomposites
The as-prepared Ag2S-hPt heterogeneous nanodimers were used as seeds for the formation of Ag2S-Au-hPt ternary nanocomposites. Typically, 50 mL of Au(III) solution in toluene was mixed with 10 mL of AgS-hPt organosol in toluene. The mixture was aged for 2 h to complete the deposition of Au on the surface of the Ag2S domain in Ag2S-hPt hetero-dimers. Additional reducing reagent was not necessary; DDA could reduce Au3+ ions in toluene effectively in the presence of Ag2S-hPt hetero-dimers. The ternary Ag2S-Au-hPt nanocomposites were precipitated by addition of methanol, centrifuged and washed twice with methanol to remove non-specifically bonded DDA, followed by re-dispersion in toluene.
Particle characterizations
Transmission electron microscopy (TEM), high-resolution TEM (HRTEM) and scanning TEM (STEM) were performed on the JEOL JEM-2100 and FEI Tecnai G2 F20 electron microscope operating at 200 kV with a supplied software for automated electron tomography. For the TEM measurements, a drop of the nanoparticle solution was dispensed onto a 3-mm carbon-coated copper grid. Excessive solution was removed by an absorbent paper and the sample was dried under vacuum at room temperature. An energy dispersive X-ray spectroscopy (EDX) analyzer attached to the TEM operating in the scanning transmission electron microscopy (STEM) mode was used to analyze the chemical compositions of the synthesized nanoparticles. UV-visible spectra of the colloidal solution of core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers and ternary Ag2S-Au-hPt nanocomposites were collected on a Hitachi U-3900 spectrophotometer. X-ray photoelectron spectroscopy (XPS) was conducted on a VG ESCALAB MKII spectrometer. Powder X-ray diffraction (XRD) patterns were recorded on a Rigaku D/Max-3B diffractometer, using Cu Kα radiation (λ = 1.54056 Å). Samples for XPS and XRD analyses were concentrated from the toluene solution of nanoparticles to 0.5 mL using flowing N2. 10 ml of methanol was then added to precipitate the nanoparticles, which were recovered by centrifugation, washed with methanol several times and then dried at room temperature in vacuum.
Electrochemical measurements
Electrochemical measurements were carried out in a standard three-electrode cell connected to a Bio-logic VMP3 (with EC-lab software version 9.56) potentiostat. A leak-free Ag/AgCl (saturated with KCl) electrode was used as the reference electrode. The counter electrode was a platinum mesh (1 × 1 cm2) attached to a platinum wire.
For the loading of the catalyst on Vulcan XC-72 carbon support, a calculated amount of carbon powder was added to the toluene solution of core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers and ternary Ag2S-Au-hPt nanocomposites. After stirring the mixture for 24 h, the Ag@Pt/C, Ag2S-hPt/C, or Ag2S-Au-hPt/C catalysts (20 wt% Pt on carbon support) were collected by centrifugation, washed thrice with methanol and then dried at room temperature in vacuum.
The working electrode was a thin layer of Nafion-impregnated catalyst cast on a vitreous carbon disk. This electrode was prepared by ultrasonically dispersing 10 mg of the carbon-supported catalysts in 10 mL of aqueous solution containing 4 mL of ethanol and 0.1 mL of Nafion solution. A calculated volume of the ink was dispensed onto the 5 mm glassy carbon disk electrode to produce a nominal catalyst loading of 20 μg cm−2 (Pt basis). The carbon electrode was then dried in a stream of warm air at 70°C for 1 h.
The room temperature cyclic voltammograms of the carbon-supported Ag@Pt, Ag2S-hPt and Ag2S-Au-hPt catalysts in argon-purged HClO4 (0.1 M) were recorded between −0.2 V and 1 V at 50 mV s−1 and used for the determination of electrochemically active surface area (ECSA) of Pt. The catalyst performance in room-temperature methanol oxidation reaction (MOR) was also measured by cyclic voltammetry. For these measurements the potential window of 0.2 V to 1 V was scanned at 20 mV s−1 until a stable response was obtained, before recording the voltammograms. The electrolyte was methanol (1 M) in perchloric acid (0.1 M). For each carbon-supported catalyst (core-shell Ag@Pt nanoparticles, Ag2S-hPt hetero-dimers, or ternary Ag2S-Au-hPt nanocomposites), the current density was normalized in reference to the ECSA to obtain the specific activities.
References
Steele, B. C. H. & Heinzel, A. Materials for fuel-cell technologies. Nature 414, 345–352 (2001).
Perry, M. L. & Fuller, T. F. A historical perspective of fuel cell technology in the 20th century. J. Electrochem. Soc. 149, S59–S67 (2002).
Gasteiger, H. A., Kocha, S. S., Sompalli, B. & Wagner, F. T. Activity benchmarks and requirements for Pt, Pt-alloy and non-Pt oxygen reduction catalysts for PEMFCs. Appl. Catal. B Environ. 56, 9–35 (2005).
Liang, H.-P. et al. Pt hollow nanospheres: Facile synthesis and enhanced electrocatalysts. Angew. Chem. Int. Ed. 43, 1540–1543 (2004).
Yang, J., Lee, J. Y., Too, H.-P. & Valiyaveettil, S. A bis(p-sulfonatophenyl)phenylphosphine- based synthesis of hollow Pt nanospheres. J. Phys. Chem. B 110, 125–129 (2006).
Yang, J., Sargent, E. H., Kelley, S. O. & Ying, J. Y. A general phase-transfer protocol for metal ions and its application in nanocrystal synthesis. Nat. Mater. 8, 683–689 (2009).
Bai, F. et al. Templated photocatalytic synthesis of well-defined platinum hollow nanostructures with enhanced catalytic performance for methanol oxidation. Nano Lett. 11, 3759–3762 (2011).
Liu, H. et al. Hollow and cage-bell structured nanomaterials of noble metals. J. Am. Chem. Soc. 134, 11602–11610 (2012).
Liu, H., Ye, F. & Yang, J. A universal and cost-effective approach to the synthesis of carbon-supported noble metal nanoparticles with hollow interiors. Ind. Eng. Chem. Res. 53, 5925–5931 (2014).
Parsons, R. & VanderNoot, T. The oxidation of small organic molecules: A survey of recent fuel cell related research. J. Electroanal. Chem. 257, 9–45 (1988).
Marković, N. M. & Ross, P. N. Surface science studies of model fuel cells electrocatalysts. Surf. Sci. Rep. 45, 117–229 (2002).
de Brujin, F. A., Dam, V. A. T. & Janssen, G. J. M. Review: Durability and degradation issues of PEM fuel cell components. Fuel Cells 8, 3–22 (2008).
Antolini, E., Lopes, T. & Gonzalez, E. R. An overview of platinum-based catalysts as methanol-resistant oxygen reduction materials for direct methanol fuel cells. J. Alloys Compd. 461, 253–262 (2008).
Cozzoli, P. D., Pellegrino, T. & Manna, L. Synthesis, properties and perspectives of hybrid nanocrystal structures. Chem. Soc. Rev. 35, 1195–1208 (2006).
Zhang, J., Sasaki, K. & Adzic, R. R. Stabilization of platinum oxygen-reduction electrocatalysts using gold clusters. Science 315, 220–222 (2007).
Yang, J. et al. Core-shell CdSe@Pt nanocomposites with superior electrocatalytic activity enhanced by lateral strain effect. J. Mater. Chem. 21, 9088–9094 (2011).
Liu, H. et al. Stellated Ag-Pt bimetallic nanoparticles: An effective platform for catalytic activity tuning. Sci. Rep. 4, 3969/1–3969/7 (2014).
Watanabe, M. & Motoo, S. Electrocatalysis by ad-atoms: Part II. Enhancement of the oxidation of methanol on platinum by ruthenium ad-atoms. J. Electroanal. Chem. 60, 267–273 (1975).
Gurau, B. et al. Structural and electrochemical characterization of binary, ternary and quaternary platinum alloy catalysts for methanol electro-oxidation. J. Phys. Chem. B 102, 9997–10003 (1998).
Iwasita, I. et al. Methanol oxidation on PtRu electrodes. Influence of surface structure and Pt-Ru atom distribution. Langmuir 16, 522–529 (2000).
Chen, A. & Holt-Hindle, P. Platinum-based nanostructured materials: Synthesis, properties and applications. Chem. Rev. 110, 3767–3804 (2010).
Nilsson, A. & Pettersson, L. G. M. [Absorbate electronic structure and bonding on metal surfaces]. Chemical bonds at surfaces and interfaces [Nilsson, A., Pettersson, L. G. M. & Nørskov, J. K. (eds.)] [78–138] (Elsevier, 2008).
Shukla, A. K. et al. An X-ray photoelectron spectroscopic study on the effect of Ru and Sn additions to platinised carbons. Appl. Surface Sci. 137, 20–29 (1999).
Yang, J. & Ying, J. Y. Nanocomposites of Ag2S and noble metals. Angew. Chem. Int. Ed. 50, 4637–4643 (2011).
Tsuji, M. et al. Stepwise growth of decahedral and icosahedral silver nanocrystals in DMF. Cryst. Growth Des. 10, 296–301 (2010).
Liu, H. et al. A core–shell templated approach to the nanocomposites of silver sulfide and noble metal nanoparticles with hollow/cage-bell structures. Nanoscale 5, 6901–6907 (2013).
Wiley, B., Herricks, T., Sun, Y. & Xia, Y. Polyol synthesis of silver nanoparticles: Use of chloride and oxygen to promote the formation of single-crystal, truncated cubes and tetrahedrons. Nano Lett. 4, 1733–1739 (2004).
Mokari, T. et al. Formation of asymmetric one-sided metal-tipped semiconductor nanocrystal dots and rods. Nat. Mater. 4, 855–863 (2005).
Costi, R. et al. Visible light-induced charge retention and photocatalysis with hybrid CdSe-Au nanodumbbells. Nano Lett. 8, 637–641 (2008).
Elmalem, E. et al. Growth of photocatalytic CdSe-Pt nanorods and nanonets. Adv. Mater. 20, 4312–4317 (2008).
Haldar, K. K., Sinha, G., Lahtinen, J. & Patra, A. Hybrid colloidal Au-CdSe pentapod heterostructures synthesis and their photocatalytic properties. ACS Appl. Mater. Interfaces 4, 6266–6272 (2012).
Lee, J.-S., Shenchenko, E. V. & Talapin, D. V. Au−PbS core−shell nanocrystals: Plasmonic absorption enhancement and electrical doping via intra-particle charge transfer. J. Am. Chem. Soc. 130, 9673–9675 (2008).
Wagner, C. D. et al. NIST Standard Reference Database 20, Version 3.2 (Web Version).
Shukla, A. K. et al. An X-ray photoelectron spectroscopic study on the effect of Ru and Sn additions to platinised carbons. Appl. Surf. Sci. 137, 20–29 (1999).
Acknowledgements
Financial support from the 100 Talents Program of the Chinese Academy of Sciences, National Natural Science Foundation of China (No.: 21173226, 21376247) and State Key Laboratory of Multiphase Complex Systems, Institute of Process Engineering, Chinese Academy of Sciences (MPCS-2012-A-11) is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
Y.F., H.L., P.W. and F.Y. performed the materials synthesis, characterization and electrochemical measurements. Q.T. and J.Y. supervised the project. J.Y. wrote the main manuscript text and all authors participated in the review of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
SREP-14-04551-SI
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Feng, Y., Liu, H., Wang, P. et al. Enhancing the Electrocatalytic Property of Hollow Structured Platinum Nanoparticles for Methanol Oxidation Through A Hybrid Construction. Sci Rep 4, 6204 (2014). https://doi.org/10.1038/srep06204
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06204
This article is cited by
-
Synthesis of Hollow Pt-Ni Nanoboxes for Highly Efficient Methanol Oxidation
Scientific Reports (2019)
-
Effects of Pt content on the crystallinity and optical properties of Ag/Pt nanoboxes: from solid to single and polycrystalline mesoporous nanostructures
Journal of Nanoparticle Research (2017)
-
Pd-dispersed CuS hetero-nanoplates for selective hydrogenation of phenylacetylene
Nano Research (2016)
-
Kinetics and Morphological Analysis of Silver Platinum Bimetallic Nanoparticles
Acta Metallurgica Sinica (English Letters) (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
