
Abstract
Designing Pt-based electrocatalysts with high catalytic activity and CO tolerance is challenging but extremely desirable for alkaline hydrogen oxidation reaction. Herein we report the design of a series of single-atom lanthanide (La, Ce, Pr, Nd, and Lu)-embedded ultrasmall Pt nanoclusters for efficient alkaline hydrogen electro-oxidation catalysis based on vapor filling and spatially confined reduction/growth of metal species. Mechanism studies reveal that oxophilic single-atom lanthanide species in Pt nanoclusters can serve as the Lewis acid site for selective OH- adsorption and regulate the binding strength of intermediates on Pt sites, which promotes the kinetics of hydrogen oxidation and CO oxidation by accelerating the combination of OH− and *H/*CO in kinetics and thermodynamics, endowing the electrocatalyst with up to 14.3-times higher mass activity than commercial Pt/C and enhanced CO tolerance. This work may shed light on the design of metal nanocluster-based electrocatalysts for energy conversion.
Similar content being viewed by others

Introduction
Hydrogen fuel cells as green yet high-efficiency energy suppliers are very attractive in contributing to fulfilling carbon neutrality, which is greatly benefited from the deployment of precious Pt electrocatalysts performing best in catalyzing anodic hydrogen oxidation reaction (HOR) due to the optimal adsorption/desorption energy toward hydrogen intermediates1,2,3. However, the large-scale implementation of conventional Pt/C electrocatalysts in alkaline HOR catalysis is beset by ultralow catalytic efficiency (e.g., two orders of magnitude decline), extortionate Pt dosage (e.g., ten times higher than that in acidic media), and poor durability in terms of weak CO tolerance and feeble stability4,5,6,7,8,9,10,11,12,13,14,15,16. Therefore, designing high-performance Pt electrocatalysts with low Pt dosage and high durability is extremely desirable for alkaline HOR, which should be the stone of killing two birds (i.e., low abundance of Pt element and unsatisfactory alkaline HOR performance).
By deciphering the alkaline HOR process2,17,18,19, it is found that the mutual competition in the adsorption-desorption between reaction intermediates (e.g., *H and *OH) and CO on the surface-active sites of electrocatalysts is the key reason for the low catalytic activity and poor CO tolerance20,21,22. Although previous researches have improved the alkaline HOR performance of Pt electrocatalysts by decreasing the size or engineering the composition/structure with other metals to conserve the Pt dosage, and regulating the d-band center to adjust the adsorption-desorption behavior of *H, *OH, and CO species, they failed to fundamentally solve the mutual competition issues of such reactant species on the active sites of Pt electrocatalysts11,23. To radically address such issues, an ideal alkaline HOR electrocatalyst (e.g., platinum group metal (PGM)-based and Ni-based) should feature the following attributes: (1) in addition to the main active sites, another type of active site with selective adsorption-desorption behavior for specific reactant species should be deployed on the electrocatalyst surface to essentially avoid the mutual adsorption-desorption competition between reactant species5,24,25,26; (2) the dissimilar active sites should be well distributed in the electrocatalyst at the atomic level and have synergistic effects with the main active sites to promote the interfacial HOR kinetics and resist CO poisoning via regulating the d-band centers, and concurrently strengthen the structural stability of the electrocatalyst7,8,17,27,28; (3) the electrocatalyst should have an ultrasmall size to maximumly uplift its atomic utilization29,30,31,32. However, such a Pt-based electrocatalyst that combines all three is not yet developed in the field.
Inspired by the conceptual design of high-performance metal nanoclusters (NCs, ≤3 nm)-based nanocatalysts with dissimilar metallic single-atom insertion33,34, we hypothesized that if we can insert highly oxophilic lanthanide single-atom into ultrasmall Pt NCs (Ln1Pt NCs for short), we may develop a high-performance electrocatalyst with both excellent catalytic activity and superior durability for alkaline HOR catalysis based on the following considerations: (1) the highly oxophilic Ln1 can serve as Lewis acid to specifically absorb OH species, while the Pt NCs could selectively absorb *H species, which prevent the mutual absorption-desorption competition issue aforementioned; (2) the Ln elements with oxophilicity and atomic-level dispersion in Pt NCs could not only adjust the d-band center to regulate the adsorption-desorption behavior of *H and CO, but also allow the fast electron transfer between dissimilar atoms as well as subsequent reaction between *OH and *H/*CO, which accelerates the HOR kinetics, ensures its structural stability through unique architecture protection, and guarantees excellent CO tolerance via prompting *CO oxidation; (3) both the single-atom Ln1 and the ultrasmall size of NCs efficiently enhance atomic utilization. However, the synthesis of such Ln1Pt NCs is very challenging using conventional wet chemistry methods because the large difference in the standard reduction potentials between Pt (1.188 V for Pt2+/Pt) and Ln (e.g., −2.522 V for La3+/La, and −2.483 V for Ce3+/Ce) usually brings about metallic phase segregation during the synthesis35, and the low reduction potentials of Ln metals are also far beyond the stable range of water36. In addition, the formation of ultrasmall Ln1Pt NCs is also hardly realized under harsh conditions (e.g., high temperature >1000 °C and strong reducing agents) due to thermodynamic incompatibility between dissimilar metals, let alone to simultaneously avoid the possible low electrochemical active surface area (ECSA), and atomic size mismatch-caused structure collapse of Ln1Pt NCs (e.g., atomic radiuses are 1.83 Å for Pt and 2.74 Å for La)37,38,39,40. Taken together, controllable synthesis of ultrasmall Ln1Pt NCs for alkaline HOR catalysis may rely on the methodological innovation, which makes up part of our motivation of this study.
Herein we report the synthesis of a series of ultrasmall Ln1Pt NCs with the insertion of highly oxophilic La, Ce, Pr, Nd, and Lu single-atom via vapor filling and spatially confined reduction of metal species inside mesoporous yet hollow carbon spheres-based nanoreactors, achieving high HOR catalytic performance in alkaline media. The key of this synthetic strategy is the atomic level mixing via vapor capillary filling as well as the efficient spatial confinement of metal species using mesoporous channels of carbon nanoreactors at a sublimation temperature, resulting in the formation of ultrasmall Ln1Pt NCs inside hollow carbon sphere (Ln1Pt@HCS for short, see Fig. 1a). Both experimental and theoretical results reveal that the as-developed Ln1Pt NCs have reduced work function (Φ) and coordination numbers (CN) due to the pronounced electron transfer/conduction nature of Ln, which enables Ln1 to serve as a Lewis acid site for selective OH− adsorption and regulates binding strength of intermediates on Pt sites, promoting the HOR kinetics and CO tolerance. As a result, the Ln1Pt@HCS yields remarkable electrocatalytic activity towards alkaline HOR (e.g., La1Pt@HCS has a mass activity of 14.3-times higher than commercial Pt/C), and simultaneously achieves enhanced durability in terms of anti-CO poisoning capability and structural stability. This study is interesting because it provides a paradigm in the design of high-performance Ln1Pt NCs-based electrocatalyst for alkaline HOR catalysis, demonstrating the synergistic power of the single atom and ultrasmall metal NCs in enhancing the catalytic activity, and deepening the fundamental understanding on the Ln/Pt alloy-directed alkaline HOR electrocatalysis.

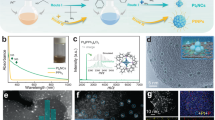
a Schematic illustration on the synthesis of La1Pt@HCS. b TEM image of HCS. c Pore size distribution of HCS estimated by DFT method. d TEM image, e SEM image, f Enlarged TEM image (inset: size distribution histogram), g HAADF-STEM image (inset: line scan of elemental profiles) of La1Pt@HCS. h Atomic-resolution HAADF-STEM image of La1Pt@HCS (inset: SAED pattern image and the atomic structure model). i Intensity profiles along lines 1 and 2 indicated in (h). j STEM-EDX mapping images of La1Pt@HCS showing distributions of C (red), Pt (green), La (blue), O (yellow), and merged elements.
Results
Material synthesis and characterization
The prerequisite of ultrasmall Ln1Pt NCs synthesis is the acquisition of hollow carbon sphere (HCS) as a nanoreactor. As illustrated in Fig. 1a, the HCS was fabricated by carbonizing core-shell resorcinol-formaldehyde resin@silica nanospheres (prepared by a hard-template method) followed by removing the silica core (see “Methods” for details)41. The obtained HCS is a particle of ~250 nm containing a regular spherical cavity surrounded by a uniform shell of ~50 nm, as evidenced by both scanning electron microscopy (SEM, Supplementary Fig. 1) and transmission electron microscopy analysis (TEM, Fig. 1b). Such an HCS can be regarded as a well-defined nanoreactor upon silica removal, consisting of mesoporous shells and pore channels with radial alignment. Moreover, the porous HCS material has a large Brunauer–Emmett–Teller surface area of over 1505.8 m2 g−1 and a high pore volume of 2.2 cm3 g−1 (Supplementary Fig. 2). Of note, the HCS also possesses tailored pore channels of 5.1 nm calculated by the density function theory (DFT) method (Fig. 1c), which would easily accommodate and confine metal species while ensuring adequate mass transport of electrolyte and gas.
With HCS nanoreactors in our hand, ultrasmall Ln1Pt NCs could be synthesized inside HCS via vapor filling of Ln and Pt precursors into mesoporous channels of HCS followed by spatially confined in-situ reduction/growth16. Taking the synthesis of La1-doped La1Pt@HCS as an example, gas-migrated Pt(acac)2 and La(acac)3 precursors were injected into the pore channels in a vacuum-sealed ampoule at a preset sublimation temperature. After successive in-situ pyrolysis, the metal precursors decomposed and reassembled into La1Pt NCs under the confinement of pore channels (Fig. 1a). The TEM and SEM images of La1Pt@HCS (Fig. 1d–f) unveil that abundant La1Pt NCs with an average size of ~2.6 nm are well distributed inside the HCS and probably confined in the pore channels rather than on the surface of HCS. The metallic composition of La1Pt@HCS was comprehensively analyzed by the high-angle annular dark-field scanning TEM (HAADF-STEM) and elemental mapping. As shown in Fig. 1g, the low-magnification HAADF-STEM image and line-scanning profile disclose that the embedded NCs consist of bimetallic alloys including Pt and La, while the aberration-corrected (AC)-HAADF-STEM image of La1Pt@HCS further reveals that La atoms are atomically dispersed (Fig. 1h) in Pt crystals, and the inserted selected area electron diffraction (SAED) pattern depicts its typical Pt crystal (inset of Fig. 1h). Along lines 1 and 2 in Fig. 1h, La1Pt@HCS delivers an obvious intensity variation of Pt and La atoms in Z contrast, as observed by the intensity profiles in Fig. 1i. Energy-dispersive X-ray (EDX) elemental mappings in Fig. 1j show even spatial distribution of Pt, La, C, and O elements. In addition, the total Pt to La ratio of La1Pt@HCS was tested to be 8.8:1 based on inductively coupled plasma optical emission spectroscopy (ICP-OES) (Supplementary Table 1).
The above-mentioned results certify the formation of single-atom La1 which is well embedded inside the ultrasmall Pt NCs in the HCS, reflecting the successful design of the synthetic strategy. Furthermore, large-scale fabrication of La1Pt@HCS could be easily realized using this approach (Supplementary Fig. 3). It should be mentioned that the mesoporous channels of HCS are crucial to the formation of such ultrasmall NCs because of their effective size confinement for metal species, which is supported by two control experiments: (1) using XC-72 carbon material (Vulcan®, BET surface area: 211.5 m2 g−1) to replace HCS but without introducing La species, resulting in the generation of large-sized Pt nanoparticles (~12.8 nm) on the XC-72 surface (Supplementary Fig. 4); (2) using commercially available porous activated carbon (Kuraray® YP-50, PAC for short; BET surface area: 1568.6 m2 g−1) to replace HCS, leading to the successful production of La1Pt NCs (~3.1 nm) inside PAC (Supplementary Fig. 5). In addition, one salient advantage of this newly developed strategy is the universal applicability, and other Ln1Pt@HCS electrocatalysts (here Ln = Ce, Pr, Nd, and Lu) can also be obtained by this method if introducing corresponding rare-earth precursors with unique 4f shell electron orbitals and high oxophilicity properties (Supplementary Figs. 6–11).
The embedding of highly oxophilic La1 single-atom in Pt NCs exerts significant influences on the work function (Φ), d-band center, and coordination numbers (CN) of Pt NCs, which are key factors affecting the HOR catalytic activity. To elucidate the impact of La1 embedding, we used La1Pt@HCS as an example to compare with the same-sized Pt NCs without La1 embedment inside HCS (Pt@HCS, and its characterization is shown in Supplementary Fig. 12). As shown in Fig. 2a, both La1Pt@HCS and Pt@HCS share the same XRD patterns consisting of the diffraction peaks of face-centered cubic (fcc) Pt crystal (JCPDS-PDF#87-0646) and the (002) graphite carbon peak belonging to HCS, which suggests that the La1 embedment with a low doping dosage (~0.98 wt.%) has a neglectable influence on the crystal structure of Pt NCs. On the other hand, the oxidation state of Pt 4f X-ray photoelectron spectra (XPS, Fig. 2b) of Pt NCs has remarkable variation upon La1 embedment, and the proportion of Pt2+ is increased from 43.8% in Pt@HCS to 56.2% in La1Pt@HCS due to slight ionization (Fig. 2b)29,42,43. In parallel, the doublet separation energy of La1Pt@HCS shown in the La 3d XPS (Fig. 2c) is reduced to 3.1 eV compared to 3.9 eV for La(OH)3 and 4.6 eV for La2O3. Of note, the decreased splitting energy of La1 in La1Pt@HCS signifies the reduction of its electron cloud density because the splitting energy is originated from spin-orbit coupling, resulting in the enhanced binding energy of La1 with reaction intermediates39,44.

a XRD patterns of La1Pt@HCS, Pt@HCS, and HCS. b Pt 4f XPS spectra of La1Pt@HCS and Pt@HCS. c La 3d5/2 XPS spectra of La1Pt@HCS, La(OH)3, and La2O3. d UPS spectra of La1Pt@HCS and Pt@HCS. e Normalized XANES at Pt L3-edge for La1Pt@HCS with Pt metal foil and PtO2 as references. f FT-EXAFS curves at Pt L3-edge, where the curve is experimental data and the circle is the best fit for La1Pt@HCS. g WT-EXAFS signals at Pt L-edge for La1Pt@HCS and the references. h Normalized XANES at La L3-edge for La1Pt@HCS with La(OH)3 and La2O3 as references. i FT-EXAFS curves at La L3-edge, where the curve is experimental data and the circle is the best fit for La1Pt@HCS. j WT-EXAFS signals at La L3-edge for La1Pt@HCS and the references.
It is well documented that the lower Φ means a higher d-band center and electron-delocalization capability, which favors the escape of electrons to intervene in the HOR process45. Figure 2d displays the Φ extracted by ultraviolet photoelectron spectroscopy (UPS), where the secondary electron cutoff energy (Ecutoff) delivers a decrease from 17.62 eV for Pt@HCS to 16.77 eV for La1Pt@HCS. Correspondingly, the Φ decreases from 4.45 eV for Pt@HCS to 3.96 eV for La1Pt@HCS. Meanwhile, the valence band maximum position (EVBM) of La1Pt@HCS shifts negatively by 0.13 eV compared to that of Pt@HCS. The valence band shifts moderately to the Fermi level upon La1 embedment, suggesting that the d-band center of La1Pt@HCS upshifts compared to that of Pt@HCS46. These results corroborate a properly enhanced OH- adsorption capacity of La1Pt@HCS, which is beneficial to accelerate the *H removal and interfacial water formation.
We also assessed the effect of La1 embedment on the atomic coordination and structural signature of La1Pt@HCS by X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements. Figure 2e presents the Pt L3-edge XANES spectra for La1Pt@HCS and the references (Pt foil and PtO2). The white line intensity peak of La1Pt@HCS is located between those of Pt foil (Pt0) and PtO2 (Pt4+), suggesting that the average oxidation state of Pt is the partial positive charge state due to the electron transfer from Pt to La–O/O species47,48, consistent with the XPS analysis (Fig. 2b). Of note, such an increased oxidation state and d orbital occupation correlate with the enhanced OH- adsorption capacity under alkaline conditions, which favors alkaline HOR kinetics12. Moreover, the Pt L-edge EXAFS spectra were obtained by Fourier transformation (FT) to elucidate bonding information (Fig. 2f). As shown, a dominant peak with the feature of the Pt foil and a second-intensive peak similar to that of the PtO2 are observed for La1Pt@HCS, which could be ascribed to Pt–Pt and Pt–O contributions, respectively. The FT-EXAFS curves at Pt L3-edge were fitted in R space (Supplementary Fig. 13), and those fitting results are summarized in Supplementary Table 2. The lower Pt-Pt average CN (5.1) and longer Pt–Pt bond length (3.05 Å) than those of Pt foils (12, 2.77 Å) reconfirm the tendency to generate weak d-orbital interactions and an upshift of the d-band center, resulting in a promoted OH- adsorption capacity. In addition, wavelet transform (WT) analysis was also conducted to obtain more intuitive bonding information (Fig. 2g), and the result discloses that the La1Pt@HCS shows both the intensity maxima of Pt foil and PtO2, which is consistent with the EXAFS result.
There may be a concern about whether the embedded La species in Pt NCs are single-atom La1 or aggregate of >2 La atoms, which could also be proven by XANES and EXAFS. As revealed by the La L3-edge XANES spectra (Fig. 2h), the white line intensities gradually decline in the order of La(OH)3, La2O3, and La1Pt@HCS, suggesting a lower La coordination number in La1Pt@HCS, which is consistent with the low coordination situation of atomically dispersed La1 within the Pt NCs40. Figure 2i shows the FT-EXAFS for La L3-edge of La1Pt@HCS, La(OH)3, and La2O3 references. The k3-weight EXAFS spectrum at La L3-edge is obtained to identify the coordination environment in R space (Supplementary Fig. 14), and those fitting results are summarized in Supplementary Table 3. The FT-EXAFS curve of La1Pt@HCS (Fig. 2i) manifests two main peaks between 1.0 and 2.5 Å, related to La–O scattering similar to that of La(OH)3 and La2O3. The La–O bond might be due to the oxidation of the single atom alloy catalyst during ex situ tests13,34. There is no La–La contributing peak at around 4.0 Å in La1Pt@HCS, validating that the La species exists in an isolated monoatomic state without long-range coordination to other La centers. This finding is consistent with the observation of La single atoms embedded within Pt NCs in the AC-HAADF-STEM image (Fig. 1h). WT-EXAFS analysis (Fig. 2j) at La L3-edge manifests one intensity maximum at ~2.5 Å related to the La–O path. It is probably reasonable to assign the second intensity peak at ~4.5 Å in the WT-EXAFS analysis to the Pt–La path. Taken together, we may conclude that single La atom is coordinated to Pt, giving rise to the Pt–La configuration. More importantly, the La1 embedment in Pt NCs could theoretically favor alkaline HOR catalysis because the electrons of La1Pt@HCS could flow from La1 into the Pt species, leaving the positively ionized donor behind as a Lewis acid site to adsorb hydroxyl species and accelerate the Volmer step of HOR44. As known, a typical HOR usually follows the Tafel (H2 + 2 M → 2M-*H)-Volmer (M − *H + OH− → M + H2O + e−) or Heyrovsky (H2 + OH− + M → M – *H + H2O + e−)-Volmer pathway (M, catalytic site; *H, adsorbed hydrogen).
Evaluation of HOR performance
As expected, the as-developed La1Pt@HCS electrocatalyst achieves high catalytic activity for alkaline HOR, which is evidenced by the HOR polarization results. The HOR polarization was tested via a rotating disk electrode method in an H2-saturated 0.1 M KOH electrolyte with Pt@HCS, Pt/XC-72, and benchmark 20% Pt/C (Pt/Ccom) as references. As shown in Fig. 3a, the HOR polarization curve of La1Pt@HCS recorded at a rotating speed of 1600 rpm manifests a sharp current response in an H2-rich environment, which elucidates that the anodic current originates from H2 oxidation considering the negligible current-voltage in N2-saturated 0.1 M KOH (Supplementary Fig. 15). Moreover, the half-wave potential (E1/2) of La1Pt@HCS is determined to be 8.4 mV (vs. RHE, all potentials in this work are given versus RHE), which is much smaller than 25.0 mV for Pt@HCS, 38.8 mV for Pt/Ccom, and 48.5 mV for Pt/XC-72 catalysts, suggesting the best kinetic activity of La1Pt@HCS for alkaline HOR catalysis. Meanwhile, the Pt@HCS delivers a sharper anodic current increment than Pt/XC-72 at the kinetic control regions, implying its better active site dispersion and higher atomic utilization triggered by the confined growth of Pt species.

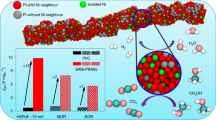
a HOR polarization curves on La1Pt@HCS, Pt@HCS, Pt/Ccom, and Pt/XC-72 in H2-saturated 0.1 M KOH with a scan rate of 5 mV s−1 at a rotating rate of 1600 rpm. b HOR polarization curves of La1Pt@HCS at varied rotating speeds (inset: corresponding Koutechy-Levich plots). c Tafel plots of the kinetic current densities. d Normalized mass activity (jk,m) at an overpotential of 25 mV (vs. RHE) and specific activity (j0,s). The error bars (standard deviations) in (d) were calculated from at least three independent testing results. e Comparison of the jk,m of La1Pt@HCS at 25 mV (vs. RHE) with that of other recently reported PGM-based electrocatalysts. f HOR polarization curves of La1Pt@HCS and Pt/Ccom before and after ADT. g Chronoamperometry (j–t) response of La1Pt@HCS and Pt/Ccom catalysts in an H2-saturated 0.1 M KOH at 60 mV (vs. RHE). h Chronoamperometry (j–t) response of La1Pt@HCS and Pt/Ccom catalysts in CO (1000 ppm)/H2-saturated 0.1 M KOH at 100 mV (vs. RHE).
The H2 mass transport as an important factor for HOR kinetics was also evaluated by analyzing the polarization process of La1Pt@HCS and reference electrocatalysts at different rotating speeds (Fig. 3b and Supplementary Fig. 16). As shown, the increased current along with the elevated rotation rate reveals the H2 mass transport, with a linear relationship between the inverse of the total current and the square root of the rotational speeds (inset in Fig. 3b). The slope of the Koutechy-Levich plot for La1Pt@HCS is determined to be 4.69 cm2 mA−1 s−1/2 at 25 mV, which is near the theoretical value of 4.87 cm2 mA−1 s−1/2 for a two-electron HOR process31. The kinetic current density (jk) of La1Pt@HCS, Pt@HCS, Pt/Ccom, and Pt/XC-72 can be extracted from the Koutechy–Levich formula to quantitatively estimate their reaction rate and activity (Fig. 3c). After normalizing the mass of Pt on La1Pt@HCS based on ICP-OES, the mass activity (jk,m) of La1Pt@HCS at 25 mV reaches 2422.38 A g−1 (Fig. 3d), which is 7.2, 14.3, and 18.0 times higher than that of Pt@HCS (338.25 A g−1), Pt/Ccom (169.05 A g−1), and Pt/XC-72 (134.29 A g−1), respectively. To the best of our knowledge, the remarkable mass activity (at 25 mV) of La1Pt@HCS outperforms that of most PGM-based electrocatalysts (Fig. 3e).
Moreover, the exchange current density (j0) for La1Pt@HCS is determined to be 20.08 mA cm−2 calculated by fitting the jk according to the Butler–Volmer formula, which is higher than that of Pt@HCS (2.61 mA cm−2), Pt/Ccom (1.55 mA cm−2), and Pt/XC-72 (1.40 mA cm−2). To quantitatively compare their intrinsic HOR activity, we calculated the specific activity normalized with the ECSA (j0,s). As the ECSA is determined to be 129.5 m2 g−1 for La1Pt@HCS, 112.3 m2 g−1 for Pt@HCS, 42.8 m2 g−1 for Pt/Ccom, and 55.1 m2 g−1 for Pt/XC-72 based on CO-stripping measurements (Supplementary Fig. 17), the j0,s of La1Pt@HCS was thus calculated to be 1.55 mA cm−2, which is 6.7, 4.3, and 6.2 times higher than that of Pt@HCS (0.23 mA cm−2), Pt/Ccom (0.36 mA cm−2), and Pt/XC-72 (0.25 mA cm−2), respectively. This result also ascertains the positive effect of spatially confined growth on Pt atom utilization/dispersion. The j0,s of some PGM-based electrocatalysts are summarized in Supplementary Table 4, which highlights the superior catalytic performance of La1Pt@HCS.
In addition to high electrocatalytic activity, the as-developed La1Pt@HCS also displays excellent durability in terms of performance stability and anti-CO poisoning capability, which is another prominent feature for advanced electrocatalysts. First, we conducted an accelerated durability test (ADT) by applying continuous potential cycling on La1Pt@HCS and Pt/Ccom catalysts in H2-saturated 0.1 M KOH. After 2000 cycles, La1Pt@HCS delivers a 3.6 mV increase in E1/2, versus 48.1 mV in Pt/Ccom (Fig. 3f), revealing the high-performance stability of La1Pt@HCS catalyst. Second, the chronoamperometry results further support the excellent performance stability of La1Pt@HCS (Fig. 3g), where La1Pt@HCS shows a current density with almost zero decay after 10 h at 60 mV, which is in stark contrast to the Pt/Ccom losing ~42% of HOR activity under the same conditions. It is worth noting that the high-performance stability of La1Pt@HCS is derived from their unique HCS architecture-mediated protection for La1Pt NCs, and we compared the surface chemical states and morphology of La1Pt@HCS before and after ADT to clarify this issue. As shown in Supplementary Fig. 18, SEM-EDS images show that the morphology and composition of the spent La1Pt@HCS are largely maintained. The invariant Pt 4f and La 3d XPS spectra of La1Pt@HCS before and after ADT corroborate its good surface chemical stability during the long-term electrochemical process (Supplementary Fig. 19). In addition, the TEM image of the spent La1Pt@HCS reveals that the La1Pt NCs are still space-confined on nanopores without apparent morphological changes (Supplementary Fig. 20). These results elucidate the superior HOR catalytic stability of La1Pt@HCS catalyst, which stems from the confined growth induced by the intrinsic porosity of HCS, effectively inhibiting the agglomeration of Pt species.
In parallel, the La1Pt@HCS also exhibits excellent long-term anti-CO poisoning ability with Pt/Ccom as a reference. It is known that CO poisoning is an intractable obstacle for anodic PGM-based electrocatalysts (especially Pt) when using fossil fuels for fuel H2 production, and the preferential CO binding on Pt surface could poison the active sites for *H adsorption/desorption. In this study, the anti-CO poisoning ability of La1Pt@HCS was evaluated by a chronoamperometry at 100 mV with 1000 ppm CO impurity. As illustrated in Fig. 3h, the La1Pt@HCS catalyst retains a high catalytic activity (current retention rate: ~61%) in the presence of CO impurity after continuously operating for ≈1000 s, whereas Pt/Ccom becomes completely inactive (current decay rate: 100%) after continuous HOR catalysis (≈650 s), corroborating the superior anti-CO poisoning capability of La1Pt@HCS comparing to Pt/Ccom. The robust anti-CO poisoning ability of La1Pt@HCS is due to that the Pt-coordinated La1 single atom with strong OH− adsorption capacity could promote the oxidation of *CO4. Taken together, the aforementioned results demonstrate that the La1Pt@HCS can serve as an efficient yet durable electrocatalyst for alkaline HOR.
Mechanism investigation
We are curious about the atomic/molecular-level catalytic mechanism of the La1Pt@HCS in alkaline HOR, and thus employed DFT simulation to approach this goal. The Pt(111) surface is selected in DFT calculations because the (111) plane possesses the lowest surface energy and the most-abundant surface sites in Pt crystal33. Firstly, the model of La1Pt@HCS was constructed by using one La atom to substitute for one Pt atom in Pt(111) (Supplementary Fig. 21) based on the XRD, STEM, and XAS data. Such a La1-embedded configuration has lower formation energy (−4.031 eV) than the La1-deposited one (fcc site: −3.151 eV, hcp site: −3.077 eV; Supplementary Fig. 22), further supporting that the La1 was embedded in Pt NCs49. With the model in hand, we analyzed all possible intermediate adsorption sites, including La top (1), Pt top (2), Pt-Pt bridge (3), Pt-La bridge (4), hcp-type hollow (5), and fcc-type hollow (6) (Supplementary Fig. 21b). For comparison, Pt@HCS was simulated using the undoped Pt(111) model (Supplementary Fig. 23), including top (1), bridge (2), fcc-type hollow (3), and hcp-type hollow (4) adsorption sites. It should be noted that only those locally stable adsorption configurations were taken into account for evaluating HOR activity, and the carbon support was not included in the modeling due to its negligible effect on the electrocatalytic activities of La1Pt@HCS and Pt@HCS. Such model simplifications could be favorable for specifically studying the modulation function of single-atom La on metallic Pt.
As depicted in Fig. 4a and Supplementary Fig. 24, the differential charge densities show that Pt-coordinated isolated La atom has a pronounced electron-deficient nature. In an alkaline environment, electron-deficient La can act as a Lewis acid site to attract lone pairs of electrons of Lewis base OH− (σ-donation)40. In the simulated 0.1 M KOH electrolyte system, a much higher OH− density is accumulated around La sites (Supplementary Fig. 25), revealing that a large number of OH− can be enriched and activated at the La site50,51. It is inferred that single-atom La-induced Lewis acid-base interactions can kinetically drive *H removal via combined *H and *OH, resulting in enhanced HOR activity52.

a Charge density difference of La1Pt NCs. Yellow, electron accumulation; cyan, electron depletion. b The PDOS diagram for the d orbital of metal surfaces on La1Pt NCs and Pt NCs. The blue dotted lines are the d-band center. c Schematic DOS diagrams illustrating the single-atom La1 doping on the d-band position of La1Pt NCs. The bond formation between the model and the adsorbates (Ads.). d Calculated OHBE on different adsorption sites for La1Pt NCs and Pt NCs. e ΔG*H on different adsorption sites for La1Pt NCs and Pt NCs. f Free energy diagram of the HOR (in eV) on La1Pt NCs and Pt NCs.
Besides, the faster HOR kinetics over La1Pt NCs can also be correlated to the change of the electronic structures incurred by the presence of La atom. The projected density of states (PDOS) of the d orbital for the metal surface in La1Pt NCs and Pt NCs were calculated (Fig. 4b), and the PDOS of La1Pt NCs system presents a high occupancy near the Fermi level, indicating that the La1 embedment increases electron transfer and conductivity, which are in good agreement with the UPS results. According to Nørskov’s d-band center theory, when the adsorbate approaches the metal surface, its orbital electronically interfaces with the metallic orbital, resulting in energy level splitting.53 The position of the antibonding orbitals created by energy level splitting determines the adsorption strength, and antibonding states above the Fermi level are advantageous for adsorbing OH- species in alkaline HOR. In the La1Pt NCs system, the d-orbital of La hybridizes with the d-orbital of Pt or the p-orbital of O, which allows electron transfer to occur because of the different electronegativity, and upshifts the d-band center from −2.75 eV of Pt NCs to −2.14 eV of La1Pt NCs (Fig. 4b). On this basis, more adsorbate antibonding states are pulled above Fermi level, corroborating a promoted interaction between adsorbates and metal atoms (Fig. 4c)54. Moreover, the OH binding energy (OHBE) information of La1Pt@HCS and Pt/Ccom can be reflected in the corresponding cyclic voltammogram curves (Supplementary Fig. 26). The La1Pt@HCS manifests lower OH- adsorption potential than Pt/Ccom, suggesting that the surfaces of La1Pt@HCS prefer to bind OH- compared to that of Pt/Ccom.
Considering that the sluggish HOR on the Pt-based catalyst in an alkaline environment is largely related to the intrinsic poor OH- adsorption on Pt (111)29, we conducted a DFT simulation to further quantify the increased OH adsorption capacity by placing OH- groups at different adsorption sites of La1Pt NCs and Pt NCs. As shown in Fig. 4d, the three most stable OH− adsorption sites are identified on the surface of La1Pt NCs and Pt NCs upon structural optimization, and the corresponding positive OHBE values on the Pt NCs are calculated to be 0.94, 0.56, and 0.62 eV, while the OH− adsorption sites on La1Pt NCs deliver negative OHBE values of −0.29, −0.36, and −0.28 eV. These distinct OHBE information reveals an enhanced interaction of the OH− with the surface of La1Pt NCs, which is consistent with the UPS and XAS results. In addition to the OHBE, the HOR catalytic activity of La1Pt NCs is also often estimated by Gibbs free energy for hydrogen adsorption (ΔG*H) because an optimal HOR catalyst should have ΔG*H close to 0 according to the Sabatier principle55,56. As displayed in Fig. 4e, the La site as a Lewis acid site, has negligible adsorption capacity for H species (ΔG*H = 1.72 eV). Besides, the hydrogen adsorption strength of other active sites in La1Pt NCs (ΔG*H = −0.37, −0.20, and −0.37 eV) is close to or slightly stronger than that of Pt NCs (ΔG*H = −036, −0.21, −0.22, and −0.19 eV), indicating that hydrogen binding energy (HBE) is not the dominant factor for boosting HOR activity in La1Pt NCs. Nevertheless, while a weaker H species binding is predicted to facilitate its oxidative removal and boost HOR kinetics, slightly enhanced *H binding in La1Pt NCs is essential to preserve adsorption sites from passivation by *OH.11 It should be noted that we further modeled carbon loaded-La1Pt NCs (La1Pt NCs@C) and -Pt NCs (Pt NCs@C, Supplementary Fig. 27). We found that both La1Pt NCs and La1Pt NCs@C show similar adsorption/desorption strength or tendency for OH (Supplementary Fig. 28a) and H intermediates (Supplementary Fig. 28b), and the same is true for Pt NCs and Pt NCs@C. These results further validate the negligible influence of carbon support on the electrocatalytic activity, highlighting the rationality of the model simplification.
We also plotted the Gibbs free energy diagram to investigate the HOR process over La1Pt NCs and Pt NCs. As shown in Fig. 4f, the Tafel step happening at the optimal H adsorption site (site H3) is the first step (Supplementary Fig. 29), in which the free energies of H2 dissociation for La1Pt NCs and Pt NCs are determined to be 0.27 and 0.18 eV with the endothermic feature, respectively. Afterward, the reaction barriers were determined from (*H + *OH) to the water formation step in the Volmer step (Supplementary Fig. 30), which are 0.42 and 0.62 eV for the La1Pt NCs and Pt NCs, respectively. The higher reaction energy barrier identifies water formation as the rate-determining step (RDS)22, and the simulation results uncover that the reaction energy of RDS was significantly lower on La1Pt NCs than on Pt NCs. We also comprehensively compared the alkaline HOR activity descriptors of the La1Pt with those of other metals and metal oxides, and found that the La1Pt performs best among them due to its relatively low energy barrier for the Volmer reaction as well as moderate HBE and OHBE which are not possessed by Pt(110), Ni, CeO2, Pd, and PtNi alloy catalysts (Supplementary Table 5). Overall, both experimental and theoretical results jointly indicate that the enhanced alkaline HOR activity originates from the tuned structural and electronic properties of the embedded La single atom inside La1Pt NCs.
As demonstrated, the weak H species adsorption capacity on rare-earth atoms cannot afford the efficient H2 adsorption/dissociation kinetics in the Tafel step, and therefore introducing the rare-earth elements with high oxophilicity into metallic Pt with the optimal ΔG*H could be a valid strategy to promote alkaline HOR activities, which will realize the low Volmer energy barrier by promoting the adsorption of OH- species and the desorption of *H57. In this way, a series of Ln1Pt@HCS catalysts (here Ln = Ce, Pr, Nd, and Lu) should all possess enhanced HOR catalytic activities due to the high oxophilicity of Ln metals. To verify this assumption, we performed a polarization test over all the Ln1Pt@HCS electrocatalysts (Supplementary Fig. 31), and the results show that all electrocatalysts have an onset potential for yielding response current as low as 0 V, indicating their similar energetics for alkaline HOR catalysis. Moreover, the E1/2 values of all Ln1Pt@HCS catalysts are concentrated at ~10.0 mV, which is significantly lower than 38.8 mV for Pt/Ccom. The increased current along with the elevated rotation rate (Supplementary Fig. 32) reveals the H2 mass transport, with a linear relationship between the inverse of total current and the square root of rotational speeds (Supplementary Fig. 33). The slopes of Koutechy-Levich plots for Ce1Pt@HCS, Pr1Pt@HCS, Nd1Pt@HCS, and Lu1Pt@HCS were determined to be 5.02, 4.95, 4.49, and 4.99 cm2 mA−1 s−1/2 at 25 mV, respectively, close to the theoretical value of 4.87 cm2 mA−1 s−1/2. Moreover, all these Ln1Pt@HCS electrocatalysts displayed remarkably enhanced CO tolerance comparing to the Pt/Ccom counterpart (Fig. 3h and Supplementary Fig. 34). DFT simulations reveal that all Ln1Pt NCs (Supplementary Fig. 35) have stronger OH- adsorption capacity on Ln sites (OHBE: −0.82 to −0.38 eV) than pure Pt NCs (OHBE: 0.55 to 0.94 eV) (Supplementary Figs. 36 and 37). Notably, the correlation between the different degrees of oxophilicity for rare-earth elements and the precise catalytic activity is not the research focus of this work but will be a topic worth investigating in the future.
These results convincedly demonstrate that embedding rare-earth single atoms into Pt NCs could yield significant electronic modulation and enhance OH− adsorption strength, resulting in favorable alkaline HOR activity through accelerated Volmer kinetics. According to the electrochemical measurements and theoretical simulations, we propose the OH--induced adsorption mechanism for alkaline HOR catalysis triggered by Pt-coordinated isolated rare-earth atoms (Supplementary Fig. 38). The introduced rare-earth single-atom, as a Lewis acid site, boosts the migration of OH− inside the alkaline environment and gives rise to an increased local OH− concentration around the Pt-Ln sites, thereby improving the hydrogen oxidation ability of metallic Pt.
Discussion
In summary, we have successfully developed a series of single-atom lanthanide (e.g., La, Ce, Pr, Nd, and Lu)-embedded Ln1Pt NCs via vapor filling and spatially confined reduction of metal species inside HCS, solving the incompatibility issue between rare-earth elements and metallic Pt, and concurrently achieving high-performance HOR catalysis in alkaline media. Mechanism studies revealed that the highly oxophilic Ln1 species could serve as the Lewis acid site for selective OH adsorption, and regulate the *H and *CO adsorption on Pt active sites via upshifting the d-band center, promoting the HOR kinetics and CO oxidation by accelerating the *H and *CO removal through OH--induced reactions. Consequently, the Ln1Pt@HCS achieved remarkable electrocatalytic activity towards alkaline HOR with a mass activity of up to 14.3-times higher than that of commercial Pt/C and enhanced durability in terms of anti-CO poisoning capability and structural stability. This study exemplifies the integration of single-atom Ln1 with ultrasmall Pt NCs for high-performance HOR catalysis and sheds light on the future design and fundamental understanding of metal NCs-based electrocatalysts for energy conversion.
Methods
Materials
Tetrapropyl orthosilicate (TPOS, 98%), ammonium hydroxide (28%–30%), resorcinol (99%), formaldehyde (37% in H2O), hydrofluoric acid (40%–45%), Platinum(II) acetylacetonate (Pt(acac)2, 97%), Lanthanum(III) acetylacetonate hydrate (La(acac)3, 98%), Cerium(III) acetylacetonate hydrate (Ce(acac)3, 99%), Praseodymium(III) acetylacetonate hydrate (Pr(acac)3, 98%), Neodymium(III) 2,4-pentanedionate (Nd(acac)3, 99%), Lutetium(III) 2,4-pentanedionate (Lu(acac)3, 98%) were purchased from Aladdin. Carbon black (Vulcan XC-72R) was bought from Carbot Co. All reagents were used as received without further purification.
Synthesis of HCS
Hollow carbon spheres (HCS) were prepared according to an improved method41. Typically, 70 mL of ethanol, 10 mL of deionized water, and 3.0 mL of ammonium hydroxide were stirred together for 10 min at room temperature, and then 3.5 mL of TPOS was added into the solution under stirring for 10 min. 2.0 mL of ethanol solution containing resorcinol (0.4 g) and formaldehyde (0.56 mL) was added to the solution. After 24 h, the SiO2@resorcinol-formaldehyde resin nanospheres were collected by centrifugation, washed with ethanol three times, and then dried at 60 °C under vacuum overnight. After that, the brown powder was annealed in Ar atmosphere at 700 °C for 5 h with a heating rate of 2 °C min−1 to produce SiO2@C. The obtained black powder was etched with 1.0 M sodium hydroxide at 70 °C for 24 h. Finally, HCS material was collected by centrifugation, washed with deionized water and ethanol three times, and dried under vacuum for 12 h.
Synthesis of La1Pt@HCS
First, 0.06 mmol of Pt(acac)2, 0.014 mmol of La(acac)3, and 35 mg of obtained HCS were sealed under vacuum at 10-5 mbar in quartz ampoule. Subsequently, the quartz ampoule was heated in a rotary oven at 140 °C and 200 °C for 48 h and 24 h, respectively. After the reaction, the ampoule was cooled rapidly. The powder was collected and washed with ethanol under sonication, then dried under vacuum for 12 h.
Synthesis of Ln1Pt@HCS (M = Ce, Nd, Pr, and Lu)
The synthesis of Ln1Pt@HCS was the same as that of La1Pt@HCS, except for the replacement of La(acac)3 by Ce(acac)3, Nd(acac)3, Pr(acac)3, and Lu(acac)3 in the first step.
Synthesis of Pt@HCS
The synthesis of Pt@HCS was the same as that of La1Pt@HCS except for the absence of La(acac)3.
Synthesis of Pt/XC-72
The synthesis of Pt/XC-72 was the same as that of Pt@HCS except for the replacement of HCS by XC-72.
Material characterizations
Scanning electron microscopy (SEM) was performed using a JSM-7900F scanning electron microscope. Transmission electron microscopy (TEM) images and low-magnification high-angle annular dark-field scanning TEM (HAADF-STEM) images were obtained with an FEL Talos F200X field emission electron microscope at an accelerating voltage of 200 kV. The signal collection duration for the HAADAF-STEM elemental maps is 10 min. Atomic resolution high magnification STEM image was obtained using FEI Themis Z aberration-corrected transmission electron microscope. Metal contents of the samples were analyzed by inductively coupled plasma optical emission spectroscopy (ICP-OES) on Thermo Scientific iCAP RRO. N2 sorption analysis was performed at 77 K with a Micromeritics ASAP 2460 analyzer. X-ray powder diffraction (XRD) test was conducted on a D8 Advance X-ray powder diffractometer equipped with a Cu Kα radiation source. X-ray photoelectron spectroscopy (XPS) was performed on a K-Alpha spectrometer from Thermo Scientific using an Al Kα photon source (hv = 1486.6 eV) at 12 kV. XAFS analyses were performed with Si(111) crystal monochromators at the BL14W beamline at the Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, China). The XAFS spectra were recorded at room temperature using a 4-channel Silicon Drift Detector (SDD) Bruker 5040. The extended X-ray absorption fine structure (EXAFS) spectra were recorded in transmission/fluorescence mode. The Φ was extracted by ultraviolet photoelectron spectroscopy (UPS) on ESCALAB Xi+ analyzer from Thermo Scientific. A He discharge lamp with the He (I) photo line (21.22 eV) was applied, and the high-binding energy secondary electron cutoff (Ecutoff) and the energy gap (ΔE) between the valence band maximum (VBM) and the fermi level were extracted from the UPS spectra44. The Φ was calculated as Eq. (1):
The VBM position was calculated as Eq. (2):
Electrochemical characterization
To prepare the electrode, 2.0 mg of catalyst, 980 μL of isopropanol, and 20 μL of 5 wt.% Nafion solution were mixed and sonicated for 1 h to prepare catalyst ink. A certain amount of the catalyst ink was taken out and coated on a glassy carbon rotating disk electrode (RDE) (diameter: 4 mm) to achieve a loading amount of 10 μgPt cm−2. 20 wt.% Pt/C was employed as a reference (10 μgPt cm−2). All electrochemical measurements were performed on a standard three-electrode system by a CHI 660d electrochemical workstation. All reference potentials have been converted to a reversible hydrogen electrode (vs. RHE) and were iR-corrected (i, current; R, resistance) for the uncompensated Ohmic drop. A graphite rod and a Hg/HgO electrode were employed as the counter electrode and reference electrode, respectively. Linear sweep voltammetry (LSV) was carried out in a 0.1 M of KOH solution saturated with H2 at various rotation rates at a sweep speed of 5 mV s−1. Cyclic voltammetry (CV) was performed in an Ar-saturated 0.1 M KOH solution at 50 mV s−1. For CO-stripping voltammetry, the electrode potential was held at 0.1 V vs. RHE for 10 min to adsorb CO on the surface of Pt. The electrode was quickly moved to a fresh 0.1 M KOH solution. Then, the adsorbed CO was stripped by scanning between 0.05 and 1.25 V (vs. RHE) at a scan rate of 50 mV s−1. Electrochemical impedance spectroscopy (EIS) was conducted with a frequency ranging from 0.1 to 105 Hz and an amplitude of 5 mV under an overpotential of 30 mV29,58.
The kinetic current density (jk) can be obtained by the Koutechy–Levich (K–L) equation:
where j and jd represent the measured current and diffusional current, respectively.
where n, F, D, v, C0, B, and ω are the number of electrons transferred (2), the Faraday constant (96485 C mol−1), the diffusion coefficient of H2 (3.7 × 10−5 cm2 s−1), the kinematic viscosity (1.01 × 10−2 cm2 s−1), the solubility of H2 (7.33 × 10−4 mol L−1), the Levich constant, and the rotating speed, respectively.
The exchange current density (j0) can be obtained by fitting jk into the Butler–Volmer (B–V) Eq. (5):
where α, R, T, and η represent the transfer coefficient, the universal gas constant (8.314 J mol−1 K−1), the operating temperature (298 K), and the overpotential, respectively.
Computational details and theoretical models
All calculations were carried out with the spin-unrestricted density functional theory (DFT) method by the DMol3 module of the Materials Studio software59,60. The exchange-correlation interactions were treated by generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional61. Grimme’s DFT-D3 corrected method was used to account for weak interactions such as long-rage van der Waals intermolecular and dispersive intermolecular interactions62. The convergence criterion was set to 10−6 au in self-consistent field computation, and the real space cutoff radius of atomic orbital was set as 4.9 Å. The geometry optimization was carried out until the convergence threshold of 1 × 10−5 Ha for energy, 2 × 10−3 Ha Å−1 for force, and 5 × 10−3 Å for displacement. The Brillouin zone was sampled with the 4 × 4 × 1 k-points for the geometry structures and energy calculations for the slab models63. The transition states were searched by using the linear synchronous transition/quadratic synchronous transit method and confirmed by the frequency calculation64.
The optimized lattice constant of bulk face-centered cubic Pt structure is 3.866 Å, which is according well to the experimental results (3.923 Å)65, suggesting the reliability of the chosen computational strategy. A (4 × 4) Pt(111) slab model of four layers was used for Pt and La1Pt, with the bottom two layers of Pt atoms fixed to mimic bulk structure. Moreover, a vacuum layer of 20 Å was set in the z-direction to avoid the interaction between the periodic layers.
The formation energies for adsorbed and embedded cases were determined by Eqs. (6) and (7):
The adsorption energy of an adsorbate (ΔE) was calculated by Eq. (8):
where Eadsorbate/sub is the total energy of the adsorbed system, Eadsorbate is the energy of an adsorbate, Esub is the energy of the substrate.
The adsorption free energies (ΔG) were determined by Eq. (9):
where ΔE, ΔZPE, and ΔS represent the binding energy, zero-point energy change, and entropy change of the adsorption of adsorbates, respectively.
The d-band center (εd) of the catalysts was represented as Eq. (10):
where ρd(E) is the density of d states at an energy level E.
Data availability
The data supporting the conclusions of this study are present in the paper and the Supplementary Information. The raw data sets used for the presented analysis within the current study are available from the corresponding authors upon request.
References
Gao, F.-Y. et al. Nickel-molybdenum-niobium metallic glass for efficient hydrogen oxidation in hydroxide exchange membrane fuel cells. Nat. Catal. 5, 993–1005 (2022).
Xiao, F. et al. Recent advances in electrocatalysts for proton exchange membrane fuel cells and alkaline membrane fuel cells. Adv. Mater. 33, 2006292 (2021).
Ramaswamy, N. & Mukerjee, S. Alkaline anion-exchange membrane fuel cells: challenges in electrocatalysis and interfacial charge transfer. Chem. Rev. 119, 11945–11979 (2019).
Duan, Y. et al. Bimetallic nickel-molybdenum/tungsten nanoalloys for high-efficiency hydrogen oxidation catalysis in alkaline electrolytes. Nat. Commun. 11, 4789 (2020).
Zhan, C. et al. Subnanometer high-entropy alloy nanowires enable remarkable hydrogen oxidation catalysis. Nat. Commun. 12, 6261 (2021).
Yang, X. et al. Ru colloidosome catalysts for the hydrogen oxidation reaction in alkaline media. J. Am. Chem. Soc. 144, 11138–11147 (2022).
Ma, M. et al. Single-atom molybdenum engineered platinum nanocatalyst for boosted alkaline hydrogen oxidation. Adv. Energy Mater. 12, 2103336 (2022).
Zhang, Y. et al. Atomically isolated Rh sites within highly branched Rh2Sb nanostructures enhance bifunctional hydrogen electrocatalysis. Adv. Mater. 33, 2105049 (2021).
Han, L. et al. Design of Ru-Ni diatomic sites for efficient alkaline hydrogen oxidation. Sci. Adv. 8, eabm3779 (2022).
Men, Y. et al. Oxygen-inserted top-surface layers of Ni for boosting alkaline hydrogen oxidation electrocatalysis. J. Am. Chem. Soc. 144, 12661–12672 (2022).
Zhu, S. et al. The role of ruthenium in improving the kinetics of hydrogen oxidation and evolution reactions of platinum. Nat. Catal. 4, 711–718 (2021).
Huang, B. et al. In-situ precise anchoring of Pt single atoms in spinel Mn3O4 for highly efficient hydrogen evolution reaction. Energ. Environ. Sci. 15, 4592–4600 (2022).
Li, M. et al. Single-atom tailoring of platinum nanocatalysts for high-performance multifunctional electrocatalysis. Nat. Catal. 2, 495–503 (2019).
Zhou, Y. et al. Lattice-confined Ru clusters with high CO tolerance and activity for the hydrogen oxidation reaction. Nat. Catal. 3, 454–462 (2020).
Du, H. et al. Unlocking interfacial electron transfer of ruthenium phosphides by homologous core–shell design toward efficient hydrogen evolution and oxidation. Adv. Mater. 34, 2204624 (2022).
He, Q. et al. Confined PdMo ultrafine nanowires in CNTs for superior oxygen reduction catalysis. Adv. Energy Mater. 12, 2200849 (2022).
Li, L. et al. Surface and lattice engineered ruthenium superstructures towards high-performance bifunctional hydrogen catalysis. Energy Environ. Sci. 16, 157–166 (2023).
Yao, Z.-C. et al. Electrocatalytic hydrogen oxidation in alkaline media: from mechanistic insights to catalyst design. ACS Nano 16, 5153–5183 (2022).
Davydova, E. S., Mukerjee, S., Jaouen, F. & Dekel, D. R. Electrocatalysts for hydrogen oxidation reaction in alkaline electrolytes. ACS Catal. 8, 6665–6690 (2018).
Koper, M. T. M. A basic solution. Nat. Chem. 5, 255–256 (2013).
Strmcnik, D. et al. Improving the hydrogen oxidation reaction rate by promotion of hydroxyl adsorption. Nat. Chem. 5, 300–306 (2013).
Su, L. et al. The Role of Discrepant reactive intermediates on Ru-Ru2P heterostructure for pH-universal hydrogen oxidation reaction. Angew. Chem. Int. Ed. 62, e202215585 (2022).
Lu, S. & Zhuang, Z. Investigating the influences of the adsorbed species on catalytic activity for hydrogen oxidation reaction in alkaline electrolyte. J. Am. Chem. Soc. 139, 5156–5163 (2017).
Qin, S. et al. Ternary nickel-tungsten-copper alloy rivals platinum for catalyzing alkaline hydrogen oxidation. Nat. Commun. 12, 2686 (2021).
Ogada, J. J. et al. CeO2 modulates the electronic states of a palladium onion-like carbon interface into a highly active and durable electrocatalyst for hydrogen oxidation in anion-exchange-membrane fuel cells. ACS Catal. 12, 7014–7029 (2022).
Luo, H. et al. Amorphous MoOx with high oxophilicity interfaced with PtMo alloy nanoparticles boosts anti-CO hydrogen electrocatalysis. Adv. Mater. e2211854 https://doi.org/10.1002/adma.202211854 (2023).
Mao, J. et al. Isolated Ni atoms dispersed on Ru nanosheets: high-performance electrocatalysts toward hydrogen oxidation reaction. Nano Lett. 20, 3442–3448 (2020).
Shan, J. et al. Integrating interactive noble metal single-atom catalysts into transition metal oxide lattices. J. Am. Chem. Soc. 144, 23214–23222 (2022).
Wang, X. et al. Atomic-precision Pt6 nanoclusters for enhanced hydrogen electro-oxidation. Nat. Commun. 13, 1596 (2022).
Zhao, T. et al. Pseudo-Pt monolayer for robust hydrogen oxidation. J. Am. Chem. Soc. 145, 4088–4097 (2023).
Liu, D.-Q. et al. Tailoring interfacial charge transfer of epitaxially grown Ir clusters for boosting hydrogen oxidation reaction. Adv. Energy Mater. 13, 2202913 (2023).
Yang, X. et al. CO-tolerant PEMFC anodes enabled by synergistic catalysis between iridium single-atom sites and nanoparticles. Angew. Chem. Int. Ed. 60, 26177–26183 (2021).
Duchesne, P. N. et al. Golden single-atomic-site platinum electrocatalysts. Nat. Mater. 17, 1033–1039 (2018).
Zheng, T. et al. Copper-catalysed exclusive CO2 to pure formic acid conversion via single-atom alloying. Nat. Nanotechnol. 16, 1386–1393 (2021).
Hu, Y., Jensen, J. O., Cleemann, L. N., Brandes, B. A. & Li, Q. Synthesis of Pt–rare earth metal nanoalloys. J. Am. Chem. Soc. 142, 953–961 (2020).
Wong, A., Liu, Q., Griffin, S., Nicholls, A. & Regalbuto, J. R. Synthesis of ultrasmall, homogeneously alloyed, bimetallic nanoparticles on silica supports. Science 358, 1427–1430 (2017).
Hu, M. et al. Surface-confined synthesis of ultrafine Pt-Rare earth nanoalloys on N-functionalized supports. Adv. Funct. Mater. 32, 2202675 (2022).
Liu, J. et al. Rare earth single-atom catalysts for nitrogen and carbon dioxide reduction. ACS Nano 14, 1093–1101 (2020).
Chen, P. et al. Rare-earth single-atom La–N charge-transfer bridge on carbon nitride for highly efficient and selective photocatalytic CO2 reduction. ACS Nano 14, 15841–15852 (2020).
Liu, X. et al. Rare earth La single atoms supported MoO3-x for efficient photocatalytic nitrogen fixation. Appl. Catal. B Environ. 301, 120766 (2022).
Wan, X.-K., Wu, H. B., Guan, B. Y., Luan, D. & Lou, X. W. Confining sub-nanometer Pt clusters in hollow mesoporous carbon spheres for boosting hydrogen evolution activity. Adv. Mater. 32, 1901349 (2020).
Gao, T. et al. Evidence of the Strong metal support interaction in a palladium-ceria hybrid electrocatalyst for enhancement of the hydrogen evolution reaction. J. Electrochem. Soc. 165, F1147–F1153 (2018).
Chen, L. et al. Promoting electrocatalytic methanol oxidation of platinum nanoparticles by cerium modification. Nano Energy 73, 104784 (2020).
Zhai, L. et al. Modulating built-in electric field via variable oxygen affinity for robust hydrogen evolution reaction in neutral media. Angew. Chem. Int. Ed. 61, e202116057 (2022).
Zhao, X. et al. Controlling the valence-electron arrangement of nickel active centers for efficient hydrogen oxidation electrocatalysis. Angew. Chem. Int. Ed. 61, e202206588 (2022).
Chen, Z. et al. Tailoring the d-band centers enables Co4N nanosheets to be highly active for hydrogen evolution catalysis. Angew. Chem. Int. Ed. 57, 5076–5080 (2018).
Liu, D. et al. Atomically dispersed platinum supported on curved carbon supports for efficient electrocatalytic hydrogen evolution. Nat. Energy 4, 512–518 (2019).
Zhu, X. et al. Intrinsic ORR activity enhancement of Pt atomic sites by engineering the d-band center via local coordination tuning. Angew. Chem. Int. Ed. 60, 21911–21917 (2021).
Sahoo, S., Dekel, D. R., Maric, R. & Alpay, S. P. Atomistic insights into the hydrogen oxidation reaction of palladium-ceria bifunctional catalysts for anion-exchange membrane fuel cells. ACS Catal. 11, 2561–2571 (2021).
Yu, X. et al. Boron-doped graphene for electrocatalytic N2 reduction. Joule 2, 1610–1622 (2018).
Li, X. et al. PdFe Single-atom alloy metallene for N2 electroreduction. Angew. Chem. Int. Ed. 61, e202205923 (2022).
Liu, S. et al. Facilitating nitrogen accessibility to boron-rich covalent organic frameworks via electrochemical excitation for efficient nitrogen fixation. Nat. Commun. 10, 3898 (2019).
Hammer, B. & Norskov, J. K. Why gold is the noblest of all the metals. Nature 376, 238–240 (1995).
Nørskov, J. K., Abild-Pedersen, F., Studt, F. & Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl Acad. Sci. USA 108, 937–943 (2011).
Wang, T. et al. Weakening hydrogen adsorption on nickel via interstitial nitrogen doping promotes bifunctional hydrogen electrocatalysis in alkaline solution. Energ. Environ. Sci. 12, 3522–3529 (2019).
Wang, X. et al. Platinum-free electrocatalysts for hydrogen oxidation reaction in alkaline media. Nano Energy 104, 107877 (2022).
Pagliaro, M. V. et al. Improving alkaline hydrogen oxidation activity of palladium through interactions with transition-metal oxides. ACS Catal. 12, 10894–10904 (2022).
Wang, X. & Xing, W. Metal-organic framework-derived nickel-based catalyst for hydrogen oxidation reaction. J. Phys Confer. Ser. 2021, 012103 (2021).
Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 113, 7756–7764 (2000).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Zhang, Q., Liu, Y. & Chen, S. A. DFT calculation study on the temperature-dependent hydrogen electrocatalysis on Pt(111) surface. J. Electroanal. Chem. 688, 158–164 (2013).
Ehrlich, S., Moellmann, J., Reckien, W., Bredow, T. & Grimme, S. System-dependent dispersion coefficients for the DFT-D3 treatment of adsorption processes on ionic surfaces. Chemphyschem 12, 3414–3420 (2011).
Chadi, D. J. Special points for Brillouin-zone integrations. Phys. Rev. B 16, 1746 (1977).
Halgren, T. A. & Lipscomb, W. N. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 49, 225–232 (1977).
Travitsky, N. et al. Pt-, PtNi- and PtCo-supported catalysts for oxygen reduction in PEM fuel cells. J. Power Sources 161, 782–789 (2006).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51877216, 52277229, 21805307, 21905300, and 22071127), National Key Research and Development of China (2022YFA1503400), the Natural Science Foundation of Shandong Province (ZR2020MB078), and Taishan Scholar Foundation (tsqn201812074).
Author information
Authors and Affiliations
Contributions
W.X. and X.Y. lead the present work. X.W. and Y.T. carried out the experiments, collected and analyzed the data. Y.T. carried out the DFT calculations. W.F., P.L., X.L., Y.C., T.C., L.Z., Q.X., and Z.Y. helped with data analysis. X.W. and X.Y. co-wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Elena Davydova, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Tong, Y., Feng, W. et al. Embedding oxophilic rare-earth single atom in platinum nanoclusters for efficient hydrogen electro-oxidation. Nat Commun 14, 3767 (2023). https://doi.org/10.1038/s41467-023-39475-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-39475-5
This article is cited by
-
Co-catalytic metal–support interactions in single-atom electrocatalysts
Nature Reviews Materials (2024)
-
Atomic interface regulation of rare-marth metal single atom catalysts for energy conversion
Nano Research (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
