
Abstract
While single cell heterogeneity is present in all biological systems, most studies cannot address it due to technical limitations. Here we describe a nano-liter droplet microfluidic-based approach for stimulation and monitoring of surfaceand secreted markers of live single immune dendritic cells (DCs) as well as monitoring the live T cell/DC interaction. This nano-liter in vivo simulating microenvironment allows delivering various stimuli reagents to each cell and appropriate gas exchanges which are necessary to ensure functionality and viability of encapsulated cells. Labeling bioassay and microsphere sensors were integrated into nano-liter reaction volume of the droplet to monitor live single cell surface markers and secretion analysis in the time-dependent fashion. Thus live cell stimulation, secretion and surface monitoring can be obtained simultaneously in distinct microenvironment, which previously was possible using complicated and multi-step in vitro and in vivo live-cell microscopy, together with immunological studies of the outcome secretion of cellular function.
Similar content being viewed by others

Introduction
The cellular phenotype is a conglomeration of multiple cellular processes, representing varying expression levels of genes and proteins that determine the cell's particular function in activities such as cellular communication, adhesion, or metabolism1,2,3,4. Average measurements of such molecular markers in cell populations obscure inherent cell-cell heterogeneity and restrict the ability to distinguish between the responses of individual cells within a sample2,5,6,7. While flow cytometry has traditionally been used to determine single cell phenotypes, it cannot provide continuous measurements of proteins in the same individual cells over time7. Recently, the development of microfluidic technology enabled analysis of cell phenomena on the single cell level8,9,10. The reported platforms, mostly based on micro well technologies, do not provide a controllable microenvironment for cell-cell interaction, however, as no reagent mixing is provided. Moreover, technical manipulation of these platforms prohibits their current use beyond the wall of the developer laboratory. In this paper, we describe a novel approach that allows stimulation and dynamic characterization of live single cells for surface and secreted protein expressions. This approach should have a broad impact on diverse biological systems for the study of cell surface and secretion proteins as potential biomarkers and targets for diagnostics and therapeutics as well as cell-cell interactions imaging. In particular, one clinical application of the described approach for monitoring live cell surface markers and secretion as well as cell-cell interactions can be applied to a DC vaccine evaluation. Evaluation of the efficacy of various therapeutic agents such (i.e. small molecules, antibodies, nucleic acid polymers) could significantly benefit from this sensitive and rapid detection methodology. The correlation between surface protein expression and secretion obtained from cells before and after treatment can be obtained in a dynamic fashion on a single cell level.
Defining the frequencies of specific cell types and states is a primary goal of ex vivo immune system diagnostics. Mature dendritic cells (DCs) play a central role in the onset of immunity or toleranceby presenting antigens to T cells11,12. The functional phenotype of mature DCs, can be characterized by their patterns of cytokine secretion and expression of differentiation/activation cell surface markers, such as co-stimulatory molecules11,12,13,14,15,16,17,18. Population-wide studies of functional phenotype mask the dynamic behavior of individual DCs and are thus often insufficient for characterizing the immuno-regulatory potential of DCs. To address this problem, we aimed to develop a droplet microfluidic platform for single live cell phenotyping. This platform enables activation and monitoring of live single cells for secretion and cell surface markers simultaneously. In particular, we applied this technology to simultaneously stimulate DC maturation with an immunological adjuvant, lipopolysaccharide (LPS) and to monitor two phenotypic markers of DCs15,17. The first of these is cluster of differentiation 86 (CD86), an important cell surface molecule that provides co-stimulatory signals necessary for T cell activation13,16,18. We also followed the maturation phenotype of the DC by measuring secreted interleukin (IL)-6, a cytokine that plays an important role in lymphocyte activation13,14,16.
Additional critical step in the generation of antigen specific immune responses that activate naïve T cells in lymph nodes is immunological synapse (IS) formation between mature DC and T cell19,20,21,22. Though attempts were made to analyze the dynamic aspects of these cell-cell interactions in microscopic imaging, a robust technique to analyze the IS formation and cell-cell interaction has lagged. Here we were able monitoring live single cell surface markers during DC/T cell IS formation in a distinct microenvironment, a feat not possible using conventional techniques.
Results
Nano-liter droplet microfluidic-based technology for stimulation and monitoring of surface and secreted markers of live single immune DCs
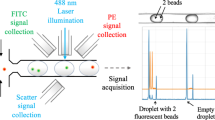
Figure 1a schematically illustrates the droplet microfluidicplatform employed by us to activate and to monitor live single cells for secretion and cell surface protein markers simultaneously. The poly(dimethylsiloxane) (PDMS)microfluidic system was fabricated using standard soft lithography methods, combining functionalities of droplet generation and an incubationchamber array of 1000 droplets (Fig. 1a, b). The generation of monodisperse droplets in a micro-channel through shearing flow at a flow-focusing zone is illustrated in Fig. 1b. In this set up, three perpendicular inlet channels form a nozzle. The individual syringe pumps were used to control flow rates of the central stream of the oil phase (1) and the left stream of DCs suspension (2) and the right stream of bioassay reagents (bead sensors and antibodies) (3) (Fig. 1a). Each generated droplet in the array contained fluorescently-labeled detection antibodies (anti-CD86-FITC Abs) generating a localized fluorescent signal on DCs expressing CD86 (Fig. 1c). In addition, each droplet contained bead sensors conjugated with capture Abs (anti-IL-6 Abs) that bind secreted IL-6 and fluorescently labeleddetection Abs (anti-IL-6-FITC Abs) that create a localized fluorescent signal on thebead surface in the droplet (Fig. 1c). The advantages of this droplets-based array technique include the physical and chemical isolation of cells in droplets eliminating the risk of cross-contamination and the fast and efficient mixing of the reagents that occurs inside droplets facilitating fast reaction rate23,24,25,26,27. More important, this nano-liter microenvironment allows delivering various stimuli and assay reagents to each cell and gas exchange to ensure viability.

(a) Illustration of the nano-liter droplet-based technology. (b) Droplet generation in the PDMS device. (c) Schematic illustration of microfluidic reaction droplet for monitoring cell surface and secretion simultaneously and the image of DC and beads co-encapsulated in droplet array.
To measure the time dependent CD86 and IL-6 expression of single DCs, the fluorescent signal generated on cell and bead surfaces was recorded each 20 min in the confined volume of droplets and analyzed by the ImageJ software (Fig. 2a). The positive signal was recorded and quantified for each cell in the droplets and compared to the background empty droplet signal (Fig. 2b). The image sequence was analyzed and the increase in intensity from each cell was plotted over time showing expression kinetics (Fig. 3a, b). After 40 minutes of incubation in LPS nano-liter droplets, the encapsulated cells started to upregulate the surface maturation marker, CD86 and reached saturation at 2 hours and 20 min (Fig. 3a). Fig. 3c describes the fluorescent signal intensity from thirty cells after 2 hours and 20 min in separate droplets, confirming the previously-described inherent CD86 marker heterogeneity11,12,13,14,15,16,17,18. Thus, this method allows us to distinguish the responses of individual cells to LPS stimulation in the confined volume of droplets. In addition to CD86, we were able to monitor LPS induced IL-6 secretion from the same encapsulated single DCs (Fig. 3b). The fluorescence signal for the detection of IL-6 on microsphere surfaces is shown in Figure 2b for single DCs. The signal was detected in droplets after 10 min in the array (Figure 3c). Droplets with LPS stimulation contained IL-6 levels that were significantly higher (three standard deviations) than droplets with no LPS, which showed negligibleIL-6 levels.

(a) DCs and bead-based sensors and reagents co-encapsulated in nano-liter reaction droplet containing anti-CD86 fluorescently tagged Abs for cell surface analysis. 1,2. Fluorescence images of droplets captured on a Zeiss 200 Axiovert microscope (a1 × 20 and a2 × 40) after 2 hours and 20 min of incubation. 3. The intensity of the fluorescence recorded for both CD86 protein and IL-6 detection measured using ImageJ software (the fluorescence intensity scale is present on the right). (b, c, d) Physical interaction and the polymerization of tubulin in live DCs that encountered a T cell and establishing signaling zone.

(a) The increase in accumulative CD86 expression over the time in response to LPS stimulation. (b) The increase in IL-6 secretion over the time in response to LPS stimulation. RS stands for relative signal. (c) Inherent CD86 marker heterogeneity.
Monitoring single cell immunological synapse (IS) formation between mature DC and T cell
Immunological synapse (IS) formation between mature DC and T cell is one of critical step in the generation of antigen specific immune responses that activate naïve T cells in lymph nodes19,20,21,22. Here we were able monitoring live single cell surface during DC/T cell IS formation in a distinct microenvironment, a feat not possible using conventional techniques. Previously described droplet microfluidic-based platform was applied to co-encapsulate pairs of cells of different types in one micro-reaction droplet (Fig. 1a). In this set up, the center stream contains oil (1), the left steam contains suspension of T cells (2) and the right steam contains DCs (3). To demonstrate the IS formation, we co encapsulated naïve T cell and LPS matured DC in nano-liter droplets of tubulin marker assay which provides green-fluorescent staining of polymerized tubulin in live cells. Fig. 2c demonstrates the physical interaction and the polymerization of tubulin in live mature DCs that encountered naïve T cell and establishing signaling zone onlive cells. We were able to monitor remodeling of the cytoskeleton and microtubule polymerization in DCs at the IS formed between co-encapsulated pairs of live DCs and CD4+ T cells in droplets.
Discussion
Altogether, our work provides a platform technology to stimulate specific cellular processes, such as DC maturation and cell-cell interactions and to measure the effects of these cellular processes, such as CD-86 and IL-6 expression, in a time dependent manner for single live cells in population. In the future, this technology could be applied to characterize functional phenotypesin various heterogeneous populations at a single cell level. Furthermore, we were able to monitor micro-size remodeling of the cytoskeleton and microtubule polymerization in DCs at the IS formed between co-encapsulated pairs of live DCs and CD4+ T cells in droplets. In addition, to demonstrate the heterogeneity in cell-cell interactions of T cell and DC populations we co encapsulate two naïve T cells and four previously LPS matured DCs in single droplets to monitor the cell-cell interaction. Figure 2d show that not all cells are interacting to create IS thus demonstrating that due to the heterogeneity in DC maturation process not all DC are able to interact and activate naive T cells. Furthermore the distinguish differences in the morphology of resting and interacting live cells are clearly can be seen in the nano-liter reaction droplets. Thus the developed approach allows to dynamically monitoring the interaction events that transpire during cell-cell communication in the immune system on a single cell level to avoid ambiguities that arise due to heterogeneous responses in cell populations.
Methods
Materials
Rat FITC-anti-CD86 antibody [GL1] was obtained from Abcam and used at concentration of 1 μg/106 cells. Biotin-anti-IL-6 Ab (504601) for bead conjugation was obtained from BioLegend and FITC-anti-IL-6 Ab (11-7061-81) for IL-6 detection was obtained from eBioscience and used at concentration of 1 μg/mL.
Microfluidic device fabrication
Microfluidic flow chambers were fabricated using soft lithography at the MGH/CEM Facility. Negative photo resist SU-8 2100 (MicroChem, Newton, MA) was deposited onto clean silicon wafers to a thickness of 150 μm and patterned by exposure to UV light through a transparency photomask (CAD/Art Services, Bandon, OR). The Sylgard 184 poly(dimethylsiloxane) (PDMS) (Dow Corning, Midland, MI) was mixed with crosslinker (ratio 10:1), poured onto the photoresist patterns, degassed thoroughly and cured for 12 hours at 75°C. Next, we peeled the PDMS devices off the wafer and bonded to glass slides after oxygen-plasma activation of both surfaces. The device is composed of a droplet forming nozzle and a storage array for 103 droplets. To improve the wetting of the channels with mineral oil in the presence (1%, w/w) of the surfactant (span80), prior to the experiments the microfluidic channels were treated with Pico-Sur™ 2 (Dolomite Microfluidics, UK) by filling the channels with the solution as received and then flushing them with air. Tygon Micro Bore PVC Tubing 100f, 0.010" ID, 0.030" OD, 0.010" Wall (Small Parts Inc, FL, USA) were connected to the channels and to the syringes. We used 1 ml syringes to load the fluids into the devices, while syringe pumps (Harvard Apparatus, USA) controlled the flow rates.
Microsphere sensors preparation
ProActive® Streptavidin Coated Microspheres (10 μm) (Bang Laboratories Inc., USA) were conjugated with the biotinylatedanti-mouse IL6 (Abcam®, MA, USA) according to the manufacturer's protocol. Unbounded active sites were blocked with StarlingBlock™ (thermo Scientific, USA) for one hour. Next, we washed the beads with the Phosphate Buffered Saline (PBS) (Sigma, USA), diluted to the final concentration 0.5 mg ml−1 and stored at 4°C.
Cell isolation and stimulation
6–8 week old female C57BL/6J mice were obtained from the Jackson Laboratories. Bone marrow dendritic cells (BMDCs) were collected from femora and tibiae and plated on non-tissue culture treated plastic dishes in RPMI medium (Gibco, Carlsbad, CA, Invitrogen, Carlsbad, CA), supplemented with 10% FBS, L-glutamine, penicillin/streptomycin, MEM non-essential amino acids, HEPES, sodium pyruvate, β-mercaptoethanol and GM-CSF (15 ng/mL; Peprotech, Rocky Hill, NJ). At day 7 floating cells were collected and used as GM-CSF derived dendritic cells. Cells were stimulated with LPS (rough, ultra-pure E. coli K12 strain LPS, 100 ng/ml; Invitrogen, San Diego, CA).
Image analysis
Fluorescence images of droplets were captured on a Zeiss 200 Axiovert microscope using an AxioCAM MRm digital camera. For the cell studies, fluorescence signals were captured separately using appropriate filter sets for FITC. Image processing and analysis was conducted using ImageJ software.
References
Bordbar, A. et al. Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol. Syst. Biol. 8, 1–12 (2012).
Katayama, H. et al. Phenotype and functional identity of GM-CSF-independent dendritic cells generated by long-term propagation of DC progenitor cells in bone marrow cells and skin langerhans cells. Transplant. Proc. 37, 17–19 (2005).
Nielsen, J. & Oliver, S. The next wave in metabolome analysis. Trends Biotechnol 23, 544–546 (2005).
Sul, J.-Y. et al. Transcriptome transfer produces a predictable cellular phenotype. Proc. Natl. Acad. Sci. USA (2009).
Fritzsch, F. S. O., Dusny, C., Frick, O. & Schmid, A. Single-Cell Analysis in Biotechnology, Systems Biology and Biocatalysis. Annu. Rev. Chem. Biomol. Eng. 3, 129–155 (2012).
Ghim, C.-M. & Almaas, E. Genetic noise control via protein oligomerization. BMC Syst. Biol. 2, 94 (2008).
Kuang, Y., Biran, I. & Walt, D. R. Simultaneously Monitoring Gene Expression Kinetics and Genetic Noise in Single Cells by Optical Well Arrays. Anal. Chem. 76, 6282–6286 (2004).
Han, Q. et al. Polyfunctional responses by human T cells result from sequential release of cytokines. Proc Natl Acad Sci U S A 109, 1607–1612 (2012).
Lu, Y. et al. High-throughput secretomic analysis of single cells to assess functional cellular heterogeneity. Anal Chem 85, 2548–2556 (2013).
Varadarajan, N. et al. A high-throughput single-cell analysis of human CD8(+) T cell functions reveals discordance for cytokine secretion and cytolysis. J Clin Invest 121, 4322–4331 (2010).
Willmann, K. & Dunne, J. F. A flow cytometric immune function assay for human peripheral blood dendritic cells. J. Leukocyte Biol. 67, 536–544 (2000).
Xu, Y., Zhan, Y., Lew, A. M., Naik, S. H. & Kershaw, M. H. Differential Development of Murine Dendritic Cells by GM-CSF versus Flt3 Ligand Has Implications for Inflammation and Trafficking. J Immunol 179, 7577–7584 (2007).
Decker, P., Kotter, I., Klein, R., Berner, B. & Rammensee, H.-G. Monocyte-derived dendritic cells over-express CD86 in patients with systemic lupus erythematosus. Rheumatology 45, 1087–1095 (2006).
Geisel, J. et al. IL-6 and Maturation Govern TLR2 and TLR4 Induced TLR Agonist Tolerance and Cross-Tolerance in Dendritic Cells. The Journal of Immunology 179, 5811–5818 (2007).
Granucci, F. et al. Early events in dendritic cell maturation induced by LPS. Microbes Infect. 1, 1079–1084 (1999).
Hegde, S., Pahne, J. & Smola-Hess, S. Novel immunosuppressive properties of interleukin-6 in dendritic cells: inhibition of NF-kappaB binding activity and CCR7 expression. The FASEB Journal (2004).
Jiang, H.-R. et al. Secretion of interleukin-10 or interleukin-12 by LPS-activated dendritic cells is critically dependent on time of stimulus relative to initiation of purified DC culture. J. Leukocyte Biol. 72, 978–985 (2002).
Park, S.-J. et al. IL-6 Regulates In Vivo Dendritic Cell Differentiation through STAT3 Activation. J Immunol 173, 3844–3854 (2004).
Chemin, K. et al. Cytokine secretion by CD4+ T cells at the immunological synapse requires Cdc42-dependent local actin remodeling but not microtubule organizing center polarity. J Immunol 189, 2159–2168 (2012).
Duchez, S., Rodrigues, M., Bertrand, F. & Valitutti, S. Reciprocal polarization of T and B cells at the immunological synapse. J Immunol 187, 4571–4580 (2011).
Einav, S. et al. Discovery of a hepatitis C target and its pharmacological inhibitors by microfluidic affinity analysis. Nat Biotechnol 26, 1019–1027 (2008).
Gunzer, M. et al. Antigen presentation in extracellular matrix: interactions of T cells with dendritic cells are dynamic, short lived and sequential. Immunity 13, 323–332 (2000).
Agresti, J. J. et al. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc. Natl. Acad. Sci. USA 107, 4004–4009 (2010).
Kiss, M. M. et al. High-Throughput Quantitative Polymerase Chain Reaction in Picoliter Droplets. Anal Chem. 80, 8975–8981 (2008).
Konry, T., Dominguez-Villar, M., Baecher-Allan, C., Hafler, D. A. & Yarmush, M. L. Droplet-based microfluidic platforms for single T cell secretion analysis of IL-10 cytokine. Biosens. Bioelectron. 26, 2707–2710 (2011).
Konry, T., Smolina, I., Yarmush, J. M., Irimia, D. & Yarmush, M. L. Ultrasensitive Detection of Low-Abundance Surface-Marker Protein Using Isothermal Rolling Circle Amplification in a Microfluidic Nanoliter Platform. Small 7, 395–400 (2011).
Koster, S. et al. Drop-based microfluidic devices for encapsulation of single cells. Lab Chip 8, 1110–1115 (2008).
Acknowledgements
AG acknowledges MGH Fund for Medical Discovery for the ECOR postdoctoral fellowship award. TK acknowledges NIH/NCI funds R21 [RM11-014] Exceptional Innovative Tools and Technologies for Single Cell Analysis. We acknowledge Nir Hacohen, Arnon Arazi, Raktima Raychowdhury from Broad Institute of MIT and Harvard for cells in this study.
Author information
Authors and Affiliations
Contributions
T.K. supervised the project, designed the experiment, conducted experiments, analysed data and wrote the manuscript, A.G. conducted experiments, analysed data and drafted the manuscript, M.Y. reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Konry, T., Golberg, A. & Yarmush, M. Live single cell functional phenotyping in droplet nano-liter reactors. Sci Rep 3, 3179 (2013). https://doi.org/10.1038/srep03179
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03179
This article is cited by
-
High-throughput spatiotemporal monitoring of single-cell secretions via plasmonic microwell arrays
Nature Biomedical Engineering (2023)
-
Single platelet variability governs population sensitivity and initiates intrinsic heterotypic responses
Communications Biology (2020)
-
Quantifying the efficacy of checkpoint inhibitors on CD8+ cytotoxic T cells for immunotherapeutic applications via single-cell interaction
Cell Death & Disease (2020)
-
Drop breakup in a symmetric T-junction microchannel under electric field
Microfluidics and Nanofluidics (2020)
-
Integrated dual-tomography for refractive index analysis of free-floating single living cell with isotropic superresolution
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
