
Abstract
Aedes (Ae.) aegypti is the primary vector for dengue viruses (serotypes1–4) and chikungunya virus. Homing endonucleases (HEs) are ancient selfish elements that catalyze double-stranded DNA breaks (DSB) in a highly specific manner. In this report, we show that the HEs Y2-I-AniI, I-CreI and I-SceI are all capable of catalyzing the excision of genomic segments from the Ae. aegypti genome in a heritable manner. Y2-I-AniI demonstrated the highest efficiency at two independent genomic targets, with 20–40% of Y2-I-AniI-treated individuals producing offspring that had lost the target transgene. HE-induced DSBs were found to be repaired via the single-strand annealing (SSA) and non-homologous end-joining (NHEJ) pathways in a manner dependent on the availability of direct repeat sequences in the transgene. These results support the development of HE-based gene editing and gene drive strategies in Ae. aegypti and confirm the utility of HEs in the manipulation and modification of transgenes in this important vector.
Similar content being viewed by others


Introduction
Aedes (Ae.) aegypti is the most important vector of arboviruses worldwide, due to its central role in the transmission of dengue viruses, yellow fever virus and chikungunya virus to human hosts1. To augment current control efforts such as source reduction and insecticide application, considerable effort has been put into the development of genetics-based strategies such as population replacement and reduction. Of these, population reduction programs using genetically sterilized mosquitoes have showed great promise2,3. In contrast, population replacement strategies have been limited to the non-transgenic introduction of beneficial Wolbachia endosymbionts4. One of the primary limitations in this regard has been the lack of experimentally validated gene drive mechanisms for Ae. aegypti, despite some dramatic success stories in both Drosophila5,6,7 and Anopheles (An.) gambiae8.
Homing endonucleases (HEs), zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), the so-called meganucleases, are able to recognize and cleave rare occurring (15–30 bp) double-stranded DNA sequences, allowing precise editing of large, complex genomes (reviewed in9,10). Whereas HEs are naturally occurring selfish elements, both ZFNs and TALENs are artificial hybrids of a tailored DNA binding domain and a non-specific nuclease domain. HEs can be divided into four distinct families based on their structure and mechanism of DNA binding and restriction (reviewed in10). Of these, the LAGLIDADG-type HEs (LHEs) such as I-SceI, I-CreI and I-AniI are by far the best characterized in terms of structural information, biochemical redesign and sequence diversity.
In most organisms, the repair of double-stranded DNA breaks (DSB) occurs through either non-homologous end-joining (NHEJ) or one of several forms of homologous recombination, such as single-strand annealing (SSA), synthesis-dependent strand invasion or gene conversion (reviewed in11). These pathways are often in competition with each other, with the choice of repair mechanism influenced by the cell cycle stage12, developmental stage5 and the presence and proximity of homologous sequences13,14,15,16,17. NHEJ-based repair may conservatively restore the parent sequence, but often results in the insertion or deletion of base pairs around the break site. SSA uses homology between two direct repeat sequences that flank the DSB to guide the repair process, with the result being a loss of all genetic information located between the repeats. In contrast, gene conversion-based repair using the sister chromatid or homologous chromosome is the most conservative, typically restoring the damaged region without error. Where repair during meiosis uses the homologous chromosome, the result is a loss of heterozygosity. Though possibly undesirable from the host organism's perspective, from the perspective of a selfish element this loss of heterozygosity represents super-Mendelian inheritance of the copied sequence.
Based on their natural tendencies to invade populations through DSB induction followed by gene conversion-based repair, HEs have been suggested as a potential gene drive system for vector-borne disease control18,19. Current models suggest that significant impacts on public health could be observed following the release of just a few HEs in relatively short timeframe (2–3 years20). Most excitingly, several groups have reported the successful establishment of an LHE-based gene drive system in both D. melanogaster5,7 and An. gambiae8 using I-SceI. An unrelated HE, I-PpoI, has been shown to induce shredding of ribosomal DNA in An. gambiae, leading to extensive sterility21 and population crashes in large cage trials22.
We have previously shown that the HEs I-SceI, I-CreI, I-AniI and I-PpoI can induce DSBs in the Ae. aegypti soma when appropriate target sites are present in the mosquito genome and that I-PpoI targets the Ae. aegypti rDNA repeats23. In this report we show that I-CreI, I-SceI and Y2-I-AniI can induce DSBs at their specific target sites in the Ae. aegypti germline using two independent transgenic strains bearing HE target sites. Of these, the efficiency of transgene excision was substantially higher for Y2-I-AniI in both cases, with 20–40% of injected survivors giving rise to progeny that had lost the target transgene. Both NHEJ- and SSA-type repair were observed, with the choice of repair associated with the presence of direct repeats in the transgene sequence. We conclude that these LHEs can edit the Ae. aegypti genome at useful frequencies and are suitable scaffolds for targeted redesign efforts and the development of HE-based gene drive systems.
Results
Development of a plasmid-based assay for HE function in Ae. aegypti embryos
Initially, we designed a series of HE-expression constructs based on the D. pseudoobscura hsp82 promoter24, used previously to successfully drive the expression of Mos1 transposase in Ae. aegypti embryos25. With the exception of I-PpoI, where significant mortality was observed, none of the other homing endonucleases tested had detectable activity (defined as excision of the EGFP reporter gene) in this system (data not shown). Therefore, we sought to identify alternative promoters with higher activity in the early embryo. Using a dual luciferase reporter assay, we examined the ability of three additional promoters, the baculovirus IE1 promoter and Ae. aegypti polyubiquitin (PUb) and UbL40promoters26, to drive gene expression at various times following injection into pre-blastoderm embryos. At early (2–4 hr); mid (12–24 hr) and late (48–72 hr) timepoints, expression from the PUb promoter significantly exceeded expression from the each of the other promoters (P < 0.05 Mann-Whitney test, Bonferroni correction based on 15 tests; Fig. 1). Maximum expression from the PUb promoter was achieved just 2–4 hr after injection, while with the IE-1 and UbL40 promoters expression did not peak until 12 hr. Therefore we placed each HE ORF downstream of the Ae. aegypti PUb promoter to analyze embryonic and germline endonuclease activity.

Transcriptional activity of IE1, UbL40, PUb and hsp82 promoters in Ae. aegypti embryos.
The ratio of Firefly (FL) to Renilla (RL) luciferase activity for each experimental promoter was compared with no-promoter control plasmid (pGL3) following injection into Ae. aegypti embryos. Error bars indicate the standard deviation amongst 3-6 biological replicates, with each replicate consisting of approximately 100 injected embryos.
To test the ability of HEs to introduce double-stranded DNA breaks in early stage mosquito embryos, we co-injected each PUb-HE expression construct with a single-stranded annealing (SSA)-dependent luciferase reporter, wherein the first 300 bp of the firefly luciferase ORF was duplicated (Fig. 2A). A series of stop codons, along with the recognition sequence for each HE, was placed in the intervening spacer region. In all three cases (Y2-I-AniI, I-SceI, I-CreI), injection of a PUb-HE expression construct resulted in a significant increase in firefly luciferase expression compared to embryos injected with a non-specific control construct, PUb-EGFP (Fig. 2B). However, expression of Y2-I-AniI induced significantly more luciferase expression than either I-SceI or I-CreI, which did not differ from each other. We conclude that all three HEs are active in the early embryos of Ae. aegypti and thus may be capable of catalyzing DSBs in the germline of this mosquito.

Embryo-based single-strand annealing (SSA) assay.
(A) Schematic representation of the SSA test construct injected into Ae. aegypti embryos along with the specified PUb-HE expression vector; successful cleavage of the HE target site followed by SSA-based repair restores the FF-luc ORF. (B) Ratio of Firefly (FL) to Renilla (RL) luciferase (24 hr) following injection into pre-blastoderm embryos. Error bars indicate the standard deviation; each point represents a group of ~100 injected embryos. Statistical significance between pairs was determined using the Mann-Whitney test; ** indicates P < 0.01.
Germline excision by homing endonucleases in line PUb-EGFP #P5
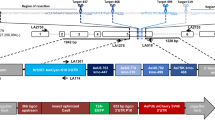
To determine whether HEs were able to catalyze the excision of Ae. aegypti genomic segments in a heritable manner, HE expression constructs were injected individually into the embryos of a transgenic strain (P5) containing a PUb-EGFP cassette flanked by the recognition sequences for each HE26, as well as a 3xP3-DsRED cassette (Fig. 3A, B). Surviving individuals were pooled, mated to an unmarked strain (khw) and offered a bloodmeal; progeny were screened as larvae for the presence/absence of each fluorescent marker (Table 1). Progeny that had lost expression of the EGFP marker were identified at varying frequencies for HEs I-CreI, I-SceI and Y2-I-AniI (Table 1). Progeny that had lost EGFP were detected in all four Y2-I-AniI pools and comprised 2–4% of the total progeny. Excision of EGFP by I-SceI (4/8 pools) and I-CreI (1/6 pools) appeared to be less efficient. No evidence of transgene excision was seen in the few survivors of I-PpoI injection, or in the absence of HE.

HE-catalyzed germline excision in Aedes aegypti transgenic line PUb-EGFP #P5.
(A) Schematic representation of the parental transgene insertion. The dual transgene construct contained two HE clusters flanking the PUb-EGFP cassette; the order of HE sites differed between the upstream (blue) and downstream (red) clusters. Connectors indicate the boundaries of excised sequence by Y2-I-AniI or I-SceI. The initial repair mechanism is indicated (NHEJ, black connector; SSA, gray connector), along with the HE used, the number of sequences obtained (n) and with the minimum number of independent occurences (shown in parentheses). (B) Mosquito larvae photographed using a DsRED (top) or EGFP (bottom) filter. DsRED/EGFP positive (R+ or G+) and negative (R− or G−) individuals are indicated; fluorescent signal in the eyes is indicated by the arrowhead. (C) Sequences obtained corresponding to NHEJ events following injection of Y2-I-AniI or I-SceI compared to a hypothetical (Hyp) sequence whereby the two I-AniI sites are cut and joined together perfectly. (D) Sequences obtained corresponding to SSA-based repair events compared to a hypothetical (Hyp) sequence where the two SV40 direct repeats are collapsed. (E) Large deletion of the 3xP3-DsRED-SV40 cassette. For C-E, numbers indicate the number of sequences obtained, with the minimum number of unique occurrences in parentheses.
Sequencing the remaining transgene sequences in the progeny that had lost EGFP confirmed complete loss of the PUb-EGFP gene cassette and revealed that both NHEJ and SSA were used as repair mechanisms to different extents (Fig. 3). For Y2-I-AniI, we obtained 47 sequences corresponding to at least nine independent events that appeared to result from NHEJ following Y2-I-AniI-induced DSBs at both the upstream and downstream recognition sites (Fig. 3A, C). Of these, 14 sequences (3 events) corresponded with perfect repair, restoring a single I-Ani recognition site. The remaining events were characterized by small deletions or insertions (Fig. 3C). In these instances, independent events were defined as having a distinct modified transgene sequence, or as being derived from independent G0 pools. The introduction of a single DSB could also be followed by SSA-based repair, resulting in the collapse of direct repeat sequences. In line P5, the largest direct repeat sequence consisted of the SV40 3'UTR (240 bp), which followed each of the EGFP and DsRED ORFs. All progeny having lost EGFP from the I-SceI experiment were found to result from the collapse of these two repeats (Fig. 3D). SSA-based collapse of SV40 repeats also was commonly observed for Y2-I-AniI. However, in this case, most of the sequenced mosquitoes contained small deletions or insertions at the remaining I-AniI site. We interpret this as the result of SSA-based repair, followed by further cleavage by Y2-I-AniI and subsequent NHEJ-based repair (Fig. 3D). While most SSA-repair utilized the SV40 repeats, we recovered at least four instances where other HE recognition sites were used in the SSA response (while the order of these sites is scrambled in each cluster, they remain organized as direct repeats). Thus, even sequences as short as 18–20 bp can direct the SSA repair response.
While almost all of the progeny recovered displaying a phenotypic change were found to be DsRED+/EGFP−, a few instances in which a DsRED marker was lost were identified. Of these, one (a single I-CreI larvae) died early in development and we were unable to determine the nature of the excised region. However, a large deletion (and small insertion) spanning the entire 3xP3-DsRED gene cassette was identified (Fig. 3E) in two individuals derived from the I-SceI experiment. As there were no sequences homologous with the transgenic construct at these locations, we classified these as NHEJ events.
Germline excision by homing endonucleases in line 3xP3-RG #P11A
Local chromosomal structure may influence the accessibility of HEs to their target sites and thus may affect the rate of DSB formation. Therefore, we repeated our experiments using a second recipient line (P11A), containing another double-marked transgene (Fig. 4A, B). Inverse PCR revealed that the P11A insertion mapped to a different genomic scaffold than line P5, though both were incorporated into the large intronic regions of protein-coding genes (Table S1). Progeny found to have lost the expression of at least one of the two markers were recovered following injection with Y2-I-AniI and I-CreI, but not with I-SceI or I-PpoI (Table 2). Interestingly, of those showing a loss of marker gene expression, injection of I-CreI resulted in mostly G+/R− progeny, while Y2-I-AniI produced mostly G−/R+ progeny. A class of progeny that had lost both marker genes was recovered only following Y2-I-AniI injection. Once again, survival of I-PpoI mosquitoes was extremely low.

HE-catalyzed germline excision in Aedes aegypti transgenic line 3xP3-RG #P11A.
(A) Transgenic construct for line P11A. Connectors indicate NHEJ (black) or SSA (gray) based repair following deletion of the intervening segment. (B) Mosquito larvae from line P11A photographed using a DsRED (top) or EGFP (bottom) filter. DsRED/EGFP positive (R+ or G+) and negative (R− or G−) individuals are indicated; fluorescent signal in the eyes is indicated by the arrowhead. Sequences obtained following NHEJ (C) or SSA (D) -based repair. For each, the number of sequenced mosquitoes (n) along with the minumum number of unique occurrences (in parentheses) are indicated.
Consistent with previous data from line P5, we observed both NHEJ (Fig. 4C) and SSA type repair (Fig. 4D) resulting in the loss of transgene segments following injection with Y2-I-AniI. Once again, when SSA-directed repair was used, most sequenced individuals contained small deletions at the remaining I-AniI recognition site, indicating additional cutting by Y2-I-AniI following the initial repair event. For Y2-I-AniI, the SSA pathway primarily utilized the SV40 direct repeats (7 events) flanking the upstream I-AniI site to direct the repair process. Two additional SSA repair events were found based on the loxP and I-SceI repeats present in the transgene construct. In contrast, all I-CreI-mediated events were found to be the result of SSA-repair resulting in the collapse of the ~260 bp 3xP3 promoter (3 events) or the 35 bp loxP sites (3 events), with no evidence for NHEJ (Fig. 4A). Attempts to identify the genetic basis for the R−/G− phenotypes seen in the Y2-I-AniI experiment were unsuccessful, as it appeared that the entire transposon construct and an unknown quantity of the surrounding chromosomal DNA had been lost. As the G1 individuals were only hemizygous for the transgene insertion, PCR using primers outside of the transgene sequence was confounded by the presence of the alternate, wild-type allele. The lack of such R−/G− individuals from the other three experiments argues against a more mundane explanation such as incomplete homozygosity in the parental strain with respect to the transgene insertion (all injections were performed from a single parental cage) and suggests HEs may also trigger larger scale deletions of chromosomal segments.
For both the P5 and P11A experiments, we calculated the minimum HE excision frequency, defined as the number of independent excision events per fertile G0 individual (Table 3). Y2-I-AniI proved the most effective in both genetic backgrounds, with 20–40% of fertile G0 individuals producing at least one offspring bearing an excision event (Table 3). While both I-SceI and I-CreI were also able to catalyze transgene excision, they did so at a reduced rate and in a manner that appeared to be more dependent on the transgene structure and/or insertion site.
Discussion
Two different transgenic lines were used to determine the ability of four HEs to perform site-specific excision of gene segments in the Ae. aegypti germline. Of these, Y2-I-AniI gave the most useful frequencies of transgene excision, followed by both I-CreI and I-SceI. Both of the target transgenes in our study were found to have integrated into the large intronic space of a gene. While HE-based editing at both of these locations was highly effective, additional target sites within tightly wound heterochromatic space would be required to determine if such local structure poses a substantial barrier to HE-based targeting. However, for most downstream applications HE-based gene targeting efforts are likely to focus on euchromatic regions.
Embryonic survival following I-PpoI injection was substantially lower (~1%) than that of the other HEs used in this study. This was not unexpected, given that I-PpoI has a substantial number of recognition sites within the 28S rDNA repeats of Ae. aegypti23 and has been shown to shred the rDNA of An. gambiae leading to early embryonic lethality21,22,27. We reasoned that the few survivors from these injections may still produce excision events due to receiving a sub-lethal dose, but this was not observed. This may be due to the fact that I-PpoI sites within the rDNA (n > 300) vastly outnumber those in the transgene (n = 2), making it impossible to titrate to a level where rDNA shredding was avoided yet recoverable cleavage events at the test locus could be obtained. Embryonic survival following injection with Y2-I-AniI, I-CreI and I-SceI ranged from 10–17%, similar to what we typically observe following injection of transposon-based DNA constructs26,28,29, suggesting that these HEs recognize few (if any) cryptic off-target sites in the Ae. aegypti genome.
Sequencing of the transgene following HE-induced DSBs revealed the footprint of both NHEJ and SSA-based repair processes. We note that gene conversion-based repair would result in the restoration of the lost transgene sequence and would be phenotypically invisible in our assay; thus all excision frequencies we report likely underestimate the magnitude of HE-induced DSB formation. The method of DNA repair we observed varied based on both the HE and the structure of the target transgene. It is well-established that these two repair methods are in competition and that the presence and proximity of direct repeats can influence the decision for repair to proceed via NHEJ or SSA (reviewed in11). Both transgenes contained a number of direct repeats that varied in length from 18–260 bp and a single DSB introduced at either cluster would leave at least one viable SSA-based repair option. For example, following I-SceI injection into line P5, we only observed SSA-based repair at the SV40 direct repeats. This suggests that only a single DSB occurred in the upstream cluster (a similar break at the downstream cluster would leave both SV40 repeats upstream of the break). Simultaneous induction of DSBs at both the upstream and downstream clusters would eliminate one of the repeats and thus might favor NHEJ as observed for Y2-I-AniI. Similarly, all I-CreI induced excision events were associated with SSA repair centered on the 3xP3 or loxP repeats, consistent with the induction of a single DSB at either the upstream or downstream clusters, respectively. Interestingly, I-CreI induced DSBs in the upstream cluster of the P11A transgene were only associated with collapse of the 3xP3 repeats, whereas this was not observed for Y2-I-AniI, whose action instead led primarily to the choice of SV40 repeats (P < 0.01, Fisher's Exact Test). One possible explanation is that the I-CreI recognition site in the upstream cluster was immediately adjacent to the second 3xP3 repeat. Thus the speed of resection and proximity of each repeat to the end of the DSB may dictate the ultimate choice of homologous sequence used for repair13,14. We note that the type of repair can also be strongly influenced by the developmental stage when DSB induction occurs5,7 and thus the expression of these same HEs at later points in development (or in the soma) might alter the chosen method of repair. Further exploration of this using the HE-based system we have developed would be an interesting platform to explore the mechanisms of DSB repair in Ae. aegypti, whose genome is rich in short repetitive elements.
The LHEs Y2-I-AniI, I-CreI and I-SceI have all been successfully redesigned through a variety of approaches to recognize new target sequences8,30,31,32,33,34,35,36. In particular, Y2-I-AniI and I-CreI were successfully altered to recognize sequences in the An. gambiae genome8. Our data indicate that the Y2-I-AniI scaffold (or its close relatives37) may be the best suited for targeted redesign experiments involving precise gene editing in Ae. aegypti. Extensive sequencing and structural analysis has revealed many more active LHE members38, providing additional scaffolds as potential starting material for redesign efforts. While testing a large number of variant LHEs or a large pool of newly described scaffolds in germline-based experiments is likely not feasible, we note that data obtained from our transient SSA assay were highly predictive of success in the more time-consuming germline experiments. Thus, we anticipate that candidate HEs based on the Y2-I-AniI or other scaffolds validated in simple yeast-based assays39 can subsequently be tested for the potential to edit the mosquito genome in a more medium to high-throughput manner (1–3 test constructs per day) compared to what is possible with germline-based experiments (2–3 months per test construct).
The recent success of TALENs in editing a wide range of genomic targets at high efficiency, combined with the almost complete modularity of TALE sequence recognition is already leading to their adoption over other technologies such as ZFNs and LHEs9,40. However, there are clear situations where LHEs remain the preferred choice due to their small size and extreme target specificity10. One such application may be the development of genetics-based control strategies for vector-borne disease agents such as dengue viruses. Such strategies depend on the ability to achieve super-Mendelian inheritance of one or more transgene sequences41. This gene drive may be coupled to an anti-pathogen gene(s), resulting in the conversion of a competent vector population to an incompetent one, or used alone to inactivate one or more essential genes, resulting in a population crash20. Effective laboratory-based gene drive systems using maternal lethality/embryonic rescue6 and LHEs5,7 have been successfully demonstrated in model systems such as Drosophila and in the malaria mosquito8, yet this has not been achieved in Ae. aegypti. We conclude that HEs such as Y2-I-AniI (or its variants) should be considered as strong candidates for evaluation in gene drive experiments in this important disease vector species.
Methods
Plasmid construction and Luciferase assays
Plasmids pGL3-IE1, pGL3-UbL40 and pGL3-PUb were described previously26. To generate pGL3-hsp82, a 992-bp region containing the Drosophila pseudoobscura hsp82 promoter was amplified by polymerase chain reaction (PCR) from pKhsp8224 using the primers 5′-TTTTCCAGTTGTTTTAATTTAACAGCAGAG-3′ and 5′-TTTTAAGCTTATGGATTTTTACCATATTATTA-3′. All PCR reactions were performed using Platinum Pfx (Invitrogen, Carlsbad, CA) as follows: 94°C for 2 mins, 94°C for 15 s, 65°C for 30 s, 68°C for 70 s, 35 cycles, 68°C for 10 mins. Amplicons were digested with HindIII and NcoI (NEB, Ipswich, MA), purified and ligated into the corresponding sites of pGL3-Basic (Promega, Madison, WI). The normalization control plasmid pRL-CMV-Renilla was purchased from Promega.
The PUb-HE (I-CreI, I-SceI and I-PpoI) expression vectors were generated using pKhsp82-HE plasmids23 as a template and primers 5′-ttttccatggTTAAATTAAAACACGGATCCATGC-3′ and R 5′-ttttgcggccgcGATCTTGATCTTCATGGTCGACG-3 in order to add NcoI and NotI restriction sites (underlined bases). Plasmid pSLfa-PUb-EGFP-SV4026 was digested with NcoI and NotI to remove the EGFP coding region; all three homing endonuclease genes were ligated into these sites to generate pSLfa/PUb-I-CreI, pSLfa/PUb-I-SceI, pSLfa/PUb-I-AniI and pSLfa/PUb-I-PpoI. pSLfa/PUb-Y2-I-AniI was produced using the QuikChange II-E Site-Directed Mutagenesis Kit (Stratagene) to introduce the F13Y and S111Y mutations described by Takeuchi et al42.
For luciferase experiments, the experimental plasmid (pGL3-PUb, pGL3-UbL40, pGL3-IE1, pGL3-hsp82 or pGL3 Basic) and normalization control plasmid (pRL-CMV) were co-injected at 0.3 μg/μl each into Aedes aegypti Liverpool strain embryos (G0). Three replicates of 100-120 embryos each were injected with each promoter construct. Injected embryos were snap-frozen in liquid nitrogen at 2, 4, 12, 24, 48 and 72 hours post injection. Embryos were homogenized in the lysis buffer provided by the manufacturer (Promega, Madison, WI). Activity of both Firefly luciferase (FF-luc) and Renilla luciferase (R-luc) were determined by using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI) with a GloMax 20/20 instrument according to the manufacturer's protocol.
Mosquito strains and microinjections
Ae. aegypti Liverpool, khW, PUb-EGFP line #P526 and transgenic line 3xP3-RG #P11A were maintained as previously described28. For embryonic microinjections, 0.3 μg/μl of pSLfa/PUb-Y2-I-AniI, pSLfa/PUb-I-SceI, pSLfa/PUb-I-CreI or pSLfa/PUb-I-PpoI in 1× injection buffer25 was injected separately into 1 hour old embryos of transgenic lines PUb-EGFP line #P5 or 3xP #-RG P11A. Eggs were hatched 5 days post injection and surviving G0 females were merged into pools of 20–25 individuals and mated to khw strain males. G0 males were mated individually to 5 khw females for 2–3 days and pooled prior to blood feeding and egg collection. G1 larvae were screened using a fluorescent Leica MZ16F microscope for presence/absence of marker genes (DsRed or EGFP).
Analysis of DSB repair events
To identify cleavage events in the transgenes from PUb-EGFP line #P5, primers 5′-CGCCACCACCTGTTCCTGTA-3′ and 5′-CTCTCAGTGCAGTCAACATGTCGAG-3′ were used to amplify the target region from genomic DNA for DsRED positive, EGFP negative individuals (G−R+). PCR conditions were: 94°C for 2 mins, 94°C for 30 s, 60°C for 30 s, 68°C for 3.5 min, 35 cycles, 68°C for 10 min using Platinum Pfx (Invitrogen, Carlsbad, CA). For G1 individuals scored as EGFP positive, DsRED negative (G+R−), primers 5′-CTTACTTCCAACTGCTCTGCGA-3′ and 5′-CTTTGTTCACTCTGAAATTTTCTCCTCTGGC-3′ were used with the same amplification conditions. To identify cleavage events in line 3xP3-RG #P11A, primers 5′-AAGTGGTGATTTTGACGTCGACGAGATCGG-3′ and 5′-TACCACCAAGCTGTCAGTTCCAAC-3′ (G−R+; 94°C for 2 min, 94°C for 30 s, 67°C for 30 s, 68°C for 2 min, 35 cycles, 68°C for 10 min) or 5′-TTGCCGGTGGTGCAGATGAACTTCAGG-3′ and 5′-CTTACTTCCAACTGCTCTGCGA-3′ (G+R−; 94°C for 2 min, 94°C for 30 s, 60°C for 30 s, 68°C for 2 min, 35 cycles, 68°C for 10 min) were used. All amplicons were gel purified and sequenced directly.
References
Halstead, S. B. Dengue. Lancet 370, 1644–1652 (2007).
Harris, A. F. et al. Successful suppression of a field mosquito population by sustained release of engineered male mosquitoes. Nat Biotechnol 30, 828–830 (2012).
Harris, A. F. et al. Field performance of engineered male mosquitoes. Nat Biotechnol 29, 1034–1037 (2011).
Hoffmann, A. A. et al. Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature 476, 454–457 (2011).
Chan, Y. S., Naujoks, D. A., Huen, D. S. & Russell, S. Insect population control by homing endonuclease-based gene drive: an evaluation in Drosophila melanogaster. Genetics 188, 33–44 (2011).
Chen, C. H. et al. A synthetic maternal-effect selfish genetic element drives population replacement in Drosophila. Science 316, 597–600 (2007).
Chan, Y. S., Huen, D. S., Glauert, R., Whiteway, E. & Russell, S. Optimising homing Endonuclease Gene Drive Performance in a Semi-Refractory Species: The Drosophila melanogaster Experience. PLoS One 8, e54130 (2013).
Windbichler, N. et al. A synthetic homing endonuclease-based gene drive system in the human malaria mosquito. Nature 473, 212–215 (2011).
Joung, J. K. & Sander, J. D. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol 14, 49–55 (2012).
Stoddard, B. L. Homing endonucleases: from microbial genetic invaders to reagents for targeted DNA modification. Structure 19, 7–15 (2011).
Huertas, P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol 17, 11–16 (2010).
Longhese, M. P., Bonetti, D., Manfrini, N. & Clerici, M. Mechanisms and regulation of DNA end resection. EMBO J 29, 2864–2874 (2010).
Agmon, N., Pur, S., Liefshitz, B. & Kupiec, M. Analysis of repair mechanism choice during homologous recombination. Nucleic Acids Res 37, 5081–5092 (2009).
Chung, W. H., Zhu, Z., Papusha, A., Malkova, A. & Ira, G. Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet 6, e1000948 (2010).
Kappeler, M., Kranz, E., Woolcock, K., Georgiev, O. & Schaffner, W. Drosophila bloom helicase maintains genome integrity by inhibiting recombination between divergent DNA sequences. Nucleic Acids Res 36, 6907–6917 (2008).
Mansour, W. Y. et al. Hierarchy of nonhomologous end-joining, single-strand annealing and gene conversion at site-directed DNA double-strand breaks. Nucleic Acids Res 36, 4088–4098 (2008).
Preston, C. R., Engels, W. & Flores, C. Efficient repair of DNA breaks in Drosophila: evidence for single-strand annealing and competition with other repair pathways. Genetics 161, 711–720 (2002).
Burt, A. Site-specific selfish genes as tools for the control and genetic engineering of natural populations. Proc R Soc Lond B Biol Sci 270, 921–928 (2003).
Burt, A. & Koufopanou, V. Homing endonuclease genes: the rise and fall and rise again of a selfish element. Curr Opin Genet Dev 14, 609–615 (2004).
Deredec, A., Godfray, H. C. & Burt, A. Requirements for effective malaria control with homing endonuclease genes. Proc Natl Acad Sci U S A 108, E874–880 (2011).
Windbichler, N., Papathanos, P. A. & Crisanti, A. Targeting the X chromosome during spermatogenesis induces Y chromosome transmission ratio distortion and early dominant embryo lethality in Anopheles gambiae. PLoS Genet 4, e1000291 (2008).
Klein, T. A., Windbichler, N., Deredec, A., Burt, A. & Benedict, M. Q. Infertility resulting from transgenic I-PpoI male Anopheles gambiae in large cage trials. Pathog Glob Health 106, 20–31 (2012).
Traver, B. E., Anderson, M. A. & Adelman, Z. N. Homing endonucleases catalyze double-stranded DNA breaks and somatic transgene excision in Aedes aegypti. Insect Mol Biol 18, 623–633 (2009).
Coates, C. J., Howells, A. J., O'Brochta, D. A. & Atkinson, P. W. The 5' regulatory region from the Drosophila pseudoobscura hsp82 gene results in a high level of reporter gene expression in Lucilia cuprina embryos. Gene 175, 199–201 (1996).
Coates, C. J., Jasinskiene, N., Miyashiro, L. & James, A. A. Mariner transposition and transformation of the yellow fever mosquito, Aedes aegypti. Proc Natl Acad Sci U S A 95, 3748–3751 (1998).
Anderson, M. A., Gross, T. L., Myles, K. M. & Adelman, Z. N. Validation of novel promoter sequences derived from two endogenous ubiquitin genes in transgenic Aedes aegypti. Insect Mol Biol 19, 441–449 (2010).
Windbichler, N. et al. Homing endonuclease mediated gene targeting in Anopheles gambiae cells and embryos. Nucleic Acids Res 35, 5922–5933 (2007).
Adelman, Z. N., Anderson, M. A., Morazzani, E. M. & Myles, K. M. A transgenic sensor strain for monitoring the RNAi pathway in the yellow fever mosquito, Aedes aegypti. Insect Biochem Mol Biol 38, 705–713 (2008).
Carpenetti, T. L., Aryan, A., Myles, K. M. & Adelman, Z. N. Robust heat-inducible gene expression by two endogenous hsp70-derived promoters in transgenic Aedes aegypti. Insect Mol Biol 21, 97–106 (2011).
Smith, J. et al. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucl. Acids Res. 34, e149 (2006).
Arnould, S. et al. Engineering of large numbers of highly specific homing endonucleases that induce recombination on novel DNA targets. J Mol Biol 355, 443–458 (2006).
Arnould, S. et al. Engineered I-CreI derivatives cleaving sequences from the human XPC gene can induce highly efficient gene correction in mammalian cells. J Mol Biol 371, 49–65 (2007).
Rosen, L. E. et al. Homing endonuclease I-CreI derivatives with novel DNA target specificities. Nucl. Acids Res. 34, 4791–4800 (2006).
Redondo, P. et al. Molecular basis of xeroderma pigmentosum group C DNA recognition by engineered meganucleases. Nature 456, 107–111 (2008).
Chen, Z. & Zhao, H. A highly sensitive selection method for directed evolution of homing endonucleases. Nucleic Acids Res 33, e154 (2005).
Doyon, J. B., Pattanayak, V., Meyer, C. B. & Liu, D. R. Directed evolution and substrate specificity profile of homing endonuclease I-SceI. J Am Chem Soc 128, 2477–2484 (2006).
Jacoby, K. et al. Expanding LAGLIDADG endonuclease scaffold diversity by rapidly surveying evolutionary sequence space. Nucleic Acids Res 40, 4954–4964 (2012).
Takeuchi, R. et al. Tapping natural reservoirs of homing endonucleases for targeted gene modification. Proc Natl Acad Sci U S A 108, 13077–13082 (2011).
Chames, P. et al. In vivo selection of engineered homing endonucleases using double-strand break induced homologous recombination. Nucleic Acids Res 33, e178 (2005).
Mussolino, C. & Cathomen, T. TALE nucleases: tailored genome engineering made easy. Curr Opin Biotechnol 23, 644–650 (2012).
James, A. A. Gene drive systems in mosquitoes: rules of the road. Trends Parasitol 21, 64–67 (2005).
Takeuchi, R., Certo, M., Caprara, M. G., Scharenberg, A. M. & Stoddard, B. L. Optimization of in vivo activity of a bifunctional homing endonuclease and maturase reverses evolutionary degradation. Nucleic Acids Res 37, 877–890 (2009).
Acknowledgements
This work was funded by the NIH-NIAID (AI071208) and by the Fralin Life Science Institute and College of Agriculture and Life Sciences at Virginia Tech. We thank members of the Adelman and Myles laboratories for technical assistance.
Author information
Authors and Affiliations
Contributions
K.M.M. and Z.N.A. designed the experiments; A.A. and M.A.E.A. performed the experiments; A.A., K.M.M. and Z.N.A. analyzed the results; A.A., K.M.M. and Z.N.A. wrote the manuscript. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Table S1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Aryan, A., Anderson, M., Myles, K. et al. Germline excision of transgenes in Aedes aegypti by homing endonucleases. Sci Rep 3, 1603 (2013). https://doi.org/10.1038/srep01603
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01603
This article is cited by
-
AePUb promoter length modulates gene expression in Aedes aegypti
Scientific Reports (2023)
-
A selectable, plasmid-based system to generate CRISPR/Cas9 gene edited and knock-in mosquito cell lines
Scientific Reports (2021)
-
Characterization of a female germline and early zygote promoter from the transcription factor bZip1 in the dengue mosquito Aedes aegypti
Parasites & Vectors (2020)
-
DNA damage response and repair in perspective: Aedes aegypti, Drosophila melanogaster and Homo sapiens
Parasites & Vectors (2019)
-
The neurotranscriptome of the Aedes aegypti mosquito
BMC Genomics (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
