
Abstract
Human induced pluripotent stem cells (hiPSCs) hold enormous potential, however several obstacles impede their translation to industrial and clinical applications. Here we describe a platform to efficiently generate, characterize and maintain single cell and feeder-free (FF) cultured hiPSCs by means of a small molecule cocktail media additive. Using this strategy we have developed an effective multiplex sorting and high-throughput selection platform where individual clonal hiPSC lines are readily obtained from a pool of candidate clones, expanded and thoroughly characterized. By promoting survival and self-renewal, the selected hiPSC clones can be rapidly expanded over multiple FF, single-cell passages while maintaining their pluripotency and genomic stability as demonstrated by trilineage differentiation, karyotype and copy number variation analysis. This study provides a robust platform that increases efficiency, throughput, scale and quality of hiPSC generation and facilitates the industrial and clinical use of iPSC technology.
Similar content being viewed by others

Introduction
Human induced pluripotent stem cells (hiPSCs) offer great promise for research and clinical applications including the modeling of human disease, drug efficacy and safety screening and ultimately as a source of autologous or allogeneic cells for regenerative medicine1,2,3,4,5,6,7,8. iPSC generation was originally demonstrated by the ectopic expression of defined transcription factors, namely Oct4, Sox2, Klf4 and cMyc9. Although many studies have further improved on this landmark innovation, hiPSC clone generation selection and characterization has generally been low throughput: with few clones being identified, cultured and characterized on an individual basis by the skilled stem cell biologist. It is clear that future applications of iPSC technology such as genetic alteration for disease correction, loci-specific modulation for reporter systems or clone selection for preferred differentiation potential will require higher throughput and more reliable methods for clone derivation and characterization. One of the hindrances to such technology advancements are that the culture systems used to date have followed those originally identified for conventional human embryonic stem cells (hESCs); i.e. clump passaging, the use of a mouse embryonic fibroblast (MEF) feeder cell layer and non-defined media including serum10,11,12,13,14,15,16. Such systems are particularly laborious and inefficient for hiPSC generation and are not applicable to defined and scalable culture for industrial or clinical use17,18,19. In addition, reprogramming of somatic cells in a feeder free (FF) culture system is an extremely inefficient process, adding a further obstacle to developing a defined system for hiPSC generation2. Recent studies have also demonstrated that hESCs and to a higher degree, hiPSCs are susceptible to genomic abnormalities with some genomic changes occurring during the reprogramming process and others occurring during extended passaging in culture17,20,21,22,23,24. Therefore, before hiPSC technology can be effectively transitioned to industrial and clinical settings, challenges pertaining to the efficiency and standardization of clone selection, characterization, scalability and genomic stability must be addressed.
In the present study, we describe a multiplex cell sorting system to allow rapid selection, characterization and expansion of hiPSC clones in feeder free (FF), single cell passage culture while maintaining pluripotent status and genomic stability. Key to this system is the identification of a small molecule cocktail of specific signaling pathway inhibitors that can be used as a media additive to support and enhance the derivation of hiPSCs in a FF culture environment. The use of this cocktail also results in hiPSCs with improved clonality and tolerance of single cell passaging. Importantly, hiPSCs generated and maintained in FF and single cell culture using this unique small molecule cocktail retain genomic stability as indicated by both karyotype analysis and copy number variation. This system has the potential to be used in concert with all reprogramming methods for the rapid and high-throughput derivation and maintenance of industrial-grade hiPSCs.
Results
Defining cell culture additives to support high-throughput iPSC generation, maintenance and expansion
Small molecule inhibitors of specific signaling pathways have been used in various aspects of iPSC generation and maintenance. To determine whether a combination of small molecules could be used to support FF culture during somatic cell reprogramming and long-term single cell culture, we investigated the inhibition of signaling pathways commonly associated with the perturbation of somatic cell reprogramming and pluripotent stem cell self-renewal and survival16,25,26,27,28,29,30,31,32,33,34,35,36,37. With an initial focus on expression of pluripotent markers such as Tra181 and the viability of cultured cells, our data demonstrated that the use of various pathway inhibitors significantly influenced the survival and maintenance of FF and single cell dissociated hiPSCs (Supplementary Fig. 1). Significant cell death was seen when hiPSCs previously generated on feeder cells were cultured on Matrigel™ using conventional medium and single cell enzymatic passaging (Supplementary Figs. 1a–c). The addition of ROCK inhibition enhanced cell viability and plating efficiency but resulted in cellular differentiation whereas MEK, TGFβ and GSK3 inhibition enabled the maintenance of pluripotency but with significant cell death (Supplementary Figs. 1a–c). However, combining the four small molecules resulted in high viability and plating efficiency of undifferentiated hiPSCs (Supplementary Figs. 1a–c). A recent high-throughput chemical screen identified Thiazovivin to be an inhibitor of ROCK activity and highly capable of promoting hESC survival30. In a direct comparison of Y27632 and Thiazovivin, it was seen that while both inhibitors of ROCK promoted cell survival, the combination including Thiazovivin better maintained the undifferentiated state (Supplementary Figs. 1d,e). The final selected combination and concentration of the small molecule media additives was termed small molecule cocktail of 4 inhibitors (SMC4), consisting of SB431542 (TGFβi), PD0325901 (MEKi), CHIR99021(GSKi) and Thiazovivin (ROCKi) (Supplementary Table 1). This combination also significantly augmented cellular reprogramming in FF culture (Supplementary Fig. 1f). This single cell culture system was therefore used as the basis for our subsequent studies to define a high-throughput iPSC generation platform.
Adaptation of hiPSCs to FF and single cell passage culture using SMC4-supplemented medium
Established, feeder-derived and clump passaged hiPSCs were adapted to single cell culture on Matrigel™-coated tissue culture plates using either conventional medium or SMC4-supplemented medium (Fig. 1a). In contrast to conventional culture, the SMC4 culture was able to support FF hiPSC colony formation while preventing cell death (Fig. 1a and Supplementary Fig. 2a). During subsequent FF culture in SMC4, individual cells readily formed colonies and maintained their undifferentiated status over multiple passages (Figs. 1b–e). Further, the adapted cells displayed effective silencing of exogenous genes and retained their genomic integrity (Figs. 1e,f). Interestingly, the hiPSCs cultured in the SMC4-supplemented media grew as a homogeneous monolayer on FF culture providing utility for future industrial applications (Fig. 1b). To determine whether the single cell and FF cultured hiPSCs were pluripotent, we removed SMC4 from culture and allowed the hiPSCs to differentiate. When SMC4 was removed from culture, cells lost their undifferentiated morphology and gained a more differentiated flat and stretched appearance (Supplementary Fig. 2b). After several weeks of differentiation, gene expression profiles and markers representative of the three somatic lineages were identified (Fig. 1g and Supplementary Fig. 2c). When introduced into the renal capsule and testis of immune compromised mice, SMC4 maintained hiPSCs gave rise to teratomas consisting of the three germ layers (Fig. 1h). Collectively, SMC4 facilitated hiPSCs adaptation to and maintenance in single cell dissociated and FF culture.

Single-cell and FF culture of adapted hiPSCs in SMC4.
(a) hiPSCs previously generated on feeder cells with conventional medium were passaged onto FF culture and maintained as single cell culture with either conventional medium or conventional medium supplemented with SMC4 (adapted hiPSC clone termed, Fate Therapeutics hiPSC clone (FTi) 60). Lower-left panel represents marker expression of generated hiPSC clone prior to adaptation. (b) Five day time course of single cell dissociated and FF cultured hiPSCs in SMC4 growing into colonies and subsequent confluent culture. (c) SSEA4 and Tra181 profiles of continuously passaged adapted FTi60 in FF and single cell culture. Profiles represent passages 2, 6 and 10, respectively. (d) FTi60 maintained in SMC4 culture for 10 passages were stained for DAPI (blue), Nanog (green) and Tra160 (red). (e) qRT-PCR for endogenous and exogenous gene expression of pluripotent markers was conducted on the original IMR90 cells (Fibroblasts), IMR90 cells infected by reprogramming factor for 4 days post infection (Day 4 P.I.), human ESCs (H1 and HUES9) and FF maintained hiPSCs in SMC4 (FTi60). Expression was normalized to Gapdh and relative within each gene group analyzed. Error bars represent standard deviation. (f) Cytogenetic analysis on 20 G-banded metaphase cells derived from passage 11 FTi60 maintained in FF culture and supplemented with SMC4. (g) FTi60 maintained in FF culture and SMC4 was induced to differentiate for 21 days and stained for markers of various lineages including: Mesoderm, alpha smooth muscle actin (αSMA); Ectoderm, Tuj1; Endoderm, Sox17. (h) Histological sections of teratoma derived from FTi60 maintained in FF culture and SMC4. Black arrows point to areas of interest: Mesoderm, smooth muscle; Ectoderm, neural rosettes; Endoderm, glands.
Single cell sorting for the derivation of FF clonal hiPSCs
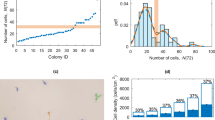
High-throughput methods of hiPSC derivation and characterization as well as clone or subclone derivation of existing or modified hiPSCs will require single cell plating of prospective lines38,39,40. To test whether the SMC4 additive supported high clonality by promoting enhanced seeding efficiency in FF culture, we flow-cytometry sorted individual cells and monitored their progress. SSEA4+/Tra181+ double positive hiPSCs were selected and plated on FF surfaces and in SMC4-supplemented medium at various cell densities, including 500 cells per well of a 6-well plate (52 cells per cm2). Within 24 hours of the sort, cell attachment and division was seen and after 7 days SSEA4+/Tra181+ derived colonies were scored for alkaline phosphotase (AP) expression (Figs. 2a–c). The sorted cells were single cell cultured on FF surfaces for an additional 5 passages and shown to be a nearly homogenous population of undifferentiated cells (Figs. 2d,e). To determine whether it was possible to derive clonal or subclonal lines, hiPSCs were sorted directly into FF 96-well plates containing SMC4. Various cell densities of hiPSCs were seeded, including 1 cell per well (3 cells per cm2) with colony formation detected within 48 hrs (Supplementary Fig. 3a). The individually sorted colonies readily expanded in FF culture and were scored for AP expression on day 8, marking a significant improvement over previously reported studies (Figs. 2f,g and Supplementary Fig. 3b)38,41. The addition of SMC4 to the medium formulation resulted in the ability to use FF culture, single cell passaging and produced hiPSCs with enhanced clonality; all characteristics required for a high-throughput iPSC generation, screening and maintenance platform.

Highly efficient single cell sorting of FF hiPSCs.
Single cells dissociated hiPSCs (FTi60) were flow-cytometry sorted for SSEA4/Tra181 dual expression and plated at various densities on FF surface coated with Matrigel and SMC4 supplemented medium. (a) Twenty-four hours post sorting, seeded cells have begun to divide and at day 7 large colonies consisting of many cells were readily observed. (b), (c) hiPSC colonies at each seeded density were stained for AP expression and scored 7 days post sort. N.C.: not counted due to confluency preventing accurate counts. (d), (e) After 5 passages post sort, approximately 1 month in culture, sorted FTi60 culture was stained for Tra160 (Red) and DAPI (Blue, lower right hand panel) and assessed for SSEA4 and Tra181 coexpression by flow-cytometry. (f) SSEA4+/Tra181+ cells of generated hiPSC clone FTi93 were sorted directly into 96 well-plates at various densities. Representative images of wells stained for AP expression 8 days post sort. (g) Wells of the sorted 96-well plates were scored for containing AP colonies on day 8. Analysis of each density was conducted in triplicate 96-well plates. Error bars represent standard deviation of triplicates.
Defining a high-throughput reprogramming platform
The advantages of the SMC4 culture were combined to develop a high-throughput method for generating FF and clonally derived hiPSCs. A scheme was devised to treat induced cells with SMC4 and select for rare individual cells that have faithfully reprogrammed as indicated by a combination of early and late pluripotency markers such as SSEA4 and Tra181 (Fig. 3)6,42. Selected cells would then be continually passaged in FF and single cell culture conditions in the presence of SMC4 and thoroughly characterized (Fig. 3). As a preliminary study, pluripotent cell enrichment was attempted using magnetic beads. IMR90 fibroblast (IMR90) cells were used as the starting cell since the generation of FF hiPSCs from this line has not previously been obtainable43. To initiate reprogramming, IMR90 cells were infected with individual lentiviruses expressing Oct4, Klf4, Sox2 and cMyc (OKSM). Cells displaying SSEA4 were enriched using magnetic bead-antibody conjugates on day 8 post infection (“Enrichment”, Fig. 3 and Supplementary Fig. 4a). The enriched cells were transferred to FF culture and supplemented with SMC4 or conventional reprogramming medium (Supplementary Fig. 4a). Consistent with previous studies, very few SSEA4/Tra181 positive colonies were observed in the presence of conventional medium in combination with a FF system (Supplementary Figs. 4a,b)43. In contrast, many SSEA4/Tra181 positive colonies were identified when the enriched cells were transferred to FF culture with SMC4 medium (average of 55-fold increase, SMC4 vs. conventional; Supplementary Fig. 4b). The hiPSC lines derived using SMC4 comprised a nearly homogeneous population of undifferentiated cells and expressed markers of pluripotency, displayed attenuation of exogenous gene activity and differentiated into cell types representing all three somatic cell lineages (Supplementary Figs. 4b–f). A similar strategy was also taken for the FF derivation of hiPSCs from adipose stem cells (Supplementary Figs. 5a–d). To further improve FF reprogramming efficiencies, we constructed 3-factor (Oct4, Klf4 and Sox2: polycistronic-OKS) and 4-factor (Oct4, Klf4, Sox2 and Myc: polycistronic-OKSM) polycistronic lentiviral vectors. These vector systems were tested in combination with the SMC4 culture system and cell selection method (Supplementary Figs. 6a,b). When we compared fibroblast reprogramming in the SMC4, FF system it was seen that the polycistronic lentiviral systems significantly improved the efficiency of colony formation over the same reprogramming factors in individual viral constructs (Supplementary Table 2). Further, the use of polycistronic vector systems and the SMC4 culture system enabled an approximate 25-fold improvement in reprogramming efficiency when compared to previously reported feeder-free and individual reprogramming systems (Supplementary Table 2)43,44.

Enhanced FF reprogramming strategy.
A schematic depiction of sorting methodologies for the enrichment and the selection of hiPSCs from a mix population. After the initiation of reprogramming and treatment with SMC4, the mixed population is sorted for established hiPSCs that express multiple markers of pluripotency including SSEA4 and Tra181 (solid line, termed Selection). Selected cells are individually sorted, for example into 96-well plates. In a high-throughput manner, clonal hiPSC are then selected, characterized and expanded. In an alternative strategy, the mixed population of reprogramming cells that have begun to express early pluripotent markers such as SSEA4 are enriched and maintained in SMC4 (dotted line, termed Enrichment). Identified hiPSC colonies are picked, characterized and expanded. In either case, the selected hiPSC lines are maintained on FF culture in SMC4 supplemented medium and expanded as clonal populations in long-term culture during routine single cell culture.
We next attempted to derive hiPSCs by directly selecting individual reprogrammed cells through cell sorting using a combination of markers at an early stage in the reprogramming process (“Selection”, Fig. 3). In a demonstration of the pharmaceutical approach for hiPSC generation, we obtained patient consented foreskin derived fibroblast cells and induced them to reprogram with polycistronic-OKSM lentivirus. Eight days after sorting and 20 days after the original initiation of reprogramming, hiPSC colonies were readily identified and picked (Supplementary Fig. 7a). After 10 passages in FF and single cell culture with SMC4, the individual colonies, Fate Therapeutics hiPSC clones (FTi) 70 and 72, were seen to have maintained their undifferentiated status, retained genomic stability and were able to give rise to all three germ layers (Supplementary Figs. 7b–d). Having confidence that an early flow-cytometry sort of double positive cells effectively leads to the derivation of hiPSCs, reprogramming was reinitiated using the polycistronic-OKSM lentivirus but with individual cells directly sorted into wells of a FF 96-well plate at 1 and 3 cells per well (Supplementary Fig. 7e). Using this platform, FF hiPSC clonal lines were derived in 96-well plates and assessed for their undifferentiated status by flow-cytometry (Supplementary Fig. 7f). On further analysis some of the clones derived in this manner contained non-clonal aberrations, suggestive of genomic instability (Supplementary Fig. 7g).
To avoid the potential selection of any non-clonal aberrations during long-term culture, we decided to repeat individual cell selection using the polycistronic-OKS lentivirus reprogramming system as it has been shown that reprogramming without cMyc improves genomic stability45. Furthermore, we coupled the reprogramming process with a multiplex platform to effectively select for the top tier clones based on selection assays of flow-cytometry, qRTPCR and immunofluorescence (Fig. 4). In an optimized multiplex protocol, reprogramming was initiated using the polycistronic-OKS lentivirus, an initial bulk sort of the SSEA4+/Tra181+ population was completed on day 20 post infection followed by resorting into 96 well-plates on day 30 (Fig. 4). In this way 50 individual clones were selected, replicate plated and characterized. Clones seen to maintain SSEA4+/Tra181+ double positive status by flow cytometry, express Nanog and attenuate expression of transgene by qRTPCR and scored positive for Oct4 and Nanog expression by immunofluorescence were chosen for expansion and further characterization (Fig. 4). Three independent patient consented fibroblast cell lines (FTC1, 5 and 7) were reprogrammed using the high-throughput multiplex methodology (Fig. 4). After this initial selection using multiplex methods of scoring pluripotency, selected clones were expanded and further characterized (Fig. 4). As seen in figure 5, FTC1 derived hiPSC clones (FTC1 clones 1 and 2) were further characterized to exhibit expression of pluripotency markers, attenuation of exogenous gene activity and de-methylation of the Oct4 promoter (Figs. 5a–c). These clones were also shown to be pluripotent by differentiating into the three somatic lineages by both in vitro differentiation and teratoma formation (Figs. 5d,e). Upon further analysis, global gene expression of SMC4 generated and cultured hiPSCs demonstrated similar gene expression of core markers of pluripotency but with further silencing of differentiated genes when compared to conventionally cultured hESC H1 and hESC HUES9 and conventionally generated and cultured hiPSCs (FTi99) from the same starting fibroblast line (Fig. 5f and Supplementary Fig. 8). High-throughput platform generated hiPSC clones from FTC5 and 7 were also thoroughly characterized and shown to express markers of pluripotency, as well to give rise to all three germ layers in vitro (Supplementary Figs. 9a,b). All selected clones were assessed for chromosomal integrity by karyotype analysis after 20 continuous single cell passages in FF and SMC4 culture: a normal karyotype was seen for both FTC1 derived clones, all four FTC5 clones and two of the four FTC7 derived clones (Fig. 6a and Supplementary Fig. 9c). Although the results suggest that the majority of derived and long-term cultured hiPSCs have maintained their genomic stability, the low frequency of acquired genomic aberrations highlights the requirement for thorough characterization of all derived and maintained hiPSC lines. To assess genomic stability at higher resolution, FTC1 clones 1 and 2 were analyzed by comparative genomic hybridization using NimbleGen 135 K array platform where copy number variation (CNV) is determined by competitive differential hybridization of 135,000 labeled probes to the genomic DNA. FTC1 clones 1 and 2 demonstrated minimal CNV with only 12 and 13 test sample CNVs, respectively (Fig. 6b). This compares favorably with the starting fibroblast line which had 14 test sample CNVs (Fig. 6b). Thus, the combination of polycistronic-OKS lentivirus induction, SMC4-supplemented medium and a multiplex characterization platform significantly enhances the kinetics of FF reprogramming while enabling the identification, selection and expansion of clonally derived and genomically stable hiPSCs in a high-throughput manner.

High-throughput FF reprogramming platform.
Polycistronic-OKS lentivirus induced fibroblast cells are sorted in a two step fashion to deliver an efficient 96-well plating platform using SMC4 supplemented medium. Fifty wells containing individual colonies are marked and expanded into 4x96-well plates to attain a total of 50 individual clones for analysis. One set is designated as the master-plate and expanded, the other three plates are processed for characterization including flow-cytometry analysis for surface marker expression including SSEA4 and Tra181, qRT PCR for expression of key markers including Nanog and transgene silencing and immunofluorescence for pluripotent markers including Oct4 and Nanog. The data panels represent snapshots of the clones surveyed (marked by wells 31–40 of the 96-well plate) during the high-throughput platform hiPSC generation of FTC5. In the qPCR panel, expression was normalized to Gapdh. Nanog expression is relative to H1 hESC while transgene expression is relative to day 4 post infection of FTC5 (Day 4 infection). Based on the characterization readouts, selected hiPSC clones are expanded for further analysis and banked. In the highlighted example, well 37 was identified as a candidate for expansion based on its multi-parameter pluripotency profile and termed FTC5 clone 1. Immuno-fluorescence images were taken at 5x magnification.

High-throughput platform, clonal and FF derivation of 3-factor (polycistronic-OKS) hiPSCs in the presence of SMC4.
(a) FTC1 clones 1 and 2 were stained for expression of pluripotent markers. (b) qRT-PCR of endogenous gene expression of pluripotent markers for foreskin fibroblast (FTC1), hESC lines (H1 and Hues9), FTC1 derived hiPSC clones (FTC1 clones 1 and 2) and day 4 post infection of FTC1 (Day 4 P.I.). Expression was normalized to Gapdh and relative within each gene group. Error bars represent standard deviation of replicates. (c) Oct4 promoter methylation status. Open circles represent unmethylated CpG islands while dark circles represent methylated CpG islands. (d) EB formation and differentiation of clones FTC1 clones 1 and 2 each at passage 10; 28 days post differentiation: Endoderm, FoxA2; Mesoderm, alpha smooth muscle actin (αSMA); Ectoderm, Tuj1. (e) Histological sections of teratoma derived from FTC1 clone 2 passage 18, generated and maintained in FF culture and SMC4. Black arrows point to areas of interest: Mesoderm, white adipose tissue; Ectoderm, neurons; Endoderm, glands. (f) Relative gene expression based on Affymetrix gene expression analysis of selected genes associated with pluripotency and lineage differentiation compared between SMC4 group (FTC1 clones 1 and 2) versus conventional culture group (H1, Hues9 and FTi99).

Genomic stability of FF and single cell cultured hiPSCs in the presence of SMC4.
(a) Karyotype summary table of high-throughput platform FTC 1, 5 and 7 hiPSC derived clones. All hiPSC clones were maintained in FF and single cell culture in the presence of SMC4 for the indicated passage prior to analysis. (b) Copy number variation as assessed by array comparative genomic hybridization. Bottom table is an interpretation summary of the data. p, passage number.
Discussion
Realizing the potential of hiPSCs for disease modeling, drug screening and ultimately disease correction and cell therapy will require several advancements in the underlying technology. Much progress has been made in advancing reprogramming technology towards potentially safer methods10,12,46,47. However, standardized methods for high throughput hiPSC generation, characterization and clonal expansion in culture environments applicable to industrial and clinical use would be a significant step for this nascent technology. Further, the maintenance or selection for pluripotent quality and genomic stability will be a requirement for all downstream applications. To this end, we aimed to define a simple multiplex approach to iPSC generation and characterization that would yield high quality and genomically stable hiPSC clones. Our initial strategy to define culture conditions that would allow FF culture and single cell passage of hiPSCs resulted in the identification of a unique combination and concentration of small molecules, SMC4 (Supplementary Table 1). The addition of this cocktail of small molecules to conventional medium facilitates enhanced FF reprogramming, in part by allowing the selection of hiPSCs from the reprogramming cell milieu using surface markers. Both hiPSC enrichment on magnetic beads and selection by flow-cytometry sorting was demonstrated, allowing identification and selection of rare and unique reprogramming events in addition to generating multiple iPSC lines from any given experiment. This is especially relevant when only operating in FF systems where reprogramming efficiencies are significantly lower. Moreover, the increased cell viability imparted by SMC4 enabled routine selection of individual hiPSCs by sorting directly into 96-well cell culture plates, resulting in the development of a high-throughput platform (Figs. 3,4). We established a multiplex method of simultaneous cell characterization and master-plate expansion and storage. This technique vastly reduces time and researcher effort in the generation of multiple iPSC lines. By using this approach a researcher is able to generate, characterize and survey multiple clones or subclones from multiple reprogramming experiments in parallel. Importantly, the hiPSC clones generated in this way were fully characterized and seen to maintain pluripotency and genomic stability. In contrast to recent studies22, high resolution comparative genomic hybridization analysis of our hiPSCs shows genomic stability, with minimal copy number variations in comparison to the starting fibroblast line even after multiple subsequent FF, single cell passages. As recent studies have continued to improve reprogramming methods by introducing safer and more efficient induction strategies48,49, our current platform has the potential to make all reprogramming strategies high-throughput, while making the reprogramming process independent of feeder cells and amenable to single cell culture. This platform is also seen to improve the efficiencies of the less effective methods for generating of hiPSCs, such as FF 3-factor reprogramming (Supplementary Table 2). Interestingly, 3-factor OKS derived hiPSCs appear to be more genomically stable than 4-factor OKSM derived hiPSCs, supporting previous findings that oncogene cMyc dysregulates cell cycle checkpoints and chromosomal stability45. It also appears that by effectively supporting hiPSCs in FF and single cell culture, there is less selection pressure for clonal outgrowth of hiPSCs that have gained an advantage through undesirable chromosomal alterations. Beyond hiPSC generation this high-throughput platform system can be applied to strategies such as disease correction, where survey through large number of lines may be required. It has also been shown that various iPSC clones have different propensities to differentiate, this high-throughput platform could be used to select clones with the highest potential for cell type specific differentiation.
There are now many techniques and methods for the derivation of pluripotent cells via the in vitro reprogramming of somatic cells. This has led to both an expansion of the field and a requirement for careful characterization and comparison of the resulting cells. Several pluripotency states may also exist adding to the complexity of analysis50. Going forward there will be a greater demand for uniformity of procedures and production for hiPSCs. SMC4 serves as a unique small molecule cocktail that alleviates the need for feeder cells, aids the reprogramming process and supports single cell culture for both long-term maintenance and processes such as flow-cytometry sorting. Furthermore, FF hiPSCs cultured in SMC4 are ideal for high-throughput screening as the cultured cells are monolayer and homogeneous. These properties combined with the high-throughput, multiplex method for hiPSC generation and subsequent characterization contribute to an “all in one” technique amenable to various pluripotent cell research and industrial applications.
Methods
Cell Culture of Pluripotent Stem Cells
Prior to FF adaptation, conventionally cultured hiPSCs were routinely maintained on feeder cells, mitomycin C treated MEF cells (Millipore) and cultured with conventional hESC medium (referred to as conventional medium in the text) containing DMEM/F12 (Mediatech), 20% v/v knockout serum replacement (Invitrogen), 1% v/v non-essential amino acids (Mediatech), 2 mM L-glutamine (Mediatech), 100 µM β-mercaptoethanol (Invitrogen) and 10 ng/mL bFGF (Invitrogen). Upon confluency, conventionally cultured hiPSCs were enzymatically dissociated using 1 mg/mL collagenase IV (Invitrogen) for 7 min at 37°C followed by mechanical dissociation into small pieces (termed as clump passaging), collected and dilute passaged 1:3–1:4 onto freshly seeded mitomycin C treated MEF cells every 5–7 days with daily addition of hESC medium. Cell cultures were maintained in a humidified incubator set at 37°C and 5% CO2. For adaptation to FF and single cell culture, upon confluency conventionally cultured hiPSCs were clump dissociated and resuspended in MEF cell conditioned medium (hESC medium conditioned on MEF cells for 24 hrs and supplemented with 10 ng/mL bFGF prior to use) and transferred to FF tissue culture plates (BD Biosciences) that were previously coated with Matrigel™ (1∶25 dilution; BD Biosciences) for 1–2 hrs in 37°C. MEF conditioned medium was changed daily. Upon confluency of greater than 90%, conditioned medium was switched to SMC4 supplemented medium [Supplementary Table 1, hESC medium supplemented with 0.4 µM PD0325901 (Biovision), 1 µM CHIR99021 (Biovision), 5 µM Thiazovivin (internally synthesized at Fate Therapeutics Inc. and purchased from Biovision) and 2 µM SB431542 (Biovision)] two hours prior to single cell dissociation, see below. Small molecules were maintained in −20°C prior to the addition to medium at a stock concentration of 5–25 mM in DMSO. All working media were maintained in 4°C for the duration of usage. In studies that included Y27632 (Ascent), 10 μM was used.
For single cell dissociation, hiPSCs were washed once with phosphate buffered saline (PBS) (Mediatech) and treated with Accutase (Millipore) for 3–5 min at 37°C followed with pipetting to ensure single cell dissociation. The single cell suspension was then mixed in equal volume with conventional medium, centrifuged at 250 g for 5 min and resuspened in SMC4 supplemented medium (Supplementary Table S1).
After resuspension in SMC4 supplemented medium, the single cells were transferred to FF tissue culture plates that were previously coated with Matrigel™ for 1–2 hrs in 37°C. In this format, cells routinely received fresh medium every other day and were passaged when confluency had reach 65–75%, which normally occurred 4–5 days post passage. With each passage cells were re-dissociated into single cells and transferred to a new tissue culture plate coated with Matrigel™ at a dilution passage of 1:8–1:15.
Induction of Reprogramming
To initiate the reprogramming process, ectopic expression of reprogramming factors were induced by lentiviral transduction. Most typically OKSM or OKS were used as indicated per each study. In most cases, the starting cells were plated at 10% confluency (i.e. 1x105 cells per well of a 6-well plate) on gelatin (Mediatech) coated surface. For the method of viral infection, freshly collected lentivirus was added to the starting cells at a dilution of 1∶2, supplemented with 4 µg/mL polybrene (Millipore) and transferred to 37°C and 5% CO2 for 8–12 hrs. After the completion of the incubation, the cells were washed three times with PBS and fed with fibroblast medium: DMEM (Mediatech), 10% FBS (Invitrogen), 1x glutamax (Invitrogen), 1x non-essential amino acids (Mediatech). Upon confluency (usually between days 4–6) the cells were dissociated with trypsin (Invitrogen), mixed with equal part fibroblast medium, centrifuged at 250 g for 5 min, resuspended in SMC4 supplemented medium and expanded 1:10 into a larger culture plate. Cultures are maintained in SMC4 until the next application.
Unique Population Enrichment
Approximately 8–12 days after the induction of reprogramming, cells were dissociated into single cells with Accutase (Millipore) and stained with various surface markers of pluripotency, markers of somatic cells and/or markers of incomplete reprogramming. Briefly, dissociated cells were resuspended in staining solution containing Hanks' Balanced Salt Solution (Invitrogen), 4% fetal bovine serum (Invitrogen) and 10 mM Hepes (Invitrogen) and kept on ice. Per recommended manufacturers' dilution, conjugated primary antibodies were added to the cell solution and incubated on ice for 15 min. The cell solution was washed and resuspended in staining buffer and maintained on ice. At this point various enrichment/depletion strategies were taken; including Fluorescent Activated Cell Sorting (BD Biosciences, see below) and Magnetic Cell Sorting (Miltenyi Biotec, see below).
Flow cytometry sorting was performed on FACS Aria II (BD Biosciences). Primary antibodies used include SSEA4-Alexa Fluor-488/555 (BD Biosciences), Tra181-Alexa Fluor-488/647 (BD Biosciences), Tra161-Alexa Fluor-488/647 (BD Biosciences). The cells were collected in SMC4 supplemented medium. The sorted cells were then centrifuged and resuspended in SMC4 supplemented medium and transferred to Matrigel™ coated tissue culture plates. For additional improvement in seeding, 5 μg/mL Fibronectin (BD Biosciences) can be added to SMC4 for the first two days. When sorted into microwells, i.e. 96 well plates, each well is prefilled with SMC4 and upon completion of the sort the plates were centrifuged for 2 min at 300 g prior to incubation. The SMC4 supplemented medium was replaced every other day except in case of 96-well plate sorting where it was replaced after 3–4 days in culture. Colony formation was typically seen 2–4 days post sort. Flow cytometry analysis was performed on Guava EasyCyte 8 HT (Millipore).
MACS Microbeads (Miltenyi Biotec) separation was performed according to protocol. Briefly, cells were dissociated into single cells and stained with appropriate FITC-conjugated primary antibodies, including SSEA4-FITC (BD Biosciences) and Tra181-FITC (BD Biosciences). Cells were then magnetically labeled with Anti-FITC Microbeads (Miltenyi Biotec). The labeled cell suspension was then loaded onto a LS MACS Column (Miltenyi Biotec). The collected cells from either positively or negatively selected fractions were centrifuged at 250 g for 5 min and resuspended in SMC4 supplemented medium and transferred to Matrigel™ (BD Biosciences) coated tissue culture plates. The following day, fresh medium is added to the culture and subsequently replaced every other day. The appearance of colonies is typically seen 2–4 days post sort.
Isolation and culture of patient consented human neonatal foreskin fibroblast cells
Protocol and donor consent form were approved by the independent Institutional Review Board of Chesapeake Research Review (Columbia, MD). The parents of 2-day old baby boy provided their written informed consent for the use of their son's foreskin biopsy for the generation of iPSCs. The tissue was collected in Hanks' balanced salt solution (HBSS), cut into small pieces (3–4 mm) and incubated with dispase (BD Biosciences) overnight at 4°C. The dermis was separated from the epidermis and cut further into 0.5 mm-pieces and 10 to 15 pieces placed in 100-mm dish. Autoclaved cover-slips were laid on top of the dermal pieces to hold them down and 6 ml of fibroblast media slowly added to the edge of the dish. Fresh medium was added every 3–4 days and floating tissue pieces were removed. After two weeks, cells were ready to collect using trypsin. Additional patient consented fibroblast lines were obtained from ZenBio Inc.
Alkaline Phosphatase Staining
Cells were fixed in 4% v/v paraformaldehyde (Alfa Aesar), washed three times with PBS and stained with Alkaline Phosphatase Staining Kit (Sigma-Aldrich). Briefly, 1 mL Sodium Nitrite Solution was added to 1 mL FRV-Alkaline Solution, mixed and incubated at 25°C for 2 min. The solution was then mixed with 45 mL of H2O followed by the addition of 1 mL Naphthol AS-BI Alkaline Solution. The alkaline-dye mixture was added to the fixed cells and incubated at 25°C for 15 min followed by a PBS wash. The cells were then scored for the presence of alkaline phosphatase.
Immunofluorescence Staining
Cells were fixed using 4% v/v paraformaldehyde (Alfa Aesar), washed three times with PBS containing 0.2% v/v Tween (PBST) (Fisher Scientific) and permeablized using 0.15% v/v TritonX-100 (Sigma-Aldrich) in PBS for 1 hr at 25°C. After permeabilization, cells were blocked with 1% v/v BSA (Invitrogen) in PBST (PBSTB) (Fisher Scientific) for 30 min at 25°C. After gentle removal of PBSTB, cells were incubated with primary antibody in PBSTB overnight at 4°C. Primary antibodies used in this study include Nanog (Abcam), Tra160 (BD Biosciences), Tra181 (BD Biosciences), SSEA4 (BD Biosciences), β-III Tubulin (R&D Systems), α-Smooth Muscle Actin (Sigma), FoxA2 (R&D Systems) and Sox17 (R&D Systems). After the overnight incubation, cells were washed three times with PBST and stained with secondary antibody (Alexa 488 or 555; Invitrogen) diluted 1:500 in PBSTB for 1 hr at 25°C. The cells were washed three times in PBST and stained with Hoechst dye (Invitrogen). Images of the stained cells were captured using the Zeiss fluorescence microscope and CCD camera.
Induction of Differentiation
hiPSC were differentiated as EBs or monolayers in differentiation medium containing DMEM/F12 (Mediatech), 20% fetal bovine serum (Invitrogen), 1% non-essential amino acids (Mediatech), 2 mM L-glutamine (Mediatech) and 100 µM β-mercaptoethanol. Briefly, for EB formation hiPSCs were single cell dissociated with Accutase (Millipore) and resuspended in differentiation medium to a final concentration of 75,000 cells/mL and 5 uM Thiazovivin was added. Cells were seeded in 100 µL/well in V-bottom 96-well non-tissue culture plate (Nunc) and centrifuged at 950 g for 5 min. The following day compact “ball-like clumps” were transfer to ultra-low binding 6-well plate (Corning) using P1000 at approximately 30–40 EBs/well. After 7 days, EBs were transferred at 1:1 to Matrigel coated 6-well plate. After 3 weeks in culture, cells were fixed and stained. For monolayer differentiation, hiPSCs were seeded in SMC4 and switched to differentiation medium the next day. Once the medium had been switched, the hiPSCs were allowed to differentiate for 14–21 days. Medium was changed every 2–3 days.
RT-qPCR Analysis
RNA was isolated using the PicoPure RNA Isolation kit (Life Technologies) and 0.5 µg RNA was used to generate first strand cDNA using the iScript cDNA Synthesis Kit (Bio-Rad). Relative gene expression levels were determined using the TaqMan Fast Universal PCR Master Mix (Applied Biosystems) and the FAM-labeled TaqMan probes listed in Supplementary Table 3.
Gene Expression Analysis
Total RNA was isolated from cells using Pico Pure RNA Isolation Kit (Life Technologies). In brief, biotinylated aRNA was prepared using the standard protocol for MessageAmp II aRNA Amplification Kit (Applied Biosystems/Ambion, Austin, TX) utilizing the optional Second Round Amplification and then transcribed into biotin labeled aRNA using MessageAmp II Biotin Enhanced Kit (Applied Biosystems/Ambion, Austin, TX) using the standard protocol. Biotin labeled aRNA was purified and fragmented according to Affymetrix recommendations. 20 µg of fragmented aRNA were used to hybridize to the Human Genome U133 Plus 2.0 chips (Affymetrix Inc. Santa Clara, CA) for 16 hrs at 45°C. The arrays were washed and stained in the Affymetrix Fluidics Station 450 and scanned using the Affymetrix GeneChip Scanner 3000 7G. The image data were analyzed using Affymetrix Expression Console software using default analysis settings. Arrays were normalized by log scale robust multi-array analysis (RMA) and visualized in Spotfire for Genomics 3.1 (Tibco Spotfire, Palo Alto, CA).
Karyotype Analysis
Cytogenetic analysis was performed on twenty G-banded metaphase cells by Cell Line Genetics (Madison, WI) or WiCell (Madison, WI).
Comparative Genomic Hybridization
High resolution comparative genomic hybridization (NimbleGen 12x135 k array; HG18 WG CGH v3.1 HX12) and subsequent copy number variation analysis was conducted by WiCell (Madison, WI). Briefly, relative copy number is determined by comparative differential hybridization of labeled genomic DNA to the 135,000 oligonucleotide whole genome tiling array.
Statistical Analysis
Student's t test was used for statistical evaluations pertaining to standard deviation. StepOne Software v2.2 (Life Technologies) was used to determine RQ minimum and maximum values (error bars).
Teratoma Formation
Teratoma grafting and analyses was conducted by Applied Stem Cells (Menlo Park, CA). Briefly, 1–5 million single cell dissociated hiPSCs were mixed in 100 uL SMC4 supplemented medium and 100 uL Matrigel and introduced to the renal capsule and testis of Beige SCID mice. The developed teratomas were harvested, sectioned and analyzed for various differentiated cell types and structures.
Accession Numbers
The GEO accession number for the Affymetrix profiling reported in this paper is GSE28815. The GEO accession numbers for CGH array reported in this paper are GSM828394, 828395 and 828396.
References
Keller, G. Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev 19, 1129–1155 (2005).
Saha, K. & Jaenisch, R. Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell 5, 584–595 (2009).
Smith, A. G. Embryo-derived stem cells: of mice and men. Annu Rev Cell Dev Biol 17, 435–462 (2001).
Kiskinis, E. & Eggan, K. Progress toward the clinical application of patient-specific pluripotent stem cells. J Clin Invest 120, 51–59 (2010).
Hanna, J. et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 133, 250–264 (2008).
Lowry, W. E. et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proc Natl Acad Sci U S A 105, 2883–2888 (2008).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Yu, J. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 (2007).
Takahashi, K. & Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Soldner, F. et al. Parkinson's disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136, 964–977 (2009).
Woltjen, K. et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 458, 766–770 (2009).
Zhou, H. et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 4, 381–384 (2009).
Zhou, W. & Freed, C. R. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 27, 2667–2674 (2009).
Nakagawa, M. et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 26, 101–106 (2008).
Thomson, J. A. et al. Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147 (1998).
Valamehr, B., Tsutsui, H., Ho, C. M. & Wu, H. Developing defined culture systems for human pluripotent stem cells. Regen Med 6, 623–634 (2011).
Mayshar, Y. et al. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 7, 521–531 (2010).
Stewart, M. H., Bendall, S. C. & Bhatia, M. Deconstructing human embryonic stem cell cultures: niche regulation of self-renewal and pluripotency. J Mol Med 86, 875–886 (2008).
Catalina, P. et al. Human ESCs predisposition to karyotypic instability: Is a matter of culture adaptation or differential vulnerability among hESC lines due to inherent properties? Mol Cancer 7, 76 (2008).
Bock, C. et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell 144, 439–452 (2011).
Gore, A. et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 471, 63–67 (2011).
Hussein, S. M. et al. Copy number variation and selection during reprogramming to pluripotency. Nature 471, 58–62 (2011).
Laurent, L. C. et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 8, 106–118 (2011).
Lister, R. et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471, 68–73 (2011).
Ying, Q. L. et al. The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 (2008).
Sato, N., Meijer, L., Skaltsounis, L., Greengard, P. & Brivanlou, A. H. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med 10, 55–63 (2004).
Chen, G., Hou, Z., Gulbranson, D. R. & Thomson, J. A. Actin-myosin contractility is responsible for the reduced viability of dissociated human embryonic stem cells. Cell Stem Cell 7, 240–248 (2010).
Ohgushi, M. et al. Molecular pathway and cell state responsible for dissociation-induced apoptosis in human pluripotent stem cells. Cell Stem Cell 7, 225–239 (2010).
Watanabe, K. et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 25, 681–686 (2007).
Xu, Y. et al. Revealing a core signaling regulatory mechanism for pluripotent stem cell survival and self-renewal by small molecules. Proc Natl Acad Sci U S A 107, 8129–8134 (2010).
Nichols, J., Silva, J., Roode, M. & Smith, A. Suppression of Erk signalling promotes ground state pluripotency in the mouse embryo. Development 136, 3215–3222 (2009).
Tsutsui, H. et al. An optimized small molecule inhibitor cocktail supports long-term maintenance of human embryonic stem cells. Nat Commun 2, 167 (2011).
Kelly, K. F. et al. beta-catenin enhances Oct-4 activity and reinforces pluripotency through a TCF-independent mechanism. Cell Stem Cell 8, 214–227 (2011).
Huangfu, D. et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol 26, 1269–1275 (2008).
Ichida, J. K. et al. A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell Stem Cell 5, 491–503 (2009).
Lin, T. et al. A chemical platform for improved induction of human iPSCs. Nat Methods 6, 805–808 (2009).
Yuan, X. et al. Combined Chemical Treatment Enables Oct4-Induced Reprogramming from Mouse Embryonic Fibroblasts. Stem Cells (2011).
Mei, Y. et al. Combinatorial development of biomaterials for clonal growth of human pluripotent stem cells. Nat Mater 9, 768–778 (2010).
Hockemeyer, D. et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 27, 851–857 (2009).
Zwaka, T. P. & Thomson, J. A. Homologous recombination in human embryonic stem cells. Nat Biotechnol 21, 319–321 (2003).
Emre, N. et al. The ROCK inhibitor Y-27632 improves recovery of human embryonic stem cells after fluorescence-activated cell sorting with multiple cell surface markers. PLoS One 5, e12148 (2010).
Chan, E. M. et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat Biotechnol 27, 1033–1037 (2009).
Sun, N. et al. Feeder-free derivation of induced pluripotent stem cells from adult human adipose stem cells. Proc Natl Acad Sci U S A 106, 15720–15725 (2009).
Sugii, S., Kida, Y., Berggren, W. T. & Evans, R. M. Feeder-dependent and feeder-independent iPS cell derivation from human and mouse adipose stem cells. Nat Protoc 6, 346–358 (2011).
Pasi, C. E. et al. Genomic instability in induced stem cells. Cell Death Differ (2011).
Damoiseaux, R., Sherman, S. P., Alva, J. A., Peterson, C. & Pyle, A. D. Integrated chemical genomics reveals modifiers of survival in human embryonic stem cells. Stem Cells 27, 533–542 (2009).
Sugii, S. et al. Human and mouse adipose-derived cells support feeder-independent induction of pluripotent stem cells. Proc Natl Acad Sci U S A 107, 3558–3563 (2010).
Warren, L. et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7, 618–630 (2010).
Carey, B. W. et al. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc Natl Acad Sci U S A 106, 157–162 (2009).
Chin, M. H., Pellegrini, M., Plath, K. & Lowry, W. E. Molecular analyses of human induced pluripotent stem cells and embryonic stem cells. Cell Stem Cell 7, 263–269 (2010).
Acknowledgements
We thank Drs. Rudolf Jaenisch, Sheng Ding, Scott Thies and John Mendlein for constructive comments and insightful discussions; Scripps Flow-Cytometry Core for their assistance and expertise in cell sorting; Applied StemCell for teratoma processing and analysis; Cell Line Genetics for karyotype analysis; WiCell for copy number variation analysis, karyotype analysis and insightful discussion.
Author information
Authors and Affiliations
Contributions
BV, RA and MR conducted the majority of the experiments including hiPSC generation and characterization. TL and DR conducted and analyzed the Affymetrix analysis. RA, MR and DS contributed ideas. BV and PF conceptualized the project and wrote the paper with contribution from all authors.
Ethics declarations
Competing interests
All authors are current fulltime employees of Fate Therapeutics, San Diego, CA.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Valamehr, B., Abujarour, R., Robinson, M. et al. A novel platform to enable the high-throughput derivation and characterization of feeder-free human iPSCs. Sci Rep 2, 213 (2012). https://doi.org/10.1038/srep00213
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep00213
This article is cited by
-
NK Cell Therapeutics for Hematologic Malignancies: from Potential to Fruition
Current Hematologic Malignancy Reports (2023)
-
Generation of T-cell-receptor-negative CD8αβ-positive CAR T cells from T-cell-derived induced pluripotent stem cells
Nature Biomedical Engineering (2022)
-
Development of an efficient single-cell cloning and expansion strategy for genome edited induced pluripotent stem cells
Molecular Biology Reports (2022)
-
Amenable epigenetic traits of dental pulp stem cells underlie high capability of xeno-free episomal reprogramming
Stem Cell Research & Therapy (2018)
-
Mesenchymal stromal/stem cell separation methods: concise review
Cell and Tissue Banking (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
