
Abstract
Multisystem Proteinopathy 1 (MSP1) disease is a rare genetic disorder caused by mutations in the Valosin-Containing Protein (VCP) gene with clinical features of inclusion body myopathy (IBM), frontotemporal dementia (FTD), and Paget’s disease of bone (PDB). We performed bone scan imaging in twelve patients (6 females, 6 males) with confirmed VCP gene mutation six (50%) of which has myopathy alone, four (33%) with both PDB and myopathy, and two (15%) were presymptomatic carriers. We aim to characterize the PDB in diagnosed individuals, and potentially identify PDB in the myopathy and presymptomatic groups. Interestingly, two patients with previously undiagnosed PDB had positive diagnostic findings on the bone scan and subsequent radiograph imaging. Among the individuals with PDB, increased radiotracer uptake of the affected bones were of typical distribution as seen in conventional PDB and those reported in other MSP1 cohorts which are the thoracic spine and ribs (75%), pelvis (75%), shoulder (75%) and calvarium (15%). Overall, we show that technetium-99m bone scans done at regular intervals are a sensitive screening tool in patients with MSP1 associated VCP variants at risk for PDB. However, diagnostic confirmation should be coupled with clinical history, biochemical analysis, and skeletal radiographs to facilitate early treatment and prevention complications, acknowledging its limited specificity.
Similar content being viewed by others

Introduction
VCP Multisystem Proteinopathy 1 (MSP1), also known as IBMPFD or VCP disease (OMIM #167,320), is a rare autosomal dominant genetic disorder that affects the muscle, bone, and the central nervous system. MSP1 is caused by missense mutations in the VCP gene on chromosome 9p13.3-12. More than 65 variants have been linked to this disease, with the R155H, R155C, and R191Q variants being the most frequent1,2,3. Clinical manifestations include progressive adult-onset proximal muscle weakness, vacuolar and inclusion body muscle pathology in 50%, VCP PDB reported in 40%, and impairments in cognitive, behavioral, and social graces associated with frontotemporal dementia in approximately 30% of MSP1 patients1,2,4,5.
PDB is the second most common bone remodeling disease after osteoporosis in the general population6. It has a general prevalence of 2.1% in people older than 40 years old, with a slightly higher occurrence in males, but present in 40% of individuals with VCP disease1. PDB is caused by abnormal bone homeostasis where disorganization of bone by overactive osteoclasts and osteoblasts leads to clinical features of bone pain, bone deformity, deafness, fractures, and rarely osteosarcoma. The detailed mechanism of PDB is currently inconclusive, however, a combination of genetic, viral, and environmental factors has been suspected. One of most important predisposing genetic factor is variants in the SQSTM1 gene that causes osteoclast activation via RANK signaling in 5–20% of PDB patients7,8. Other causative genes of PDB include VCP, PFN1, OPTN, CSF1, TNFRSF11A, and TM7SF49,10,11,12. Elevation of bone turnover biochemical markers such as serum alkaline phosphatase (ALP), are associated13, however, normal ALP levels are observed in early or localized PDB disease.
In MSP1, PDB is reported in approximately 40% of patients14. PDB is often asymptomatic and is found incidentally in imaging and lab studies, however bone pain can be observed in 30% of MSP1 patients5. PDB can manifest as the first symptom in 4.9% of patients15. Co-presentation of myopathy and PDB as the initial manifestation occurs in 16% of MSP1 patients1. There are no studies comparing the biochemical or clinical differences between the PDB in MSP1 and conventional PDB. The mechanism of PDB pathogenesis however is similar since previous studies have shown that pathogenic variants in VCP can upregulate the NF-kB signaling pathway leading to upregulated osteoclastogenesis and bone resorption7 and can therefore contribute to PDB16,17,18. Similarly, report of VCP’s role in osteoblast activity involves complex regulation of bone morphogenetic protein (BMP) receptors via the VCP mediated ubiquitin/protein degradation system which may play a part in PDB pathogenesis19,20. Despite the lack of treatment options for the neuromuscular and neurological manifestations of MSP1, treatment of PDB with bisphosphonates is very effective in alleviating bone pain, and preventing deformity and other comorbidities.
Generally, plain radiographs are used to diagnose PDB due to their ability to show cortical thickening, bone expansion, and bone deformity21,22. While plain radiographs have high specificity, their sensitivity for the disease is relatively low. Alternatively, skeletal scintigraphy or bone scans prove to be more sensitive diagnostic tests compared to radiographs, especially in assessing regions of asymptomatic disease, albeit with lower specificity23. Since technetium-99m tracers (Tc-99m) are absorbed intensely in areas with increased osteoblastic activity, it is used to identify bones at risk for local complications23. Early stages of PDB present as osteolytic lesions or advancing lytic wedges in long bones on plain radiographic imaging, while sclerotic changes associated with thickened trabeculae and cortices, bone expansion, and deformity are seen later. Currently, most bone scan data in MSP1 patients is scattered in different case reports. Thus far, no formal bone scan analysis on the disease extent has been performed. Here, we present bone scan findings from twelve patients with MSP1 from one clinical center and systematically reviewed the anatomically affected areas. We also discuss guidelines for diagnostic testing and treatment of PDB.
Patients and methods
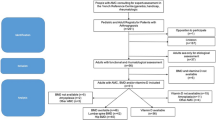
We recruited twelve patients (6 females and 6 males) from seven families with MSP1 and confirmed VCP gene mutation at the UC Irvine Medical Center (Table 1). Our patient cohort included affected individuals with VCP associated IBM only, both IBM and PDB, and carriers. Prior diagnosis of PDB was made by their physicians based on elevated serum ALP, with or without bone pain, and imaging findings. The carriers were diagnosed by presymptomatic testing for the familial variants. This study was approved by UC Irvine Institutional Review Board (IRB 2007-5832). All patients signed an informed consent. All methods were performed in accordance with the relevant guidelines and regulations.
All laboratory findings from the patient’s blood and imaging presented here were performed at UC Irvine Medical Center, Orange, CA. Bone scintigraphy used to survey the whole skeleton and assess the extent of disease uses a radiotracer like technetium-99m diphosphonate (Tc-99m) to identify areas of increased bone activity. All participants received an intravenous injection of technetium-99m and were imaged after 3 hours for whole-body imaging in the anterior and posterior projections. Radiograph imaging was performed on the same day of the pertinent area if an abnormality was identified on the bone scan. All bone scans and radiographs were read and interpreted by a blinded board-certified radiologist, and a second review included the senior author. Assessment of PDB was correlated with clinical history, lab testing and radiographic findings. All statistical analysis was conducted using Microsoft Excel version 16.71 and Prism version 9.5.1, and p-values ≤ 0.05 was considered statistically significant.
Results
Clinical characteristics are summarized in Tables 1 and 2. Four (33.3%, ID 1–4) had both myopathy and PDB at the time of visit with a mean age of PDB onset at 39.3 years [range, 38–45 years], and a mean age of myopathy onset at 43.0 years [range, 33–49 years]. Six (50%, ID 5–10) had myopathy only at the time of visit with a mean age of onset at 39.8 years [range, 25–51 years], in addition to two pre-symptomatic carriers (16.7%, ID 11–12). Two patients with myopathy only were diagnosed with PDB at the time of visit (ID 6 and 10) with bone scans and subsequent correlation with radiographs and lab findings. Bone scan findings in different anatomical locations for all individuals diagnosed with PDB are summarized in Table 3. Degenerative joint disease (DJD) was noted on bone scan in seven individuals including the two pre-symptomatic individuals. Only one patient known to have PDB had bone pain attributable to the PDB. No controls were included in this study because of the risks of exposure to radiation, as requested by the IRB. The two initially pre-symptomatic individuals later developed myopathy at age 37 respectively (ID 11 and 12), and also had cardiomyopathy associated with a pathogenic c.177_187del variant in the MYBPC3 gene24.
Spine and ribs
Six (50%) MSP1 patients had spine and rib involvement (Figs. 1, 2 and 3). Three patients had thoracic spine involvement while two had lumbar spine involvement with abnormal focal uptakes. Cervical spine vertebral uptake was seen in one patient. All six patients had one or multiple abnormal focal rib uptake. One presymptomatic patient (ID 12) had increased uptake at the anterior aspect of the right seventh rib attributed to DJD and not considered PDB.

Whole body 99mTc-MDP bone Scan and Radiographic findings in patient 1 (ID 1). A 60-years-old female with back pain and leg pain, limb girdle distribution of myopathy and Paget disease revealed (a) increased uptake in the skull, thoracic and lumbar vertebrae on whole-body bone scan, increased uptake in the knees due to associated arthritis, (b) radiograph images reveal corresponding bone lesions of the skull bilaterally (blue arrows), (c) thoracic T10 vertebrae (orange arrow) and lumbar L3 vertebrae (yellow arrow).

Whole body 99mTc-MDP bone scan and multiple spot views images of patient 2 (ID 2). A 43-years-old male with inclusion body myopathy, and previously diagnosed Paget’s disease revealed positive tracer uptake in the spine (blue arrow), left iliac bone (orange arrow) and right ankle (green arrow).

Whole body 99mTc-MDP bone scan images and multiple spot views of patient 4. A 49-years-old male with inclusion body myopathy, and Paget’s disease revealed positive bone scan findings of the left mandible (blue arrow), left hemipelvis (green arrow), multiple levels of the thoracic and lumbar spine (purple arrow) and proximal left fibula (yellow arrow).
Pelvis
Focal uptake in the pelvic region was seen in five (42%) MSP1 patients (Figs. 1, 2, 3 and 4). Acetabulum uptake was observed in three patients, and two had increased uptake in the ischium. Three patients had additional tracer uptake in hemipelvis areas including the sacrum, iliac crest and iliac brim, and superior pubic ramus.

Whole body 99mTc-MDP bone scan and skull radiographs in two newly diagnosed PDB in myopathy only patients. (a) A 28-years-old female (ID 6) with increased uptake in the skull on the bone scan corresponding to a mixed lytic and sclerotic bone lesion in the left parietal calvarium on the AP and lateral views of the plain radiographs (purple arrows). (b) A 38-years-old female (ID 10) with increased uptake in the right ischium and acetabulum on whole-body bone scan (orange arrows).
Skull
Calvarium uptake was observed in three (25%) MSP1 patients (Figs. 1 and 4a). Increased uptake was seen in the frontoparietal, parietal calvarium bones, and mandibles.
Shoulder, clavicle, sternum, and manubrium
Five (42%) patients had increased tracer uptake in the shoulder and clavicle region. Increased activity was seen in the sternoclavicular joints in two patients. Three patients also had an increased radiotracer uptake in the manubriosternal joint. One presymptomatic patient had increased tracer uptake in the clavicle due to DJD.
Lower extremity
Increased uptake in the lower extremity regions was observed in three (25%) MSP1 patients. One patient had increased uptake in the patella and ankle region (Fig. 2). One patient had increased uptake in the proximal femur, midfoot, and first toe. Another patient had increased uptake in the proximal left fibula (Fig. 3).
Degenerative joint disorder (DJD) changes
Degenerative bone changes were noted on bone scan findings in three of the six patients with myopathy and PDB, three with myopathy only and one pre-symptomatic carrier. In PDB patients, DJD was seen in the lumbar and thoracic spine, clavicle, and scapula. DJD affected areas such as clavicle, bilateral shoulders, sternoclavicular joints, and left patella were seen in one pre-symptomatic carrier.
Laboratory results
ALP levels were elevated in only one of the four patients (234 IU/L) who had myopathy and PDB at the time of visit, and three had normal values (mean 99.6 IU/L, normal 44–147 U/L). The newly diagnosed PDB patients also did not exhibit elevated ALP levels (mean 80 IU/L). Two patients with myopathy and PDB had elevated CPK levels (mean > 250 IU/L, normal 20–200 IU/L), all the other individuals had normal levels.
Treatment
Half of our patients (6/12) were not taking any form of medications nor supplements. Among those taking medications 4/6 (66.7%) were receiving daily intake of multivitamins and only one (16.7%) was receiving bisphosphonate (ID 4, zoledronic acid). Other medications reported by patients included analgesics, anesthetics, anticholinergics, selective serotonin reuptake inhibitor, diuretics, and anti-lipidemics.
Discussion
Typical manifestation of pathogenic variants of the VCP gene (MSP1) includes progressive axial and proximal muscle weakness, PDB starting in their thirties respectively, and FTD starting in their fifties1. Compared to conventional PDB with an approximate mean onset age of 50 years, MSP1 patients have an earlier mean age of onset of PDB in their late thirties as was noted in our cohort15. This is the first effort to formally analyze bone scan findings from MSP1 patients to ascertain and characterize PDB disease. Farpour et al. previously only assessed plain radiographs from MSP1 patients22.
Bone pain is typically the most common symptom (52–74%) followed by bone deformity (18–22%), deafness (8–9%) and fractures (6–9%) in PDB found in the general population25, however, we only found that one individual had associated pain in our cohort. Serum ALP level was previously considered the first line biochemical screening in high risk PDB population due to its widespread availability and affordability. However, serum ALP screening may not have high enough clinical sensitivity and specificity in the diagnosis of early or small Paget disease lesions21. As noted in our cohort of subjects, only one individual with active PDB were identified with an elevated ALP level.
Interestingly two myopathy patients were diagnosed with PDB as a result of the testing at the time of visit despite having normal ALP levels and no bone pain. PDB was observed in the left parietal calvarium in ID 6 while PDB was observed in the right ischium and acetabulum in ID 10 (Fig. 4). Subsequent plain radiograph imaging was performed after increased bone scan focal uptakes were observed. The increased radiotracer uptake of the affected bones found in our study such as the spine, pelvis, and skull are of typical distribution as seen in conventional PDB. Among the six patients diagnosed with PDB, three patients showed increased activity in the skull, none of which showed the ring-like pattern on the margin of the lesions typically seen in conventional PDB. Three MSP1 patients also had increased uptake of the lower extremities as was noted in other types of MSPs26,27. On radiographs, we observed both lytic and blastic or sclerotic lesions on the skull radiographs for patient 1 (ID 1), and more sclerotic appearance of the skull radiographs on patient 6 (ID 6).
Although these may be a consequence of aging, osteoarthritis is seen in approximately 73% of those suffering from general PDB, and commonly manifests in joints adjacent to the Pagetic bone16. In our study degenerative bone changes were noted on bone scan in three patients with PDB, three myopathy alone and one pre-symptomatic carrier. The degenerative changes were seen in the bone or a nearby bone in three patients in which PDB was seen. PDB may cause subchondral bone expansion leading to the narrowing of joint spaces and increased joint pressure promoting cartilage necrosis17. Our cohort also showed increased activity in the thoracic region: ribs, manubrium, and the sterno/acromio clavicular joints, which are commonly attributable to degenerative diseases28,29. The increased mechanical joint stress in osteoarthritis can induce osteoblastic response leading to asymmetrical Tc-99m uptake18. Studies have shown that trauma from sports, especially leading to non-union fractures can show persistent radiotracer uptake30,31. In addition, areas of repeated stress can show increased uptake such as the patellar tendon insertion to the tibial tuberosity32. Two of our patients (ID 7–8) reported trauma history however increased uptake in the relevant affected regions were excluded in the analysis.
There is currently no definitive treatment for the neuromuscular and neurological manifestations of MSP1. However, treatment with bisphosphonates such as alendronate and zoledronic acid has been noted to provide relief from bone pain in the majority of patients with PDB14,33,34. In our cohort, only one patient (ID 4) reported to be taking bisphosphonates who had five active PDB regions and an ALP level of 234 IU/L. Bone scans in addition to ALP levels can be effective in monitoring bone osteoclast reactivation in patients following treatment34. Future studies including bone-specific alkaline phosphatase (BALP), procollagen type I intact N-terminal propeptide (P1NP), and collagen type 1 C-telopeptide (CTX) may provide data on their usefulness in diagnosing and monitoring of PDB in this at-risk population13. Treatment comprises of bisphosphonates including zoledronic acid, pamidronate, risedronate and alendronic acid, and can help in mitigating symptoms of bone pain. Treatment response and recurrence can be monitored with raised ALP or increased uptake on bone scan. Vitamin D deficiency should be corrected prior to administration of bisphosphonates, to mitigate against hypocalcemia14. The ZiPP trial in presymptomatic patients with SQSTM1 pathogenic variants will determine if early treatment with zoledronic acid can prevent PDB35. A similar treatment trial in VCP MSP1 patients has the potential to prevent the occurrence of PDB in this at-risk population.
Our study has some limitations. Due to the rarity of disease and burden of travel to our center, the cohort size is relatively small. In addition, MSP1 patients have significant variability and expressivity. Nevertheless, VCP MSP1 is a rare disease, and this series represents a significant addition to the literature. Recent studies have reported intergenerational change of PDB phenotype in general population and those with SQSTM1 mutations20,36. In our study, we did not characterize these changes as our cohort is small although we anticipate a subsequent study characterizing the severity of PDB phenotype in MSP1 patients. As a technique, nuclear imaging can be time-consuming, but is useful in screening asymptomatic bone sites due to its high sensitivity.
Positive findings on bone scans, in conjunction with the clinical history and assessment and correlation with other imaging modalities such as skeletal radiography is considered very suspicious of the diagnosis of PDB in this high-risk population. Our study shows that bone scan imaging can be used in conjunction with clinical history, laboratory testing and plain radiographs to ascertain affected Pagetic bones in asymptomatic patients with MSP1 due to its high detection sensitivity. Early surveillance, treatment, and monitoring are paramount to prevent the serious complications of PDB in MSP1 patients.
Data availability
All data presented in this study are available from the corresponding author on request.
References
Schiava, M. et al. Genotype–phenotype correlations in valosin-containing protein disease: A retrospective muticentre study. J. Neurol. Neurosurg. Psychiatry 93(10), 1099–1111. https://doi.org/10.1136/jnnp-2022-328921 (2022).
Al-Obeidi, E. et al. Genotype-phenotype study in patients with VCP valosin-containing protein mutations associated with Multisystem Proteinopathy. Clin. Genet. 93(1), 119–125. https://doi.org/10.1111/cge.13095 (2018).
Columbres, R. C. A. et al. Novel variants in the VCP gene causing Multisystem Proteinopathy 1. Genes. 14(3), 676. https://doi.org/10.3390/genes14030676 (2023).
Kimonis, V. E. et al. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet. Med. Off. J. Am. Coll. Med. Genet. 2(4), 232–241. https://doi.org/10.1097/00125817-200007000-00006 (2000).
Kimonis, V. Inclusion body myopathy with paget disease of bone and/or frontotemporal dementia. In: GeneReviews® (eds Adam, M. P., Ardinger, H. H., Pagon, R. A. et al.) (University of Washington, Seattle, 1993). Accessed 19 Jul 2021. http://www.ncbi.nlm.nih.gov/books/NBK1476/
Valenzuela, E. N. & Pietschmann, P. Epidemiology and pathology of Paget’s disease of bone–a review. Wiener Med. Wochenschr. 167(1–2), 2–8. https://doi.org/10.1007/s10354-016-0496-4 (2017).
Sabharwal, R. et al. An insight in to Paget’s disease of bone. Niger. J. Surg. 20(1), 9–15. https://doi.org/10.4103/1117-6806.127098 (2014).
Ralston, S. H. & Layfield, R. Pathogenesis of Paget disease of bone. Calcif. Tissue Int. 91, 97–113. https://doi.org/10.1007/s00223-012-9599-0 (2012).
Hsu, E. Paget’s disease of bone: Updates for clinicians. Curr. Opin. Endocrinol. Diabetes Obes. 26(6), 329–334. https://doi.org/10.1097/MED.0000000000000503 (2019).
Merlotti, D. et al. Mutation of PFN1 gene in an early onset, polyostotic Paget-like disease. J. Clin. Endocrinol. Metab. 105(8), dgaa252. https://doi.org/10.1210/clinem/dgaa252 (2020).
Wei, Z. et al. Mutations in profilin 1 cause early-onset Paget’s disease of bone with giant cell tumors. J. Bone Miner. Res. 36(6), 1088–1103. https://doi.org/10.1002/jbmr.4275 (2021).
Ralston, S. H. & Albagha, O. M. Genetics of Paget’s disease of bone. Curr. Osteoporos. Rep. 12(3), 263–271. https://doi.org/10.1007/s11914-014-0219-y (2014).
Shankar, S. & Hosking, D. J. Biochemical assessment of Paget’s disease of bone. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 21(Suppl 2), P22–P27. https://doi.org/10.1359/jbmr.06s204 (2006).
Korb, M. et al. Development of a standard of care for patients with valosin-containing protein associated Multisystem Proteinopathy. Orphanet. J. Rare Dis. 17, 23. https://doi.org/10.1186/s13023-022-02172-5 (2022).
Kimonis, V. E. et al. Clinical studies in familial VCP myopathy associated with Paget disease of bone and frontotemporal dementia. Am. J. Med. Genet. A. 146A(6), 745–757. https://doi.org/10.1002/ajmg.a.31862 (2008).
Shaker, J. L. Paget’s disease of bone: A review of epidemiology, pathophysiology and management. Therap. Adv. Musculoskel. Dis. 1(2), 107–125. https://doi.org/10.1177/1759720X09351779 (2009).
Altman, R. D. Articular complications of Paget’s disease of bone. Semin. Arthritis Rheum. 23(4), 248–249. https://doi.org/10.1016/0049-0172(94)90048-5 (1994).
Van der Wall, H. & Fogelman, I. Scintigraphy of benign bone disease. In Seminars in Musculoskeletal Radiology https://doi.org/10.1055/s-2008-1060332 (2007)
Li, H. et al. VCP/p97 increases BMP signaling by accelerating ubiquitin ligase Smurf1 degradation. FASEB J. 33, 2928–2943. https://doi.org/10.1096/fj.201801173R (2019).
Dessay, M. et al. Clinical phenotype of adult offspring carriers of the pPro392Leu mutation within the SQSTM1 gene in Paget’s disease of bone. Bone Rep. 13, 100717. https://doi.org/10.1016/j.bonr.2020.100717 (2020).
Ralston, S. H. et al. Diagnosis and management of Paget’s disease of bone in adults: A clinical guideline. J. Bone Miner. Res. 34(4), 579–604. https://doi.org/10.1002/jbmr.3657 (2019).
Farpour, F. et al. Radiological features of Paget disease of bone associated with VCP myopathy. Skeletal. Radiol. 41(3), 329–337. https://doi.org/10.1007/s00256-011-1193-4 (2012).
Cook, S. J. & Wall, C. Paget’s disease of bone: A clinical update. Aust. J. Gen. Pract. 50(1–2), 23–29. https://doi.org/10.31128/AJGP-10-20-5690 (2021).
Choy, N., Wang, S., Abbona, P., Leffler, D. & Kimonis, V. Severe cardiomyopathy associated with the VCP p.R155C and c.177_187del MYBPC3 gene variants. Eur. J. Med. Genet. 65(6), 104480. https://doi.org/10.1016/j.ejmg.2022.104480 (2022).
Lombardi, A. F., Aihara, A. Y., Fernandes, A. D. R. C. & Cardoso, F. N. Imaging of Paget’s disease of bone. Radiol. Clin. North Am. 60(4), 561–573. https://doi.org/10.1016/j.rcl.2022.02.005 (2022).
Waggoner, B. et al. Heterogeneity in familial dominant Paget disease of bone and muscular dystrophy. Am. J. Med. Genet. 108(3), 187–191 (2002).
Kim, H. J. et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause Multisystem Proteinopathy and ALS. Nature 495(7442), 467–473. https://doi.org/10.1038/nature11922 (2013).
Fink-Bennett, D. M. & Shapiro, E. E. The angle of louis. A potential pitfall (“louie’s hot spot”) in bone scan interpretation. Clin. Nucl. Med. 9(6), 352–354 (1984).
Fogelman, I. & Maisey, M. An Atlas of Clinical Nuclear Medicine. Dunitz/C.V. Mosby.
Gandhi, S. J. & Rabadiya, B. Bone scan in detection of biological activity in nonhypertrophic fracture nonunion. Indian J. Nucl. Med. 32(4), 326–329. https://doi.org/10.4103/ijnm.IJNM_50_17 (2017).
Günalp, B. et al. Role of bone scanning in the management of non-united fractures: A clinical study. Eur. J. Nucl. Med. 19, 845–847. https://doi.org/10.1007/BF00168158 (1992).
Gnanasegaran, G., Cook, G., Adamson, K. & Fogelman, I. Patterns, variants, artifacts, and pitfalls in conventional radionuclide bone imaging and SPECT/CT. In Seminars in Nuclear Medicine Vol. 39, No. 6, pp. 380–395 (WB Saunders, 2009).
Tao, X. et al. Clinical characteristics and pathogenic gene identification in Chinese patients with Paget’s disease of bone. Front. Endocrinol. 13, 850462. https://doi.org/10.3389/fendo.2022.850462 (2022).
Durgia, H. et al. Response to zoledronic acid in patients with active Paget’s disease of bone: A retrospective study. Indian J. Endocrinol. Metab. 23(1), 117. https://doi.org/10.4103/ijem.IJEM_327_18 (2019).
Cronin, O. et al. Zoledronate in the prevention of Paget’s (ZiPP): Protocol for a randomised trial of genetic testing and targeted zoledronic acid therapy to prevent SQSTM1-mediated Paget’s disease of bone. BMJ Open 9(9), e030689. https://doi.org/10.1136/bmjopen-2019-030689 (2019).
Michou, L. & Orcel, P. The changing countenance of Paget’s Disease of bone. Jt. Bone Spine 83(6), 650–655. https://doi.org/10.1016/j.jbspin.2016.02.011 (2016).
Acknowledgements
The authors would like to thank the patients, their families/relatives and their healthcare providers for their participation and contribution to these studies.
Funding
This study was supported by the National Institute of Health (AR AR050236 R01 and R56 to VK), and the Institute of Clinical and Translational Science (ICTS), University of California Irvine.
Author information
Authors and Affiliations
Contributions
Study design, R.C.A.C. and V.K. First draft preparation, R.C.A.C., S.D., L.G. and V.K. Data curation and analysis, R.C.A.C., S.D., L.G. and V.K. All authors revised the paper critically for intellectual content and approved the final version. All authors agree to be accountable for the work and to ensure that any questions relating to the accuracy and integrity of the paper are investigated and properly resolved.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Columbres, R.C.A., Din, S., Gibbs, L. et al. Bone scan findings of Paget’s disease of bone in patients with VCP Multisystem Proteinopathy 1. Sci Rep 14, 5917 (2024). https://doi.org/10.1038/s41598-024-54526-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54526-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
