
Abstract
Fungal pathogens are considered as serious factors for deadly diseases and are a case of medical concern. Invasive fungal infections also complicate the clinical course of COVID-19, leading to a significant increase in mortality. Furthermore, fungal strains' multidrug resistance has increased the demand for antifungals with a different mechanism of action. The present study aimed to identify antifungal compounds targeting yeast topoisomerase II (yTOPOII) derived from well-known human topoisomerase II (hTOPOII) poisons C-1305 and C-1311. Two sets of derivatives: triazoloacridinones (IKE1-8) and imidazoacridinones (IKE9-14) were synthetized and evaluated with a specific emphasis on the molecular mechanism of action. Our results indicated that their effectiveness as enzyme inhibitors was not solely due to intercalation ability but also as a result of influence on catalytic activity by the formation of covalent complexes between plasmid DNA and yTOPOII. Lysine conjunction increased the strength of the compound's interaction with DNA and improved penetration into the fungal cells. Triazoloacridinone derivatives in contrast to starting compound C-1305 exhibited moderate antifungal activity and at least twice lower cytotoxicity. Importantly, compounds (IKE5-8) were not substrates for multidrug ABC transporters whereas a derivative conjugated with lysine (IKE7), showed the ability to overcome C. glabrata fluconazole-resistance (MIC 32–64 µg mL−1).
Similar content being viewed by others
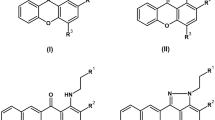

Introduction
In the last few years increasing importance is attached to problems caused by fungal pathogens. The diagnosis of fungal infections is not easy, and the therapy is complicated1. The fungistatic effect of fluconazole and its frequent use led to the increased resistance of Candida spp. strains2,3. Among 5 million fungal species, around 300 cause human diseases4. Fungemia is mainly caused by Candida spp. (60–80% cases)5, of which C. albicans constitutes 35% to 60% of isolates6,7. The C. albicans strain is a natural microflora in humans, but under certain conditions, it can pose a serious threat to health due to the possibility of causing opportunistic infections. There are no available vaccines against C. albicans infection and the preventability of invasive candidiasis is low. The C. albicans strain was included in the Critical Priority Group on the first WHO fungal priority pathogens list issued to highlight the problem of growing fungal resistance and to strengthen the global response to fungal infections8. Several antifungal compounds are highly effective in some cases, however, they have limitations in use: long half-life, widespread in tissues, and high toxicity9. The antifungal medications currently available for the treatment of invasive fungal infections belong to only four drug classes (azoles, echinocandins, pyrimidines, and polyenes), whereas two of which target ergosterol, a component of the fungal cell membrane10. Right now, there are known cases of fungi resistant to all approved oral drugs11. Therefore, a novel antifungal drug with different mechanisms of action is urgently needed.
Research indicates that fungal topoisomerases are crucial for the survival of some fungal strains, mammalian and fungal enzymes show diversity in terms of molecular structures and properties as well as differences in sensitivity to some human topoisomerase II (hTOPOII) inhibitors12. For example, previous research showed that Candida TOPOII demonstrates less sensitivity for m-AMSA (Amsacrine; 4′-(9-Acridinylamino)methanesulfon-m-anisidide, acridine-compound) and etoposide than human enzyme while podophyllotoxins are inactive against mammalian enzyme but inhibit the activity of fungal TOPOII13. Also, our results obtained for antitumor imidazoacridinone C-1311 and triazoloacridinone C-1305, whose molecular mechanism of action is related to hTOPOII poisoning14,15, indicated that the fungal enzyme is much less sensitive to both compounds than the human one16. Moreover, our preliminary studies showed that acridine/acridone derivatives may effectively inhibit yeast TOPOII (yTOPOII), which could be related to their antifungal activity16,17,18. However, due to the existence of fungal cell wall and differences in membrane permeability, some yTOPOII inhibitors like 5-[(2-aminoethyl)amino]-8-hydroxy-6H-imidazo[4,5,1-de]acridin-6-one (previous described as Compound 116), C-1305, C-1311 cannot pass through the fungal cell wall and access their molecular target inside the cell. With this in mind, we have decided to modify the structures of the previously studied compounds16 including C-1305, C-1311, as well as C-1330, and more thoroughly analyzed the properties of a new group of triazoloacridinones (IKE1-8) C-1305 derivatives and imidazoacridinones (IKE9-14) C-1311 derivatives in terms of their influence on yTOPOII activity as well as their ability to affect fungal growth. Modifying the structure of a potential drug compound by conjugation with lysine is also proposed as an efficient approach to increase the ability to penetrate the fungal cell and decrease cytotoxicity19,20.
Results
Our preliminary studies indicated that targeting yTOPOII by acridine/acridone derivatives might not always result in an antifungal effect. Unfortunately, due to the presence of a fungal cell wall and variances in membrane permeability, certain potent yTOPOII inhibitors face challenges in reaching their intended molecular target16,17,18. Unfavorable properties can be eliminated by modifying the lead structure for example by changing the lipophilicity or combining it with amino acids, which increases their bioavailability and decreases cytotoxicity17,19,20,21. An example of such an approach is the modification of nitroacridine derivative by lysine moiety that led to increased activity of the compound against fluconazole-resistant fungal cells17. Thus, the compounds analyzed by us exhibit variations in lipophilicity (having OCH3 or CH3 R groups instead of OH, as seen in C-1305 and C-1311 compounds, that cannot penetrate the fungal cell) and feature different substituents on the nitrogen atom (R1, R2: ethyl groups rather than methyl, similar to compound C-1330 which effectively enters the fungal cell22). Additionally, some of them, incorporating lysine conjugation, are proposed as an efficient strategy to enhance fungal cell penetration while minimizing cytotoxicity. The structures of derivatives that were synthesized in this study are divided into two groups: C-1305 analogs (triazoloacridinones group I) and C-1311 derivatives (imidazoacridinones group II) (Table 1).
Synthesis
Triazolo- and imidazoacridinone derivatives with 1-diaminoalkyl substituent with two or three methylene groups between two amino centers, and with the distal amino group bearing Lys were synthesized. The first group, 5-[(aminoalkyl)amino]-8-substituted-6H-[1,2,3]triazolo[4,5,1-de]acridin-6-one presented in Figure S1 (collected in Supplementary Information, SI) (IKE1-4) and C-1305 were prepared according to the previously reported procedure23,24. Acridone derivatives with a side chain with an unsubstituted amino terminal group (IKE5-6), were obtained by condensation of 1-chloroacridinone derivatives with an excess of the corresponding amine in DMA (N,N-Dimethylacetamide), and at the next stage these compounds were condensed with protected amino acid in the presence of coupling agents in DMSO/TEA (Triethylamine) and then deprotection of nitrogen atoms (IKE7-8).
The second group of synthesized compounds were imidazoacridinone derivatives (IKE9-14) presented in Figure S1 (collected in Supplementary Information, SI). Compounds C-1311, C-1330, IKE9-10, and IKE12-13 were obtained from 1-chloro-7-substituted-4-nitro-9(10H)-acridinone, through the substitution with an excess of aliphatic amines, reduction and cyclization of the resulting derivatives. This method of synthesis was described previously25,26. Compounds IKE10 and IKE13 were used as a starting material for the synthesis of derivatives IKE11 and IKE14. The procedure was the same as for the analogous triazoloacridinone derivatives. All derivatives, except for C-1305, C-1311, C-1330, and IKE12 are the newly synthesized ones14,15,16.
Inhibition of yeast topoisomerase II in vitro
Inhibition of the catalytic activity of yeast topoisomerase II was analyzed with the use of two methods: the relaxation of supercoiled plasmid DNA as well as decatenation of kinetoplast DNA in vitro. With the use of the second method, we exclude the possibility of tricking the activity of the enzyme by drug only through direct drug/DNA interaction (intercalation of the drug to DNA) which can result in the unwinding of DNA27.
The relaxation of supercoiled plasmid DNA by yeast enzyme was studied in the presence of different concentrations of compounds with the use of a gel-based assays that allow to determine IC50 (Table 2).
The IC50 values determined for IKE1-IKE14 against yeast topoisomerase II were compared with those determined for starting compounds C-1305 and C-1311 , which are well-known human topoisomerase II (hTOPOII) poisons that inhibit the enzyme through the formation of TOPO II-stabilizing complexes, along with intercalation with DNAs14,15. The ability of both derivatives to inhibit yTOPOII relaxation activity was also previously established16. The most effective inhibitors towards yeast topoisomerase II synthesized by us were IKE5 from the group (I) and IKE13 from the group (II). As previously reported, IC50 for C-1305 and C-1311 against human topoisomerase II-mediated relaxation was determined at a concentration of 2.5 μM and 6.5 μM, respectively14,15. In our hands, the yeast equivalent of this enzyme was much less sensitive to both anticancer compounds (Table 2). Moreover, yTOPOII-relaxation inhibitory activities of the compounds IKE5 and IKE13 were found to be in accordance with the results when kinetoplast DNA was used as a substrate for decatenation (Fig. 1A). Both compounds IKE5 and IKE13 were more effective in inhibiting decatenation than the other derivatives (Fig. 1B). Although no complete inhibition of decatenation activity was observed, in the presence of compounds IKE1, IKE3, IKE7, IKE8, IKE9, and IKE12 as well as IKE14 more decatenation products (partially decatenated mini circles) were observed than for positive control. Results indicated some decatenation activity perturbations.

Inhibition of the catalytic activity of purified yeast topoisomerase II by selected compounds as measured decatenation of kinetoplast DNA (catenated). The influence of (a) IKE5, IKE13 at 1, 10, 50, 150 µM and (b) IKE1, IKE3, IKE7, IKE8, IKE9, IKE12 and IKE14 at 50, 150 µM concentration on the yeast topoisomerase II decatenation ability were tested. Kinetoplast DNA (-yTOPOII) was decatenated by yeast topoisomerase II in the absence (+ yTOPOII) or presence of etoposide (lane 3, 150 µM) or selected compounds. DNA was separated in a 1% agarose gel. The data shown are typical of three independent experiments. The gels were cropped to improve the clarity and conciseness of the presentation. Original gels are presented in Figure S4 (SI).
Studies using purified topoisomerase II from yeast confirmed the ability of IKE5 and IKE13 to inhibit the catalytic activity of topoisomerase II.
To analyze whether newly synthesized compounds, similar to the 5-[(2-aminoethyl)amino]-8-hydroxy-6H-imidazo[4,5,1-de]acridin-6-one (IKE12) and C-1311 (starting compound)16, may also influence the formation of covalent DNA–yeast topoisomerase II complexes we have performed linearization assay tests (Fig. 2).

The influence of IKE3, IKE5, IKE7, IKE9, IKE12, IKE13, and IKE14 on the formation of covalent DNA-yeast topoisomerase II complexes. Supercoiled pBR322 plasmid DNA (lane 2, -yTOPOII) was incubated with purified yeast topoisomerase II in the absence (lane 1, + yTOPOII) or in the presence of 200 µM m-AMSA (lane 4) or with 0.25 µM or 10 µM of selected compounds (lanes 5–17). After forming DNA/yeast topoisomerase II complexes, proteinase K was used for digestion. Subsequently, the various topological forms of DNA were separated in a 1.2% agarose gel containing 0.1 µg mL−1 ethidium bromide. The data shown are typical of three independent experiments. Lane 3, linearized pBR322 DNA. SC, supercoiled DNA; R, relaxed DNA; L, linear DNA; N, nicked circular DNA. The gel was cropped to improve the clarity and conciseness of the presentation. Original gels are presented in Supplementary Fig. S4.
As we demonstrated earlier, compounds IKE12 and C-1311 (the starting compound) with a similar structure to the investigated, impact the formation of covalent complexes between pBR322 plasmid DNA and yeast topoisomerase II16. The same mode of action is also known for compound C1305 (the starting compound), inhibiting human topoisomerase II15. As it was expected all analyzed by us compounds indicated the formula of yeast enzyme-stabilizing complex. However, this effect was reduced compared to IKE1216.
Selected compound’s interaction with DNA
The ambiguous results obtained for decatenation assay prompted us to conduct additional experiments on the possible compounds intercalation role in yeast topoisomerase II inhibition by performing DNA unwinding assays (Fig. 3).

DNA Unwinding Assay. Supercoiled (or relaxed) pBR322 plasmid DNA (lane 1,—Wheat germ TOPO I) was incubated in the absence (lane 2, + Wheat germ TOPO I) or presence of analyzed compounds IKE3, IKE5, IKE7, and IKE13 prior to incubation with wheat germ TOPO I. The ability to intercalate into DNA was investigated. m-AMSA was used to show the ability of intercalation, etoposide was used as an example of a non-intercalator. SC, supercoiled DNA; R, relaxed DNA; T, DNA topoisomers. DNA was separated in a 1% agarose gel. The data shown are typical of three independent experiments. The gels were cropped to improve the clarity and conciseness of the presentation. Original gels are presented in Supplementary Fig. S4.
As indicated in Fig. 3, selected compounds IKE3, IKE5, IKE7, and IKE13 inhibited TOPO I-mediated DNA unwinding in a dose-dependent manner. Both derivatives' behaviour seemed to be the same as for m-AMSA, an eminent DNA intercalating human TOPOIIα poison29. Thus, the probable mode of action of analyzed by us derivatives in the context of the importance of the intercalation step could be similar.
Due to the probable role of intercalation of analyzed compounds into DNA in enzyme inhibition, we have decided to perform a more detailed analysis of selected compounds' interaction with calf thymus DNA (CT-DNA) by UV–Vis absorption spectroscopy. The effect of varying concentrations of CT-DNA on the absorption spectra of IKE1 and IKE8 is shown in Fig. 4 and for the other studied compounds IKE3, IKE7, IKE9, IKE14 is shown in Figs. S14–S15 of SI. With an increasing concentration of CT-DNA, the absorption bands of the compounds were affected, resulting in the tendency of hypochromism. The strong absorption of these compounds in the near UV region (240–325 nm) is attributed to the aromatic system30. The binding compounds to CT-DNA caused hypochromism. This hypochromic effect is thought to be due to the interaction between the electronic states of the intercalating chromophore and those of the DNA bases and caused the conformational changes of CT-DNA31,32,33 Hypochromism results from the contraction of the helix of CT-DNA. It was observed that a continuous decrease in the absorbance of acridines was followed by the gradually increasing concentration of CT-DNA in the solution. A slight bathochromic shift (3–5 nm) was observed with increasing concentrations of CT-DNA for IKE1, IKE8, and IKE14 (Figs. S7, S10 and S13 of SI). These spectral characteristics (Table S1 of SI) could indicate their stronger binding to CT-DNA than those observed for IKE7, IKE3, and IKE9, respectively. Changes in the position of analyzed compounds' absorption bands as a result of their interaction with DNA are presented in Table S1.

Absorption spectra of IKE1 (a) and IKE8 (b) in the presence of increasing amounts of CT-DNA. Arrows indicate that absorbance changes upon increasing CT-DNA concentrations. Inset: plot of Ao/(A–Ao) = f(1/[DNA]) established as a result of both compounds’ interactions through titration with CT-DNA in Tris–HCl buffer (5 mM/50 mM NaCl; pH 7.43).
Taking all results together for compounds analyzed by us we can observe intercalator-induced topoisomerase II-mediated DNA cleavage.
Antifungal activity
IKE1-IKE14 derivatives were tested for their in vitro antifungal activity against five reference strains. Minimal inhibitory concentrations (MICs) of the studied compounds determined by the microplate serial dilution method are shown in Table 3. To establish the possible mode of action we analyzed the killing activity and determined the minimal fungicidal concentrations (MFCs) for selected derivatives as well as performed some flow cytometry analysis with propidium iodide uptake as a cell viability indicator (Fig. 5a).

(a) Representative histograms and quantification of flow cytometry analysis of fungi labeled with 20 µg mL−1 of propidium iodide to detect membrane permeability in S. cerevisiae ATCC 9763 cells after treatment with IKE7, IKE14, C-1305, and C-1311 at concentration 64 µg mL−1. Error bars represent the mean ± (SD) of the data collected from three independent events. To determine the statistical significance, a one-way analysis of variance (ANOVA) test was conducted. (b) Fluorescence microscopic analysis of uptake and accumulation of IKE7 and C-1305 in S. cerevisiae ATCC 9763 cells. Cells were suspended in phosphate-buffered saline and incubated in the presence of compounds in 100 µM concentration for an appropriate period. Scale bars correspond to 20 µm. (c) Representative histograms and quantification of flow cytometry analysis for the accumulation of IKE7 and C-1305 at concentration 64 µg mL-1 in S. cerevisiae ATCC 9763 cells. Incubation times of 15 and 60 min were considered. Error bars represent the mean ± (SD) of the data collected from three independent events. To assess statistical significance, a one-way analysis of variance (ANOVA) test was performed.
In contrast to initial compounds C-1305 and C-1311 some newly synthetized derivatives exhibited moderate antifungal activity. Comparison of triazoloacridinones (IKE1-8) and imidazoacridinones (IKE9-14) antifungal activity showed better activity of the former. Our results also indicated that the mode of action of those compounds is fungicidal. The most active compounds from the first group were chosen to test antifungal activity against C. glabrata clinical strains, fluconazole-resistant34. Results showed that the addition of l-lysine moiety to the structure of IKE7 compound improved its antifungal activity and allowed to overcome fluconazole resistance (Table 4).
Our results also indicated that the higher antifungal activity of IKE7 against fluconazole-resistant clinical strains might be related to the inability to efflux drugs by multidrug-resistance pumps. We used S. cerevisiae AD mutants modified by deleting seven multi drug resistance (MDR) transporters and their transformants overexpressing genes encoding CDR1, CDR2, and MDR1 C. albicans drug-efflux pumps35,36. Derivatives without lysine conjunction IKE1-3 did not show any activity against S. cerevisiae AD mutants at the tested concentration whereas MIC values for IKE5, 6 were in the range of 64–32 µg mL−1. Interestingly, the MIC values for compounds modified by lysine conjunction, IKE7, and IKE8, were lower. Furthermore, both the parental strain S. cerevisiae AD1-8u− and the pump-expressing strains exhibited similar susceptibility to IKE7 and IKE8 compounds, indicating that the antifungal activity was not affected by the expression of certain efflux pumps in the strains (Table 5).
Further analysis indicated that the antifungal activity of IKE7 was related to compound accumulation in the cells (Fig. 5b,c).
The influence on fungal virulence factors
The ability of fungal cells to perform morphological transformation as well as biofilm formation on abiotic (prostheses, catheters) and biotic (mucous membranes) surfaces are important factors for virulence37,38. To check whether the presence of analyzed compounds affects C. albicans ATCC 10231 yeast-to-mycelium transformation, fungal cells were incubated under conditions stimulating this process and in the presence of selected derivatives. The results were evaluated by light microscopy, only hyphal inhibitors (IKE1, IKE3, IKE7, IKE9, IKE14) and controls are shown (Fig. S16 of SI). Our results indicated that compounds IKE1, IKE3, and IKE7 were able to completely block hyphal formation in Spider medium. To examine the inhibitory effects against C. albicans biofilms, we used a microtiter-plate biofilm model (Fig. S17 of SI). Compounds C-1330, IKE1, IKE2, and IKE9 were the only ones that inhibited C. albicans ATCC10231 biofilm formation. Surprisingly, although synthesized by us derivatives exhibited no antifungal activity against C. albicans (MICs > 64 μg mL−1, Table 3) our results indicated that in the presence of selected compounds, some candidal virulence factors might be strongly affected.
Cytotoxicity of selected derivatives
To understanding the balance between the effectiveness of the analyzed compounds against fungi and their impact on non-target human cells, selected derivatives were screened for their in vitro antiproliferative activity against mammalian cell lines, including normal human embryonic cells (HEK293), and human liver cancer cells (HepG2) using colorimetric MTT ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. The results were calculated after 72 h of treatment and expressed as a half-inhibitory growth concentration (EC50) (Table 6).
Compounds differ in their cytotoxic activity against human cells. Initial compounds, C-1305, C-131116 showed the greatest cytotoxicity whereas the most effective antifungal compound synthetized by us, IKE7 exhibited the lowest suppressive effect on HEK293 cells. To estimate the selectivity concerning mammalian cells mycostatic selectivity index (MSI) for S. cerevisiae cells was calculated as the ratio of EC50–MIC90 values after converting the MIC90 value to micromolar concentrations. The results are presented in Table 7.
Positive mycostatic selectivity index values (MSI = EC50/MIC90) were obtained for all selected triazoloacridinone derivatives. The highest level of selectivity was obtained for IKE7 and IKE8.
Discussion
The emergence of drug resistance among various fungal pathogens has highlighted the urgent need to explore alternative approaches to drug design. While topoisomerases II have been recognized as molecular targets for several chemotherapies, their potential as targets against fungal genera has not been explored. Moreover, the barrier of the fungal cell wall blocks the possibility of using the already discovered, effective fungal enzyme inhibitors.
The present study aimed to identify antifungal compounds targeting yeast topoisomerase II (yTOPOII) derived from human topoisomerase II (hTOPOII) poisons C-1305 and C-1311. Both starting compounds are known as cleavable, complex stabilizing drugs. Our previously published results obtained for C-1311 and IKE12 also indicated the same mechanism of action for yeast enzyme16. Accordingly to newly synthetized compounds the formation of cleavable complexes between yeast topoisomerase II and pBR322 plasmid DNA was also observed. Although all analyzed by us derivatives induced fewer cleavable complexes than m-AMSA or IKE12 it seemed that the mechanism of action could be the same. The ability of selected compounds to intercalate with DNA was confirmed by performing DNA unwinding assays as well as a detailed UV–Vis spectral analysis. Interestingly, the stimulation of the CT-DNA interaction affinity was proved in the case of derivatives with the lysine moiety. The mentioned conclusion was formulated based on the detailed comparative analyses of the results reported herein. The significantly higher values of binding constants (Kbind) established spectrophotometrically (Table 8), for derivatives IKE8, IKE7, and IKE14 bound CT-DNA in comparison to those values of Kbind determined for their analogs (IKE1, IKE3, IKE9), unequivocally prove stronger interaction mode of the first ones to bind with CT-DNA.
Based on the data collected in Table 8, the specific relation can be formulated. IKE8 derivative interacted with CT-DNA 6 times more strongly than the compound IKE1. IKE7 showed 2 times stronger affinity than IKE3. Interestingly, the Kbind values obtained for pair IKE9 and IKE14 interacted with a biomolecule, identifying 60 times stronger IKE14-CT-DNA adduct formation. UV–Vis spectroscopy experiments showed bathochromic shifts and hypochromic effects of the absorption spectra of analyzed triazolo- and imidazoacridinones after interaction with CT-DNA, indicative of IKE derivatives being deeply buried in the CT-DNA structure which was also proved by the high Kbind values determined. The constant ionic strength and pH (7.43) in the experimental procedure led to indicating that all compounds investigated behaved as CT-DNA intercalators. Due to the Kbind values reported, DNA was confirmed as a cellular target for all compounds studied. Finally, we found that the intercalation model best suited to reproduce our registered experimental UV–Vis data for all compounds was in analogy to well-known C-1305 and its affinity to CT-DNA39. Moreover, we suggested that for IKE8, IKE7, and IKE14 this mode of binding with CT-DNA is also supported electrostatically. Thus, for derivatives modified by lysine conjunction (showed higher Kbind values) the hybrid type of interactions were suggested: electrostatic as well as intercalation with CT-DNA binding. On the other hand, even 60 times stronger binding affinity with CT-DNA determined for IKE14 than IKE9 does not result in increased inhibitory activity against yTOPOII. Thus, one may conclude that intercalation ability is not the only reason for being effective as an enzyme inhibitor.
The most effective yTOPOII relaxation activity inhibitors are IKE5 and IKE13, although those compounds' antifungal effect seems to be moderate (IKE5) or very low (IKE13). Additionally IKE5 was not active against fluconazole-resistant C. glabrata cells. Preliminary structure–activity relationships (SARs) show a correlation between in vitro antifungal activities and chemical structures. Overall, compounds containing triazole ring attached to an acridinone core (triazoloacridinones) inhibited fungal growth better than the corresponding analogues contain an imidazole ring (imidazoacridinones). Moreover, the introduction of lysine moiety within the diaminoalkyl side chain of triazoloacridinones resulted in greater antifungal activity and decreased cytotoxicity. The demonstrated killing activity of the examined derivatives, which increases the likelihood of quickly defeating the infection and decreases the probability of persistent or recurrent infection and resistance development, is also their undoubted advantage. Moreover, triazoloacridinone combined with lysine moiety (IKE7) exhibited antifungal activity against C. glabrata fluconazole-resistant cells and was not recognised as a substrate by C. albicans CDR1p, CDR2p, and MDR1p efflux pumps. Lysine conjunction also resulted in increased uptake and accumulation of IKE7 in fungal cells.
Our results indicate the possibility of using triazoloacridinone derivatives conjugated with lysine moiety as efficient antifungal agents capable of overcoming fluconazole resistance in C. glabrata. However, further research is required to enhance their selectivity. Compound IKE7 is the most interesting, for which a higher level of MSI using HEK293 cells was obtained.
Materials and methods
Chemical synthesis
All compounds described in this work were obtained as hydrochlorides, with a purity of 95–99%. The confirmation of their structures relied on spectral techniques: mass spectrometry ESI–MS, as well as proton and carbon magnetic resonance. The compounds’ purity was determined through the application of thin-layer chromatography (TLC) and high-performance liquid chromatography. All of the synthesized derivatives are hygroscopic. The determination of melting points was conducted using a Stuart SMP30 capillary apparatus, and the values obtained were left uncorrected. The 1H NMR and 13C NMR spectra were captured using a Varian VXR-S spectrometer operating at a frequency of 500 MHz. Chemical shifts are expressed in δ units, measured in parts per million (ppm), and referenced downfield from internal tetramethylsilane (TMS). The NMR abbreviations utilized in this study include: br.s for broad signal, s for singlet, d for doublet, dd for doublet of doublets, t for triplet, q for quartet, quint for quintet, and m for multiplet. The elemental analyzes' outcomes for individual elements were found to be within a range of ± 0.4% compared to the corresponding theoretical values.
General procedure for the synthesis of IKE1-IKE6
In a dry 3 mL of DMA suspension, 4.7 mmol of the corresponding amine was introduced. The mixture was then stirred and heated at 60 °C for 3 h after adding 5-chloro-8-methyl-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one (1.5 mmol). Following the reaction, 115 mL of chloroform and water mixture 15:8 (by volume) was combined, and the resulting mixture was vigorously shaken. After separating the organic layer, an additional 75 mL of water was added, and the solution was acidified using l-lactic acid. Subsequently, the water layer was separated, and 1 M NaOH was added to make it basic. The solution was then extracted with chloroform. The organic layer obtained from the extraction was evaporated and dissolved in methanol. Eventually, it was prepared as dihydrochlorides by adding an ether/HCl mixture.
5-[-(3′-Dimethylamino)propylamino]-8-methyl-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one (IKE1).
3-(Dimethylamino)-1-propylamine and 5-chloro-8-methyl-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one was used;
Yield 72%; m.p. 180–181 °C;
1H NMR (DMSO-d6) δ: 10.98 (br.s, 1H, NH+), 9.26 (t, J = 5.8 Hz, 1H, 5-NH), 8.25–8.29 (m, 2H), 8.04 (s, 1H), 7.76 (d, J = 8.3 Hz, 1H), 7.16 (d, J = 9.2 Hz, 1H), 3.66 (q, J = 6.7 Hz, 2H, NHCH2CH2), 3.18 (m, 2H, CH2CH2N(CH3)2), 2.76 (s, 6H, N(CH3)2), 2.51 (s, 3H, CH3Ar), 2.12 (quint, J = 7.3 Hz, 2H, NHCH2CH2CH2N(CH3)2).
13C NMR (DMSO-d6) δ: 177.42, 152.73, 136.94, 135.61, 135.42, 133.16, 132.00, 129.33, 127.46, 124.90, 115.73, 112.41, 99.84, 54.51, 42.43, 34.48, 24.31, 21.40.
ESI–MS m/z: [M + H]+ calcd. for C19H21N5O1 335.4, found 336.2
HPLC 96.34%
5-[-(3′-Diethylamino)propylamino]-8-methyl-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one (IKE2).
3-(Diethylamino)-1-propylamine and 5-chloro-8-methyl-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one was used;
Yield 69%; m.p. 194–195 °C;
1H NMR (DMSO-d6) δ: 10.42 (br.s, 1H, NH+), 9.37 (t, J = 6.3 Hz, 1H, 5-NH), 8.39 (d, J = 8.3 Hz, 1H), 8.35 (d, J = 9.3 Hz, 1H), 8.19 (s, 1H), 7.84 (d, J = 9.7 Hz, 1H), 7.25 (d, J = 9.3 Hz, 1H), 3.69 (q, J = 6.8 Hz, 2H, NHCH2CH2), 3.10–3.18 (m, 6H, CH2CH2N(CH2CH3)2), 2.54 (s, 3H, CH3Ar), 2.10 (quint, J = 7.3 Hz, 2H, NHCH2CH2CH2N(CH2CH3)2), 1.22 (t, J = 7.3 Hz, 6H, NHCH2CH2CH2N(CH2CH3)2).
13C NMR (DMSO-d6) δ: 177.51, 155.78, 136.99, 135.68, 135.45, 133.12, 132.05, 129.40, 127.54, 124.95, 115.78, 112.44, 99.87, 48.31, 46.65, 29.49, 23.54, 21.40, 8.95.
ESI–MS m/z: [M + H]+ calcd. for C21H25N5O1 363.4, found 364.3
HPLC 98.98%
5-[-(3′-Dimethylamino)propylamino]-8-methoxy-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one (IKE3).
3-(Dimethylamino)-1-propylamine and 5-chloro-8-methoxy-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one was used;
Yield 77%; m.p. 190–191 °C;
1H NMR (DMSO-d6) δ: 10.82 (br.s, 1H, NH+), 9.29 (t, J = 6.1 Hz, 1H, 5-NH), 8.36 (d, J = 8.8 Hz, 1H), 8.30 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 2.9 Hz, 1H), 7.56 (dd, J1 = 2.9 Hz, J2 = 8.8 Hz, Hz, 1H), 7.18 (d, J = 9.3 Hz, 1H), 3.93 (s, 3H, OCH3), 3.68 (q, J = 6.8 Hz, 2H, NHCH2CH2), 3.18 (m, 2H, CH2CH2N(CH3)2), 2.77 (s, 6H, CH2CH2N(CH3)2), 2.12 (quint, J = 7.8 Hz, 2H, NHCH2CH2CH2N(CH3)2).
13C NMR (DMSO-d6) δ: 176.89, 158.16, 152.77, 135.33, 131.39, 129.39, 129.30, 126.24, 122.43, 117.42, 112.27, 109.40, 99.87, 56.15, 54.53, 42.46, 24.29.
ESI–MS m/z: [M + H]+ calcd. for C19H21N5O2 351.4, found 352.1
HPLC 96.85%
5-[-(3′-Diethylamino)propylamino]-8-methoxy-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one (IKE4)
3-(Diethylamino)-1-propylamine and 5-chloro-8-methoxy-6H-[1–3]-triazolo[4,5,l-de] acridin -6-one was used;
Yield 52%; m.p. 232–233 °C;
1H NMR (DMSO-d6) δ: 10.13 (br.s, 1H, NH+), 9.37 (m, 1H, 5-NH), 8.44 (d, J = 8.8 Hz, 1H), 8.36 (d, J = 8.8 Hz, 1H), 7.80 (d, J = 2.0 Hz, 1H), 7.62 (dd, J1 = 2.4 Hz, J2 = 8.8 Hz, Hz, 1H), 7.24 (d, J = 8.8 Hz, 1H), 3.95 (s, 3H, OCH3), 3.60–3.71 (m, 2H, NHCH2CH2), 3.09–3.16 (m, 6H, CH2CH2N(CH2CH3)2), 2.08–2.11 (m, 2H, NHCH2CH2CH2N(CH2CH3)2), 1.22 (t, J = 7.3 Hz, 6H, NHCH2CH2CH2N(CH2CH3)2).
13C NMR (D2O) low solubility δ: 174.19, 156.35, 151.32, 132.78, 127.92, 129.30, 126.07, 123.07, 115.36, 111.76, 106.57, 96.12, 55.00, 48.88, 39.54, 23.01, 8.22.
ESI–MS m/z: [M + H]+ calcd. for C21H25N5O2 379.4, found 380.1
HPLC 97.88%
5-[-(3′-Aminopropyl)amino]-8-methoxy-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one (IKE5)
1,3-Diaminopropane and 5-chloro-8-methoxy-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one was used;
Yield 64%; m.p. 138–139 °C;
1H NMR (DMSO-d6) δ: 9.36 (t, J = 6.1 Hz, 1H, 5-NH), 8.42 (d, J = 8.8 Hz, 1H), 8.34 (d, J = 9.3 Hz, 1H), 8.08 (br.s, 3H, NH3+), 7.78 (d, J = 2.4 Hz, 1H), 7.61 (dd, J1 = 2.9 Hz, J2 = 8.8 Hz, 1H), 7.21 (d, J = 9.3 Hz, 1H), 3.94 (s, 3H, OCH3), 3.68–3.72 (m, 2H, NHCH2CH2CH2NH2), 2.91–2.94 (m, 2H, NHCH2CH2CH2NH2), 1.97–2.03 (m, 2H, NHCH2CH2CH2NH2).
13C NMR (DMSO-d6) δ: 177.11, 158.37, 152.51, 134.94, 130.84, 129.02, 128.19, 125.87, 121.89, 116.56, 111.28, 108.50, 99.07, 54.86, 48.22, 39.57, 37.04, 26.95.
ESI–MS m/z: [M + H]+ calcd. for C17H17N5O2 323.3, found 324.2
HPLC 98.6%
5-[-(3′-Aminopropyl)amino]-8-methyl-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one (IKE6)
1,3-Diaminopropane and 5-chloro-8-methyl-6H-[1–3]-triazolo[4,5,l-de]acridin-6-one was used;
Yield 60%; m.p. 212–213 °C;
1H NMR (DMSO-d6) δ: 9.31 (t, J = 6.1 Hz, 1H, 5-NH), 8.29–8.34 (m, 2H), 8.15 (br.s, 3H, NH3+), 8.11 (s, 1H), 7.79 (dd, J1 = 1.9 Hz, J2 = 8.3 Hz, 1H), 7.18 (d, J = 9.3 Hz, 1H), 3.66–3.70 (m, 2H, NHCH2CH2CH2NH2), 2.91–2.95 (m, 2H, NHCH2CH2CH2NH2), 2.52 (s, 3H, CH3-Ar), 1.98–2.04 (m, 2H, NHCH2CH2CH2NH2).
13C NMR (DMSO-d6) δ: 177.30, 152.65, 136.80, 135.46, 135.33, 133.04, 131.88, 129.17, 127.34, 124.78, 115.60, 112.30, 99.74, 36.84, 27.28, 21.40.
ESI–MS m/z: [M + H]+ calcd. for C17H17N5O1 307.3, found 308.1
HPLC 98.5%
General procedure for the synthesis of IKE7-IKE8
Example synthesis IKE7
To a stirred solution of N-BOC-Lys(Z)-OH (1 mmol) in DMSO (Dimethylsulfoxide) (10 mL) 1 mmol of NHS (N-hydroxysuccinimide), 1 mmol DCC (N,N′-dicyclohexylcarbodiimide), 0.65 mmol triazoloacridinone derivative (IKE5) in the presence of a few drops of TEA. After 24 h of stirring at room temperature, the reaction mixture was filtered, washed with DMSO, and then water was added to the filtrate. The precipitate was filtered off and then dried. Yield 86% (0.37 g). The obtained compound was dissolved in 5 mL of glacial AcOH and 1.5 mL of 33% HBr in acetic acid was added. At ambient temperature, the reaction mixture was stirred for a duration of 0.5 h and then the resulting mixture was poured into approximately 15 mL of diethyl ether and stirred again for another 0.5 h. The precipitate was filtered off, washed with ether, and dried. The residue was purified by column chromatography on silica gel. The initial eluent was CHCl3/MeOH/NH3 (15:1.5:0.15 by volume) and then (10:2:0.2 by volume), (10:3:0.2 by volume). The purified product in the form of a base, was dissolved in methanol (10 mL) and acidified with HCl/diethyl ether. Next, it was precipitated out with diethyl ether.
N′-{3-[(8-methoxy-6-oxo-6H-[1,2,3]triazolo[4,5,1-de]acridin-5-yl)amino]propyl} lysinamide (IKE7)
Yield 51% m.p. 159–160 °C;
1H NMR (DMSO-d6) δ: 9.42 (t, J = 6.1 Hz, 1H, 5-NH), 8.87 (t, J = 5.4 Hz, 1H, NHC(O)), 8.45 (d, J = 8.8 Hz, 1H), 8.35 (d, J = 9.3 Hz, 1H), 8.32 (br.s, 3H, NH3+), 7.98 (br.s, 3H, NH3+), 7.81 (s, 1H), 7.62 (dd, J1 = 2.9 Hz, J2 = 9.3 Hz, 1H), 7.21 (d, J = 9.2 Hz, 1H), 3.96 (s, 3H, OCH3), 3.78–3.85 (m, 2H, NHCH2CH2CH2NHC(O)CH(NH2)), 3.63–3.67 (m, 1H, NHCH2CH2CH2NHC(O)CH(NH2)), 3.27–3.33 (m, 2H, NHCH2CH2CH2NHC(O)CH(NH2)), 2.76–2.80 (m, 2H, CHCH2CH2CH2CH2NH2), 1.85–1.92 (m, 2H, NH2CH2CH2CH2NH2), 1.73–1.80 (m, 2H, CHCH2CH2CH2CH2NH2), 1.56–1.62 (m, 2H, CHCH2CH2CH2CH2NH2), 1.35–1.41 2.76–2.80 (m, 2H, CHCH2CH2CH2CH2NH2).
13C NMR (DMSO-d6) δ: 176.85, 169.10, 158.125, 152.91, 135.18, 131.39, 129.39, 129.30, 126.26, 122.41, 117.37, 112.27, 109.40, 99.26, 56.14, 52.43, 40.68, 38.64, 36.62, 30.74, 29.15, 26.69, 21.70.
ESI–MS m/z: calcd. for C23H29N7O3 451.5, found [M + H]+ 452.2, [M − H]−450.2
HPLC 99.2%
N′-{3-[(8-methyl-6-oxo-6H-[1,2,3]triazolo[4,5,1-de]acridin-5-yl)amino]propyl} lysinamide (IKE8)
The method of synthesis was similar to that in the case of derivative IKE7: derivative IKE6 was used; Yield 57%; m.p. 128–129 °C;
1H NMR (DMSO-d6) δ: 9.41 (t, J = 6.1 Hz, 1H, 5-NH), 8.81 (t, J = 5.8 Hz, 1H, NHC(O)), 8.41 (d, J = 8.3 Hz, 1H), 8.35 (d, J = 9.2 Hz, 1H), 8.28 (br.s, 3H, NH3+), 8.21 (s, 1H), 7.93 (br.s, 3H, NH3+), 7.85 (dd, J1 = 1.9 Hz, J2 = 8.3 Hz, 1H), 7.20 (d, J = 9.3 Hz, 1H), 3.76–3.80 (m, 1H, NHCH2CH2CH2NHC(O)CH(NH2)), 3.60–3.66 (m, 2H, NHCH2CH2CH2NHC(O)CH(NH2)), 3.28–3.31, (m, 2H, NHCH2CH2CH2NHC(O)CH(NH2)), 2.76–2.80 (m, 2H, CHCH2CH2CH2CH2NH2), 1.87–1.91 (m, 2H, NH2CH2CH2CH2NH2), 1.74–1.77 (m, 2H, CHCH2CH2CH2CH2NH2), 1.57–1.60 (m, 2H, CHCH2CH2CH2CH2NH2), 1.36–1.39 (m, 2H, CHCH2CH2CH2CH2NH2).
13C NMR (DMSO-d6) δ: 177.35, 169.05, 152.83, 136.85, 135.51, 135.26, 133.10, 131.94, 129.30, 127.42, 124.86, 115.67, 112.36, 99.66, 52.44, 40.69, 38.69, 36.67, 30.77, 29.16, 26.74, 21.72, 21.41.
ESI–MS m/z: calcd. for C23H29N7O2 435.5, found [M + H]+ 436.2, [M − H]− 434.1
HPLC 98.3%
General procedure for the synthesis of IKE9-10 and IKE12-13
The synthesis procedure was based on the previously described one for 5-[(2-aminoethyl)amino]-8-hydroxy-6H-imidazo[4,5,1-de]acridin-6-one16 compound. In summary, the corresponding amine (13.34 mmol) was added to the suspension of 1-chloro-7-substituted-4-nitro-9(10H)-acridone derivatives (3.44 mmol) in 20 mL of DMSO, and the solution was stirred at room temperature for a duration of 2.5 h. Following this, approximately 50 mL of water was added, and the reaction mixture was subjected to stirring for an additional 0.5 h. The precipitate obtained was collected through filtration and then transferred into water, where it was acidified using dilute hydrochloric acid and stirred for 0.5 h. The undissolved material was filtered out, and the resulting solution was evaporated to a reduced volume. To precipitate the product, acetone (approximately 100 mL) was employed, and the precipitate was subsequently filtered. The obtained derivative, along with 10% Pd/C in catalytic quantities and 40 mL of 96% formic acid, was subjected to hydrogenation by passing gaseous hydrogen through them at room temperature for a period of 24 h. After this time, the catalyst was separated by filtration. To the filtrate, 2–3 mL of concentrated HCl was added, and the mixture was heated at 110 °C for 24 h. Following this step, the formic acid was evaporated, and the resulting residue was heated for 3 h in a mixture of water and methanol with a 1:1 ratio (approximately 50 mL). The solvent was evaporated, and the residue was dissolved in methanol and then acidified with concentrated hydrochloric acid. The product was finally crystallized from diethyl ether.
5-{[(2-dimethylamino)ethyl]amino}-8-methoxy-6H-imidazo[4,5,1-de]acridin-6-one (IKE9)
N,N-Dimethylethylenediamine and 1-chloro-7-methoxy-4-nitro-9(10H)-acridone was used;
Yield 71%; m.p. 283–284 °C;
1H NMR (DMSO-d6) δ: 10.10 (s, 1H, N10CH), 9.01–9.12 (m, 1H, 5-NH), 8.45 (d, J = 9.3 Hz, 1H), 8.12 (d, J = 9.3 Hz, 1H), 7.81 (d, J = 2.9 Hz, 1H), 7.62 (dd, J1 = 2.9 Hz, J2 = 9.3 Hz, 1H), 7.22 (d, J = 9.3 Hz, 1H), 3.93 (s, 3H, OCH3), 3.93–3.95 (m, 2H, NHCH2CH2N(CH3)2), 3.38–3.41 (m, 2H, NHCH2CH2N(CH3)2), 2.88 (s, 6H, N(CH3)2).
13C NMR (DMSO-d6) δ: 177.78, 157.81, 149.43, 136.08, 131.91, 129.15, 128.37, 126.53, 122.46, 118.77, 109.38, 108.37, 102.64, 56.18, 55.03, 42.91, 37.98.
ESI–MS m/z: [M + H]+ calcd. for C19H20N4O2 336.3, found 337.1
HPLC 98.1%
5-[(2-aminoethyl)amino]-8-methyl-6H-imidazo[4,5,1-de]acridin-6-one (IKE10)
Ethylenediamine and 1-chloro-7-methyl-4-nitro-9(10H)-acridone was used;
Yield 61%; m.p. 264–265 °C;
1H NMR (DMSO-d6) δ: 9.16 (s, 1H, N10CH), 9.02 (m, 1H, 5-NH), 8.30 (d, J = 8.3 Hz, 1H), 8.15 (s, 1H), 7.97 (d, J = 8.8 Hz, 1H), 7.73 (d, J = 8.3 Hz, 1H), 6.84 (d, J = 9.2 Hz, 1H), 3.36–3.41 (m, 2H, NHCH2CH2NH2), 2.88–2.90 (m, 2H, NHCH2CH2NH2), 2.51 (s, 3H, Ar-CH3).
13C NMR (DMSO-d6) δ: 177.98, 149.60, 136.23, 135.99, 135.33, 132.88, 132.14, 128.54, 128.14, 127.25, 125.09, 116.88, 108.26, 102.93, 37.98, 21.30.
ESI–MS m/z: [M + H]+ calcd. for C17H16N4O1 292.3, found 293.1
HPLC 98.9%
5-[(2-aminoethyl)amino]-8-hydroxy-6H-imidazo[4,5,1-de]acridin-6-one (IKE12)
Ethylenediamine and 1-chloro-7-hydroxy-4-nitro-9(10H)-acridone was used;
Yield 59%; m.p. 332–334 °C;
1H NMR (DMSO-d6) δ: 10.27 (s, 1H, N10CH), 9.11–9.21 (m, 1H, 5-NH), 8.34 (d, J = 9.3 Hz, 1H), 8.10 (d, J = 8.8 Hz, 1H), 7.98 (br.s, 3H, NH3+), 7.77 (s, 1H), 7.41 (dd, J1 = 2.9 Hz, J2 = 8.8 Hz, 1H), 7.23 (d, J = 9.3 Hz, 1H), 3.77–3.80 (m, 2H, NHCH2CH2NH2), 3.09–3.13 (m, 2H, NHCH2CH2NH2).
13C NMR (DMSO-d6 + TFA) δ: 175.41, 155.52, 150.06, 130.73, 127.74, 125.20, 124.22, 123.99, 121.70, 117.84, 116.87, 111.85, 110.34, 99.33, 39.37, 37.50.
ESI–MS m/z: calcd. for C16H14N4O2 294.3, found [M + H]+ 295.1, [M-H]- 293.1
HPLC 98.5%
5-[(2-aminoethyl)amino]-8-methoxy-6H-imidazo[4,5,1-de]acridin-6-one (IKE13)
Ethylenediamine and 1-chloro-7-methoxy-4-nitro-9(10H)-acridone was used;
Yield 63%; m.p. 336–337 °C;
1H NMR (DMSO-d6) δ: 9.57 (s, 1H, N10CH), 8.97 (m, 1H, 5-NH), 8.41 (d, J = 8.8 Hz, 1H), 8.25 (br.s, 3H, NH3+), 8.04 (d, J = 9.3 Hz, 1H), 7.75 (d, J = 2.9 Hz, 1H), 7.58 (dd, J1 = 2.9 Hz, J2 = 8.8 Hz, 1H), 7.06 (d, J = 8.8 Hz, 1H), 3.93 (s, 3H, OCH3), 3.78–3.85 (m, 2H, NHCH2CH2NH2), 3.08–3.11 (m, 2H, NHCH2CH2NH2).
13C NMR (DMSO-d6) δ: 177.61, 157.72, 149.68, 135.95, 131.91, 129.11, 128.51, 128.39, 126.49, 122.31, 118.69, 109.34, 108.09, 102.60, 56.14, 38.00, 36.95.
ESI–MS m/z: [M + H]+ calcd. for C17H16N4O2 308.3, found 309.1
HPLC 96.5%
General procedure for the synthesis of IKE11 and IKE14
Example synthesis IKE11
To a stirred solution of N- BOC-Lys(Z)-OH (1.23 mmol) in DMSO (15 mL) 1.23 mmol of NHS, 1.23 mmol DCC, 0.81 mmol imidazoacridinone derivative (IKE10) in the presence of a few drops of TEA. After 24 h of stirring at room temperature, the reaction mixture was filtered, washed with DMSO, and then water was added to the filtrate. The precipitate was filtered off and then dried. Yield 80%. The obtained compound was dissolved in 5 mL of glacial AcOH and 1.5 mL of 33% HBr in acetic acid was added. The reaction mixture was stirred at room temperature for 0.5 h. The resulting mixture was poured into diethyl ether (~ 15 mL) and then stirred for 0.5 h. The precipitate was filtered off, washed with ether, and dried. The residue was purified by column chromatography on silica gel. The initial eluent was CHCl3/MeOH/NH3 (15:1.5:0.15 by volume) and then (10:2:0.2 by volume), (10:3:0.2 by volume). The purified product in the form of a base, was dissolved in methanol (10 mL) and acidified with HCl/diethyl ether. Next, it was precipitated out with diethyl ether.
N-{2-[(8-methyl-6-oxo-6H-imidazo[4,5,1-de]acridin-5-yl)amino]ethyl}lysinamide (IKE11)
Yield 41%; m.p. 148–149 °C;
1H NMR (DMSO-d6) δ: 9.32 (s, 1H, N10CH), 8.94 (t, J = 5.8 Hz, 1H, NHC(O)), 8.97 (m, 1H, 5-NH), 8.37 (d, J = 8.2 Hz, 1H), 8.30 (br.s, 3H, NH3+), 8.18 (s, 1H), 8.04 (d, J = 8.8 Hz, 1H), 7.97 (br.s, 3H, NH3+), 7.79 (d, J = 8.6 Hz, 1H), 7.01 (d, J = 9.0 Hz, 1H), 3.35–3.76 (m, 5H, NHCH2CH2NHC(O)CH(NH2)), 2.73–2.77 (m, 2H, CHCH2CH2CH2CH2NH2), 2.52 (s, 3H, Ar-CH3), 1.70–1.80 (m, 2H, CHCH2CH2CH2CH2NH2), 1.53–1.59 (m, 2H, CHCH2CH2CH2CH2NH2), 1.36–1.40 (m, 2H, CHCH2CH2CH2CH2NH2).
13C NMR (DMSO-d6) δ: 177.80, 169.51, 150.10, 136.49, 135.60, 135.26, 132.72, 131.87, 127.76, 127.27, 125.18, 116.99, 108.70, 102.39, 52.42, 49.04, 42.00, 38.56, 30.72, 26.69, 21.64, 21.32.
ESI–MS m/z: calcd. for C23H28N6O2 420.5, found [M + H]+ 421.2, [M-H]- 419.2
HPLC 98.4%
N-{2-[(8-methoxy-6-oxo-6H-imidazo[4,5,1-de]acridin-5-yl)amino]ethyl}lysinamide (IKE14)
The method of synthesis was similar to that in the case of derivative IKE11: derivative IKE13 was used; Yield 45%; m.p. 164–165 °C;
1H NMR (DMSO-d6) δ: 9.31 (s, 1H, N10CH), 8.98 (m, NHC(O)), 8.93 (t, J = 5.4 Hz, 1H, 5-NH), 8.43 (d, J = 9.1 Hz, 1H), 8.29 (br.s, 3H, NH3+), 8.04 (d, J = 9.1 Hz, 1H), 7.94 (br.s, 3H, NH3+), 7.81 (d, J = 2.7 Hz, 1H), 7.60 (dd, J1 = 2.9 Hz, J2 = 9.1 Hz, 1H), 7.00 (d, J = 9.0 Hz, 1H), 3.94 (s, 3H, OCH3), 3.72–3.80 (m, 1H, NHCH2CH2NHC(O)CH(NH2)), 3.58–3.63 (m, 2H, NHCH2CH2NHC(O)CH(NH2)), 3.38–3.42 (m, 2H, NHCH2CH2NHC(O)CH(NH2)), 2.64–2.75 (m, 2H, CHCH2CH2CH2CH2NH2), 1.70–1.81 (m, 2H, CHCH2CH2CH2CH2NH2), 1.52–1.58 (m, 2H, CHCH2CH2CH2CH2NH2), 1.36–1.39 (m, 2H, CHCH2CH2CH2CH2NH2).
13C NMR (DMSO-d6) δ: 177.38, 169.52, 157.86, 150.17, 135.48, 131.54, 128.87, 127.89, 126.57, 122.29, 118.79, 109.21, 108.60, 102.01, 56.18, 52.41, 42.05, 38.55, 30.71, 26.68, 21.62.
ESI–MS m/z: [M + H]+ calcd. for C17H16N4O2 364.4, found 437.2
HPLC 98.0%
Microbiological tests
Microorganisms strains and growth conditions
The following fungal strains were used: Candida albicans ATCC 10,231, Candida glabrata ATCC 90,030, Candida krusei ATCC 6258, Candida parapsilosis ATCC 22,019, Saccharomyces cerevisiae ATCC 9763, Candida albicans clinical isolates40,41: B3, B4, Gu4, Gu5, F2, F5, Candida glabrata clinical isolates34: CZD 373, CZD 377, CZD 513, Gd 310, Saccharomyces cerevisiae strains35,36,42: AD-MDR1-GFP, AD1-8u−, AD-CDR1-GFP, AD-CDR2-GFP. Fungal strains were routinely grown over 18 h at 30 °C in YPG liquid medium (1% m/V yeast extract, 1% m/V peptone, 2% m/V glucose) in a shaking incubator (INFORS HT Bottmingen, Switzerland). For growth on solid media, 1.5% m/V agar was added to the YPG medium. For antimicrobial activity assays RPMI-1640 (Sigma-Aldrich, St. Louis, MO, USA) medium buffered to pH 7.0 was used for all strains except for Saccharomyces cerevisiae mutants AD-MDR1-GFP, AD1-8u−, AD-CDR1-GFP, AD-CDR2-GFP. Saccharomyces cerevisiae mutants were grown in 0.67% m/V yeast nitrogen base medium without amino acids, folic acid, p-aminobenzoic acid, with ammonium sulphate (MP Biomedicals, Irvine, CA, USA) supplemented with 2% m/V glucose, 0.192% m/V yeast synthetic drop-out medium supplement without uracil (Sigma-Aldrich, St. Louis, MO, USA) and 0.0076% m/V uracil (Sigma-Aldrich, St. Louis, MO, USA).
Antifungal activity assays
Determination of in vitro antifungal activity was performed according to the modified M27-A3 specified by the CLSI43. Strains (104 CFU (colony-forming unit) mL−1) were grown in the appropriate medium in a 96-wells microplate for 24 h (48 h for S. cerevisiae mutants) at 30 °C with or without tested compounds. The optical denticity was measured at 600 nm by a microplate reader (TECAN Spark 10 M, Grödig, Austria). The Minimum Inhibitory Concentration (MIC) was determined as the lowest concentration of the drug showing a minimum 90% reduction in turbidity compared to the drug-free control.
The Minimum Fungicidal Concentration (MFC) was defined as the lowest concentration of the test compound in which no recovery of microorganisms was observed. The MFC was determined as described previously17 by spot assay.
Accumulation of the compounds in fungal cells
The accumulation of the compounds in fungal cells was examined with the Olympus BX-60 fluorescence microscope (λex = 470–490 nm, λem ≥ 500 nm, lens × 40) equipped with the Olympus XC50 digital camera (Olympus Corporation, Tokyo, Japan). The overnight culture of S. cerevisiae ATCC 9763 in YPG medium was subjected to two rounds of washing using sterile phosphate-buffered saline (PBS) and resuspended to achieve a cell density of 2 × 107 CFU mL−1. Cells were incubated with IKE5, IKE7, and C-1305 compounds at 100 µM concentration for 10–60 min. Following the designated time period, a 1 mL aliquot of the cell suspension underwent a triple-washing procedure using sterile phosphate-buffered saline (PBS). Cells were resuspended in 40 µL 90% (by volume) glycerol and 10% (by volume) of tenfold concentrated PBS. Then 3 µL of inoculum was transferred to a microscopic slide.
Morphological changes of fungi
Candida albicans ATCC 10,231 cells were cultured in 24-well plates with a glass bottom and incubated with either DMSO or the investigated compounds in RPMI-1640 + 10% (by volume) fetal bovine serum or Spider medium (1% m/V nutrient broth (Difco), 1% m/V D-mannitol (Sigma-Aldrich, St. Louis, MO, USA), 0.2% m/V K2HPO4, pH 7.2) at 37 °C for 3 h. Following the incubation period, the cells were washed three times with PBS and imaged using a light microscope. Specifically, an LSM 800 inverted laser-scanning confocal microscope from Carl Zeiss (ZEISS, Göttingen, Germany), equipped with a × 63 1.4-NA Plan Apochromat objective from Carl Zeiss, was utilized for imaging.
The effect of analyzed compounds on C. albicans biofilm formation
Candida albicans biofilm formation was analyzed with the use of a microtiter plate-based method according to the previously described protocol17.
Flow cytometry analysis
To evaluate the change in fungal membrane permeability, the propidium iodide influx assay was utilized. The Saccharomyces cerevisiae ATCC 9763 cells were cultured in a YPG medium at a concentration of 106 CFU mL−1. These cells were then exposed to the investigated compounds for 60 min. After the incubation period, the cells were collected and washed twice with PBS buffer. Subsequently, the cells were resuspended and incubated with 20 µg mL−1 of propidium iodide in PBS. Additionally, the accumulation of the compounds within the cells was measured after 15 and 60 min by assessing the intensity of green fluorescence. A cell suspension was analyzed using the Guava easyCyte 8 cell sorter (Merck Millipore, Hayward, CA, USA)), and the data were processed using FlowJo v10.8.0 software (BD Life Sciences, Franklin Lakes, NJ, USA). Each experiment was independently repeated three times.
Topoisomerase assays
Yeast topoisomerase II relaxation assay and inhibition
The inhibition of yeast topoisomerase II was analyzed according to the relaxation assay method described previously.
Formation of cleavable complexes in vitro
Cleavage assay of yeast topoisomerase II was performed according to the method described previously16.
Decatenation assay
Yeast topoisomerase II (Saccharomyces cerevisiae, Inspiralis Limited, UK) ability to decatenate kinetoplast DNA (kDNA) from Crithidia fasciculata was examined according to a decatenation assay kit from Inspiralis (Inspiralis Limited, Norwich, UK). In summary, for the experimental procedure, a mixture containing 200 ng of kDNA, 1 mM ATP (Inspiralis Limited, UK), and varying concentrations (ranging from 1 to 150 µM) of the analyzed compounds was prepared with reaction buffer (comprising 4 mM Tris–HCl at pH 7.9, 40 mM KCl, 2 mM MgCl2, and 0.8% (by volume) glycerol). The enzymatic reaction was initiated by introducing the enzyme and allowed to proceed at a temperature of 30 °C for a duration of 30 min. Termination of the reaction was achieved by adding 40% (m/V) sucrose, along with 100 mM Tris–HCl at pH 8, 10 mM EDTA, and 0.5 mg mL−1 of Bromophenol Blue. For the subsequent steps, a two-step extraction was performed using chloroform:isoamyl alcohol (in a ratio of 24:1, by volume) and butanol water. The resulting mixtures were then analyzed on a 1% (w/v) agarose gel in 1 × TAE buffer (Tris–acetate-EDTA buffer, NZYTech, Lisbona, Portugal), with an electrophoresis time of 4 h at 4.5 V cm−1. The gel was immersed in GelRed 3 × staining solution (Biotium, Fremont, CA, USA) for a duration of 30 min. Subsequently, it was captured using the Gel Doc XR + Gel Documentation System (Bio-Rad: Hercules, CA, USA) to obtain photographs.
Unwinding assay
The kit from Inspiralis (Inspiralis Limited, Norwich, UK) was utilized to assess the compounds' capacity to intercalate into the DNA, which subsequently resulted in the unwinding of the DNA strands. Briefly, supercoiled or relaxed pBR322 plasmid (250 ng) was combined with the tested compounds at concentrations ranging from 1 to 100 µM. This mixture was then supplemented with the reaction buffer comprising 25 mM Tris–HCl (pH 7.9), 0.5 mM EDTA, 0.5 mM DTT (ditiotreitol), 10% (by volume) glycerol, and 25 mM NaCl. The assay was initiated by introducing Wheat germ topoisomerase I, and the mixture was then incubated for 30 min at 37 °C. To stop the reaction, 10 µL of water and 25 µL of butanol were added, followed by centrifugation at 16,100 xg for 1 min. Further, 40% (m/V) sucrose, 100 mM Tris–HCl at pH 8, 10 mM EDTA, and 0.5 mg mL−1 of Bromophenol Blue were added, and extraction with chloroform:isoamyl alcohol (in a ratio of 24:1, by volume) was performed. The mixtures were then analyzed on a 1% (m/V) agarose gel in 1 × TAE buffer for 18 h at 1.1 V cm−1. GelRed 3 × staining solution (obtained from Biotium, Fremont, CA, USA) was used to stain the gel for 30 min, and the gel was photographed using the Gel Doc XR + Gel Documentation System (Bio-Rad: Hercules, CA, USA).
Antiproliferative activity determination
Cell culture
HEK-293 (human embryonic kidney) and HEPG2 (human liver cancer) cell lines were obtained from ATCC (Manassas, Virginia, USA). HEK-293 cells were cultured in Dulbecco's Modified Eagle Medium (DMEM), while HEPG2 cells were cultured in Minimum Essential Medium Eagle (MEM). Both cell lines received a supplement of 10% (by volume) fetal bovine serum, 2 mM L-glutamine, and antibiotics: penicillin at a concentration of 62.6 µg mL−1 and streptomycin at a concentration of 40 µg mL−1. The cells were cultivated in a humidified environment with 5% CO2 and 95% air. Regular screening for Mycoplasma was carried out to ensure the integrity of the cell lines. All the reagents utilized in this study were purchased from Corning unless otherwise stated.
Cell viability
The assessment of antiproliferative activity was carried out using the (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich in St. Louis, MO, USA). In summary, cells were seeded in 96-well culture plates and allowed to adhere overnight. The compounds under investigation were dissolved in DMSO and added to the wells, resulting in a final DMSO concentration of 1% (by volume). Different concentrations of the compounds were tested. After 72 h of incubation under standard culture conditions, 20 µL of MTT solution (4 µg mL−1 in PBS) was added to each well, and the plates were further incubated for 3 h. Subsequently, the medium was removed, and the formazan crystals were dissolved in DMSO. The absorbance of the resulting solution was measured at 540 nm using a microplate reader (Asys UVM 340 Microplate Reader, Biochrom, Cambridge, UK). Each experiment was independently performed three times in triplicate.
Statistical analysis
Statistical analysis was conducted using GraphPad Prism 9 software. Uniform significance levels were applied consistently across the entire manuscript as follows: ns (not significant) for p > 0.01, * for p < 0.01, ** for p < 0.001, *** for p < 0.0001, and **** for p < 0.00001. Statistical significance was determined by comparing the results to the DMSO-treated control using one- or two-way ANOVA.
Acridine derivatives interactions with Calf thymus DNA
Chemicals and UV–Vis measurements preparation of stock solutions
The electronic spectra were recorded on an Evolution 300 spectrophotometer (Thermo Fischer Scientific, Waltham, MA, USA) in the range of 200–600 nm in the case of UV detection of intermolecular interactions with biomolecules together with a spectral band with a 2 nm width. Calf Thymus DNA (CT-DNA) was dissolved in Tris–HCl buffer (pH 7.43 ± 0.01) at 4 °C for 24 h with occasional stirring to ensure the formation of a homogeneous and stable biomolecule pre-solution. The final concentration of the stock deoxyribonucleic acid solution was determined spectrophotometrically at 260 nm by using a molar extinction coefficient of 6600 cm−1 M−1 for CT-DNA44. The concentration of freshly prepared CT-DNA dissolved in biological buffer (Tris–HCl 5 mM /50 mM NaCl; pH 7.43) was spectrophotometrically verified. Special attention was focused on the absorbances’ values ratio to be able to confirm the purity of the CT-DNA45. The A260/A280 ratio for CT-DNA was 1.84 which proves that the biomolecule studied treated as acridines’ cellular target was sufficiently free from proteins (Figs. S5, S8, and S11, SI). In the next step. the spectrophotometric titrations were performed at room temperature by gradually increasing the CT-DNA concentration added to individual acridines’ studied. After each addition of an amount of CT-DNA solution (470 μM), electronic absorption spectra of specific adducts formed were recorded by spectrophotometric titrations. Importantly, note that the dilution effects related to the measurement procedure used were strictly eliminated at the preliminary stage of the results processing.
Methodology used in binding studies
The binding ability of six acridine derivatives to interact with Calf thymus DNA (CT-DNA, Sidma-Aldrich; catalog No. D8661-1ML) was investigated by spectrophotometric titrations. The UV absorbance at 260 nm of a diluted solution (1/20) of CT-DNA used in our experiments was measured to be 3.102 (path the length equal 1 cm) and the final concentration of the stock CT-DNA solution was calculated to be 0.47 mM CT-DNA in Tris–HCl. The initial concentrations of all acridine samples were prepared in Tris–HCl buffer [μM]: [IKE1] = 93; [IKE8] = 71; [IKE3] = 79; [IKE7] = 35; [IKE9] = 146; [IKE14] = 91. The time dependency of interaction for each object was different and was established based on the preliminary kinetic measurements (Figs. S6, S9, S12 SI). The CT-DNA solution then was added dropwise to the individual acridine solution. The stability of the binding properties of the compounds studied toward CT-DNA was examined by taking spectra after the time described in SI, and the same results were obtained for repetition made. Importantly, the preliminary step of results elaboration included the dilution effect elimination. Based upon the variation in absorbance, the association/binding constants of these interactions with CT-DNA were determined according to the Benesi–Hildebrand Eq. (1) presented below46:
where A0 and A are the absorbances of the acridine derivative in the absence and presence of CT-DNA, respectively; εG and εH–G are the absorption coefficients of the acridine derivative unbounded and its adduct with CT-DNA, respectively. Thus, the double reciprocal plot of A0/(A–A0) versus 1/[DNA] is linear and the binding constant (Kbind) was estimated based on the linear fit parameters established as the ratio of the intercept to the slope47,48.
Statistical analysis
All experiments were carried out in duplicates, in two independent experimental sets. The means ± SD were used in the statistical analysis of the data and the graphics.
Data availability
Data supporting the reported results are presented in this manuscript and its supplemental material. Raw data supporting our results are also available as data sets: Gabriel, I., Paluszkiewicz, E., Kozłowska-Tylingo, K., Roślik, M., & Rząd, K. (2021). Determination of the minimum inhibitory concentration of C-1305 derivatives (IKE1-IKE8) against Candida strains [Data set]. Gdańsk University of Technology. https://doi.org/https://doi.org/10.34808/47q7-jg74; Gabriel, I., Paluszkiewicz, E., Kozłowska-Tylingo, K., & Rząd, K. (2021). IKE1-IKE3 (C-1305 derivatives) inhibitory effect of the Yeast Topoisomerase II relaxation activity (Version 2.0) [Data set]. Gdańsk University of Technology. https://doi.org/https://doi.org/10.34808/j3t1-qg11; Rząd, K., Panuś, O., Gabriel, I., Paluszkiewicz, E., & Kozłowska-Tylingo, K. (2023). Determination of the minimum inhibitory concentration of C-1330 derivatives (IKE9-IKE14) against Candida strains [Data set]. Gdańsk University of Technology. https://doi.org/https://doi.org/10.34808/wx9v-ck79;
References
Staniszewska, M. et al. Pathogenesis and treatment of infections. Postep. Mikrobiol. 53, 229–240 (2014).
Skrodenienė, E., Dambrauskienė, A. & Vitkauskienė, A. Susceptibility of yeasts to antifungal agents in Kaunas University of Medicine Hospital. Med. 42, 294–299 (2006).
Gualco, L. et al. Antifungal resistance in Candida spp. isolated in Italy between 2002 and 2005 from children and adults. Int. J. Antimicrob. Agents 29, 179–184 (2007).
Perfect, J. R. The antifungal pipeline: A reality check. Nat. Rev. Drug Discov. 16, 603–616 (2017).
Biliński, P., Seferyńska, I. & Warzocha, K. Diagnosis and treatment of systemic fungal infections in oncohematology. Onkol. w Prakt. Klin. 4, 15–24 (2008).
Tadec, L. et al. Epidemiology, risk factor, species distribution, antifungal resistance and outcome of Candidemia at a single French hospital: A 7-year study. Mycoses 59, 296–303 (2016).
Turner, S. A. & Butler, G. The Candida pathogenic species complex. Cold Spring Harb. Perspect. Med. 4, 4-a019778 (2014).
WHO fungal priority pathogens list to guide research, development and public health action. Geneva: World Health Organization (Licence: CC BY-NC-SA 3.0 IGO. Licence: CC BY-NC-SA 3.0 IGO; 2022).
Bondaryk, M., Kurzątkowski, W. & Staniszewska, M. Antifungal agents commonly used in the superficial and mucosal candidiasis treatment: Mode of action and resistance development. Postep. Dermatol. I Alergol. 30, 293–301 (2013).
Gray, K. C. et al. Amphotericin primarily kills yeast by simply binding ergosterol. Proc. Natl. Acad. Sci. 109, 2234–2239 (2012).
Wiederhold, N. P. Antifungal resistance: current trends and future strategies to combat. Infect. Drug Resist. 10, 249–259 (2017).
Kondaka, K. & Gabriel, I. Targeting DNA topoisomerase II in antifungal chemotherapy. Molecules 27, 7768–7783 (2022).
Shen, L. L. & Fostel, J. M. DNA topoisomerase inhibitors as antifungal agents. Adv. Pharmacol. 29, 227–244 (1994).
Skladanowski, A., Plisov, S. Y., Konopa, J. & Larsen, A. K. Inhibition of DNA topoisomerase II by imidazoacridinones, new antineoplastic agents with strong activity against solid tumors. Mol. Pharmacol. 49, 772–780 (1996).
Lemke, K., Poindessous, V., Skladanowski, A. & Larsen, A. K. The Antitumor triazoloacridone C-1305 is a topoisomerase II poison with unusual properties. Mol. Pharmacol. 66, 1035–1042 (2004).
Rząd, K. et al. The effect of conjugation with octaarginine, a cell-penetrating peptide on antifungal activity of imidazoacridinone derivative. Int. J. Mol. Sci. Artic. 22, 13190 (2021).
Rząd, K., Paluszkiewicz, E. & Gabriel, I. A new 1-nitro-9-aminoacridine derivative targeting yeast topoisomerase II able to overcome fluconazole-resistance. Bioorg. Med. Chem. Lett. 35, 127815 (2021).
Gabriel, I., Rząd, K., Paluszkiewicz, E. & Kozłowska-tylingo, K. Antifungal activity of Capridine β as a consequence of its biotransformation into metabolite affecting yeast topoisomerase II activity. Pathogens 10, 189 (2021).
Arifian, H., Maharani, R., Megantara, S., Gazzali, A. M. & Muchtaridi, M. Amino-acid-conjugated natural compounds: Aims designs and results. Molecules 27, 7631–7665 (2022).
Qadir, M. A., Ahmed, M. & Khaleeq, A. Synthesis, antibacterial and antifungal possession of amino acids containing sulfonamide moieties. Pak. J. Pharm. Sci. 29, 1609–1613 (2016).
Lin, S. et al. Semisynthesis and biological evaluation of xanthone amphiphilics as selective, highly potent antifungal agents to combat fungal resistance. J. Med. Chem. 60, 10135–10150 (2017).
Taraszkiewicz, A., Szewczyk, G., Sarna, T., Bielawski, K. P. & Nakonieczna, J. Photodynamic inactivation of Candida albicans with imidazoacridinones: Influence of irradiance, photosensitizer uptake and reactive oxygen species generation. PLoS One 10, 1–19 (2015).
Cholody, W. M., Martelli, S. & Konopa, J. 8-Substituted 5-[(aminoalkyl)amino]-6H-v-triazolo[4,5,1-de]acridin-6-ones as potential antineoplastic agents. Synthesis and biological activity. J. Med. Chem. 33, 2852–2856 (1990).
Capps, D. B., Dunbar, J., Kesten, S. R., Shillis, J. & Werbel, L. M. 2-(Aminoalkyl)-5-nitropyrazolo[3,4,5-kl]acridines, a new class of anticancer agents. J. Med. Chem. 35, 4770–4778 (1992).
Cholody, W. M., Martelli, S., Paradziej-Lukowicz, J. & Konopa, J. 5-[(Aminoalkyl)amino]imidazo[4,5,1-de]acridin-6-ones as a novel class of antineoplastic agents. Synthesis and biological activity. J. Med. Chem. 33, 49–52 (1990).
Konieczny, M. T. & Konopa, J. Acridone Derivatives and Preparation of 8-Hydroxy-Imidazoacridinone Derivatives (EP 0 929 552 B1) (BTG International Limited; 1998).
Zeman, S. M. & Crothers, D. M. Simultaneous measurement of binding constants and unwinding angles by gel electrophoresis. Methods Enzymol. 340, 51–68 (2001).
Gabriel, I., Paluszkiewicz, E., Kozłowska-Tylingo, K. & Rząd, K. IKE1-IKE3 (C-1305 derivatives) inhibitory effect of the Yeast Topoisomerase II relaxation activity (Version 2.0). Gdańsk Univ. Technol. https://doi.org/10.34808/j3t1-qg11 (2021).
Ketron, A. C., Denny, W. A., Graves, D. E. & Osheroff, N. Amsacrine as a topoisomerase II poison: Importance of Drug–DNA Interactions. NIH Public Access 51, 1730–1739 (2012).
Shahabadi, N., Kashanian, S., Khosravi, M. & Mahdavi, M. Multispectroscopic DNA interaction studies of a water-soluble nickel(II) complex containing different dinitrogen aromatic ligands. Transit. Met. Chem. 35, 699–705 (2010).
Gonzalez-Ruiz, V. et al. Biomedical engineering trend, research and technologies. In InTech India 65 (2011).
Ju, C. C., Zhang, A. G., Yuan, C. L., Zhao, X. L. & Wang, K. Z. The interesting DNA-binding properties of three novel dinuclear Ru(II) complexes with varied lengths of flexible bridges. J. Inorg. Biochem. 105, 435–443 (2011).
Fukuda, R., Takenaka, S. & Takagi, M. Metal ion assisted DNAinteraction of crown ethar-linked acridine derivatives. J. Chem. Soc. Chem. Commun. https://doi.org/10.1039/C39900001028 (1990).
Szweda, P. et al. Mechanisms of azole resistance among clinical isolates of Candida glabrata in Poland. J. Med. Microbiol. 64, 610–619 (2015).
Shukla, S. et al. Functional characterization of Candida albicans ABC transporter Cdr1p. Eukaryot. Cell 2, 1361–1375 (2003).
Sharma, M. et al. Curcumin modulates efflux mediated by yeast ABC multidrug transporters and is synergistic with antifungals. Antimicrob. Agents Chemother. 53, 3256–3265 (2009).
Kumamoto, C. A. & Vinces, M. D. Contributions of hyphae and hypha-co-regulated genes to Candida albicans virulence. Cell. Microbiol. 7, 1546–1554 (2005).
Fanning, S. & Mitchell, A. P. Fungal biofilms. PLoS Pathog. 8, e1002585 (2012).
Lemke, K. et al. Induction of unique structural changes in guanine-rich DNA regions by the triazoloacridone C-1305, a topoisomerase II inhibitor with antitumor activities. Nucleic Acids Res. 33, 6034–6047 (2005).
Franz, R. et al. Multiple molecular mechanisms contribute to a stepwise development of fluconazole resistance in clinical Candida albicans strains. Antimicrob. Agents Chemother. 42, 3065–3072 (1998).
Franz, R., Ruhnke, M. & Morschhauser, J. Molecular aspects of fluconazole resistance development in Candida albicans. Mycoses 42, 453–458 (1999).
Pasrija, R., Banerjee, D. & Prasad, R. Structure and function analysis of CaMdr1p, a major facilitator superfamily antifungal efflux transporter protein of Candida albicans: Identification of amino acid residues critical for drug/H+ transport. Eukaryot. Cell 6, 443–453 (2007).
CLSI. Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard—Third Edition. CLSI document M27-A3. (Clinical and Laboratory Standards Institute; 2008).
Sastri, C. V., Eswaramoorthy, D., Giribabu, L. & Maiya, B. G. DNA interactions of new mixed-ligand complexes of cobalt(III) and nickel(II) that incorporate modified phenanthroline ligands. J. Inorg. Biochem. 94, 138–145 (2003).
Reichmann, M. E., Rice, S. A., Thomas, C. A. & Doty, P. A further examination of the molecular weight and size of desoxypentose nucleic acid. J. Am. Chem. Soc. 76, 3047–3053 (1954).
Sirajuddin, M., Ali, S. & Badshah, A. Drug–DNA interactions and their study by UV–Visible, fluorescence spectroscopies and cyclic voltametry. J. Photochem. Photobiol. B Biol. 124, 1–19 (2013).
Mech-Warda, P. et al. Low-molecular pyrazine-based DNA binders: Physicochemical and antimicrobial properties. Molecules 27, 3704–3726 (2022).
Chylewska, A. et al. Photosensitive and pH-dependent activity of pyrazine-functionalized carbazole derivative as promising antifungal and imaging agent. Sci. Rep. 10, 11767–11780 (2020).
Acknowledgements
The authors would like to thank Piotr Szweda from the Gdansk University of Technology for sharing the C. glabrata clinical isolates strains.
Funding
This research was funded by Gdansk University of Technology, ARGENTUM—‘Excellence Initiative—Research University’ program, grant number DEC-24/2020/IDUB/I.3.3.
Author information
Authors and Affiliations
Contributions
Conceptualization, I.G., K.R.; Funding acquisition, K.R.; Investigation, K.R., E.P., A.K., M.O, A.C., A.M.D., and I.G.; Methodology, K.R., E.P., A.K., M.O, A.C., A.M.D., K.K.-T., and I.G.; Project administration, I.G.; Supervision, I.G.; Writing—original draft, I.G., KR., A.C., A.M.D.; Writing—review and editing, I.G, KR. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rząd, K., Gabriel, I., Paluszkiewicz, E. et al. Targeting yeast topoisomerase II by imidazo and triazoloacridinone derivatives resulting in their antifungal activity. Sci Rep 14, 3594 (2024). https://doi.org/10.1038/s41598-024-54252-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54252-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
