
Abstract
The Houston toad (Bufo [= Anaxyrus] houstonensis) is an endangered amphibian with a small geographic range. Land-use changes have primarily driven decline in B. houstonensis with population supplementation predominant among efforts to reduce its current extinction risk. However, there has been historic uncertainty regarding the evolutionary and conservation significance of B. houstonensis. To this end, we used 1170 genome-wide nuclear DNA markers to examine phylogenetic relationships between our focal taxon, representatives of the Nearctic B. americanus group, and B. nebulifer, a sympatric Middle American species. Phylogenetic analyses indicate B. houstonensis is a taxon that is distinct from B. americanus. We corroborated such genetic distinctiveness with an admixture analysis that provided support for recent reproductive isolation between B. americanus and B. houstonensis. However, ABBA-BABA tests for ancient admixture indicated historic gene flow between Nearctic species while no signal of historic gene flow was detected between Nearctic and Middle-American species. We used an admixture analysis to recognize four Management Units (MU) based on observed genetic differentiation within B. houstonensis and recommend captive propagation, population supplementation, and habitat restoration efforts specific to each MU. Our results re-affirm the evolutionary novelty of an endangered relict.
Similar content being viewed by others

Introduction
Amphibians are recognized as one of the most imperiled vertebrate groups, with nearly a third of all species threatened with extinction, and they also represent the largest proportion of data deficient species1,2. Various threats to amphibians have been documented (e.g., habitat loss, climate change, and infectious diseases), with synergies between extrinsic threats and amphibian ecological/life history traits resulting in higher extinction risks for large-bodied amphibians with small geographic ranges3,4,5.
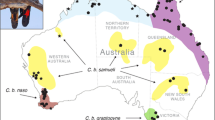
Houston toads (Bufo [= Anaxyrus] houstonensis) serve as an example of an amphibian taxon with a small geographic range and high risk of extinction6,7,8. Historical classification of North American toads based on morphological similarity placed B. houstonensis in the Bufo americanus species group along with Bufo americanus, Bufo terrestris, Bufo hemiophrys, Bufo microscaphus, Bufo woodhousii, Bufo fowleri, and Bufo velatus9,10. Additionally, B. houstonensis is considered a late Pleistocene relict. Populations of B. houstonensis were left behind in Texas after a widely distributed parental species (i.e., a precursor to present day American Toads [B. americanus]) experienced a large range expansion to the north, following retreat of the Wisconsin glaciation9. The contemporary range of B. houstonensis encompasses nine counties (i.e., Austin, Bastrop, Burleson, Colorado, Lavaca, Lee, Leon, Milam, and Robertson; Fig. 1) in Southeast and Central Texas11; however, it was historically present but extirpated from Brazos, Freestone, Fort Bend, Grimes, Harris, and Liberty counties. Land-use changes have predominantly driven their decline given that the toad is a habitat specialist with a strong preference for deep, sandy soils that are currently most often associated with forest cover6,12,13,14. Owing to continued population declines since B. houstonensis was first described15, it was designated as an endangered species on national and international lists7,8.

(A) Map of sampling localities relative to known distribution for 8 American Toads (Bufo [= Anaxyrus] americanus), 48 Houston toads (B. houstonensis), 18 Woodhouse’s Toads (B. woodhousii), and 19 Gulf Coast Toads (B. nebulifer). (B) The study included 48 samples of B. houstonensis from Bastrop, Lee, Austin, Leon, Milam, and Robertson counties within species range. © 2020 Texas Department of Transportation, U.S. Census, TomTom North America, Inc., U. S. Department of Agriculture, National Agricultural Statistics Service, and Global Biodiversity Information Facility (https://www.gbif.org/species/2422886; 2422872; 5217017; 2422404).
Despite long-term, broad-scale declines and being the first amphibian species to be listed as Federally Endangered, there have been few attempts to characterize genetic variation within B. houstonensis. McHenry’s16 range-wide study of population genetic structure in B. houstonensis recovered nine, largely geographically concordant clusters (i.e., five in Bastrop County and one each in Austin, Milam, Colorado, and Leon Counties) that did not exhibit reciprocal monophyly of mtDNA haplotypes but differed significantly in nuDNA allele frequencies. Further, both mtDNA sequences and microsatellite genotypes showed that B. houstonensis from Austin County were highly differentiated from all other B. houstonensis16. McHenry also reported low levels of contemporary genetic connectivity but considerable range-wide allelic diversity that may have been due to a larger historical species range and higher historical population connectivity. As a result, management recommendations were to, at minimum, supplement Austin and Bastrop populations while protecting and restoring dispersal corridors to enhance future population connectivity16.
Further, there has been historic uncertainty over the taxonomic status of B. houstonensis since these toads are only slightly differentiated in morphology and mating call from B. americanus17,18,19. Phylogenetic relationships within the Bufo americanus species group have focused on more widely distributed species while B. houstonensis has received considerably less attention20,21,22. Phylogenetic analyses that included B. houstonensis corroborated its placement within the B. americanus group but did not resolve relationships between B. houstonensis and B. americanus16,23,24. These previous analyses were based on mtDNA. Variation in nuclear and mitochondrial genes may be strikingly different for a single species for a variety of reasons such as sex-biased dispersal, incomplete lineage sorting, and asymmetric introgression. The mode of transmission of mitochondrial (i.e., maternal) and nuclear (i.e., biparental) genes further explains differences in sensitivity of each marker to fluctuations in demography and spatial range25. Given such potential for mitonuclear discordance, we sought to resolve these taxonomic ambiguities using several independent markers. Information regarding the genetic distinctiveness of B. houstonensis is critical to establishing whether costly propagation and supplementation efforts continue to be warranted.
Here we present a high-resolution genomic analysis of B. houstonensis, two closely related toad species in the B. americanus group, and a sympatric Middle American toad species (Bufo [= Incilius] nebulifer) to primarily reconcile uncertainties in the taxonomic status of B. houstonensis. We applied a double digest restriction-site associated DNA sequencing (ddRADseq) approach for genome-wide sampling of several nuclear loci and reconstructed phylogenetic relationships using a matrix of concatenated loci. Additionally, we sought to use our increased density of marker sampling to consolidate McHenry’s16 assessment of intraspecific genetic variation in B. houstonensis.
Results
Phylogenetic relationships and divergence times among Nearctic and Middle American taxa
We obtained a total 11,563,297 raw reads with 124,337 ± 74,570 (Mean ± SD) reads per sample (Table S1). We observed a maximum individual pairwise data-missingness of 0.99 across clustering thresholds with Pearson’s coefficient of correlation between data-missingness and genetic distance ranging from 0.53 to 0.70. Specifically, we observed a very low proportion of shared loci between our Nearctic and Middle American species, with the level of such missing data independent of the clustering threshold used to establish homology among reads (Figs. S1–S12). We observed, on average, ~ 4% (n = 50) of all loci recovered in B. nebulifer samples were shared with Nearctic study species. Within our Nearctic study group, on average, ~ 60% (n = 729) of loci were shared among species. For our entire dataset, we retained 1170 loci with a total 39,616 Single Nucleotide Polymorphisms (SNPs). Our sequence matrix of 91 samples had 557,972 sites with 49.80% missing sites.
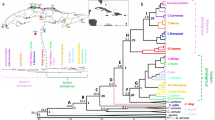
The Bayesian consensus tree for our entire dataset (Fig. 2) showed the greatest genetic divergence between Nearctic and Middle American species, while the second largest genetic divergence was between B. woodhousii and B. houstonensis. In comparison, B. houstonensis and B. americanus were less genetically divergent but each species grouped in distinct clades (Fig. 2).

Bayesian majority rule consensus tree generated from concatenated ddRAD sequence supermatrix with 1170 loci from 91 individuals that includes 6 American Toads (Bufo [= Anaxyrus] americanus), 48 Houston toads (B. houstonensis), 18 Woodhouse’s Toads (B. woodhousii), and 19 Gulf Coast Toads (B. nebulifer). Branch lengths between Nearctic and Middle American species (denoted in green) indicate the greatest divergence. The next largest genetic divergence was between B. woodhousii (denoted in red) and B houstonensis. In comparison, B. houstonensis (denoted in blue) and B. americanus (denoted in black) were less genetically divergent but each species grouped in distinct clades.
Genotype structure and ancient admixture
From all individual alignments, we analyzed 151,115,496 sites and retained 292,117 sites after filtering. For the subset of B. americanus and B. houstonensis alignments, we analyzed 91,537,049 sites and retained 107,509 sites after filtering. With alignments of B. houstonensis alone, we analyzed 84,066,898 sites and retained 92,228 sites after filtering. We observed separation between B. americanus and B. houstonensis with the first principal component (PC1) accounting for 18.6% of total variation in our dataset while the second principal component accounted for 2.3% of total variation in our dataset. Additionally, structure within populations of B. houstonensis and B. americanus was indicated along both principal components (Fig. 3a). Similarly, we saw separation among populations of B. houstonensis from Bastrop, Lee, Austin, and Leon Counties, with PC1 and PC2 accounting for 19.3% and 2.5% of total variation in our dataset (Fig. 3b). Admixture analysis between B. americanus and B. houstonensis supported K = 4 (i.e., highest ΔK value; Fig. S18) grouping levels, with B. americanus assigning to one genomic cluster, and three other genomic clusters identified in B. houstonensis (Fig. 3c). Admixture analysis within B. houstonensis supported K = 4 (i.e., highest ΔK value; Fig. S19) grouping levels, with B. houstonensis from Austin, Leon, Lee, and Bastrop Counties assigning to one genomic cluster each (Fig. 3d). Individuals sampled from Milam County (n = 2) showed mixed ancestry from three and four genomic clusters while the lone individual sampled from Robertson County showed shared ancestry with toads from all four genomic clusters.

(A) Principal component analysis using 107,509 sites from 8 American Toads (Bufo [= Anaxyrus] americanus) and 48 Houston toads (B. houstonensis). The first two components of this PCA accounted for 20.9% of the variation in our dataset. (B) Principal component analysis using 92,228 sites from 48 Houston toads (Bufo [= Anaxyrus] houstonensis). The first two components of this PCA accounted for 21.8% of the variation in our dataset. (C) Admixture analysis of 8 American Toads (Bufo [= Anaxyrus] americanus) and 48 Houston toads (B. houstonensis) distinguished between B. americanus and B. houstonensis. (D) Admixture analysis of 48 Houston toads (Bufo [= Anaxyrus] houstonensis) identified 4 genomic clusters at the top level of hierarchical structure, enabling delineation of one management unit each from Bastrop, Lee, Austin, and Leon Counties.
In six gene flow models that used B. americanus as the sister group (H1), we observed a significantly higher ABBA count in one model that indicated gene flow between B. houstonensis (H2) and B. woodhousii (H3; Z = 14.54; Table 1, Fig. 4). We did not observe significant gene flow between B. americanus (H1) and B. nebulifer (H3) or between B. houstonensis (H2) and B. nebulifer (H3; Z = 0.68; Table 1, Fig. 4). Similarly, we did not find support for significant gene flow between B. woodhousii (H2) and B. nebulifer (H3; Z = 1.01; Table 1, Fig. 4). In the remaining three models, we observed significantly higher BABA counts. Two models indicated significant gene flow between B. americanus (H1) and B. houstonensis (H3; Z = − 355.05; Table 1; Fig. 4) and B. americanus (H1) and B. woodhousii (H3; Z = − 189.58; Table 1; Fig. 4) relative to B. nebulifer (H2). One model supported significant gene flow between B. houstonensis (H3; Z = − 55.85; Table 1; Fig. 4) and B. americanus (H1) relative to B. woodhousii (H2).

Z-scores from ABBA-BABA tests of ancient gene flow between populations of Nearctic American Toads (Bufo [= Anaxyrus] americanus), Houston toads (B. houstonensis), Woodhouse’s Toads (B. woodhousii), and Middle American Gulf Coast Toads (B. nebulifer). Significant gene flow between taxa is indicated in red font. We aligned sequences to a common toad (Bufo bufo) reference genome and assumed allele states in this reference sequence were ancestral states. We used 292,117 sites and corroborated ancient gene flow between Nearctic species (i.e., B. americanus, B. houstonensis, and B. woodhousii) but found no support for gene flow between Nearctic and Middle American (i.e., B. nebulifer) species.
Three of the four models that used B. houstonensis as the sister group (H1) showed significantly higher BABA counts. We found support for significant gene flow between B. americanus (H3; Z = − 338.61; Table 1, Fig. 4) and B. houstonensis (H1) as well as between B. woodhousii (H3; Z = − 240.64; Table 1, Fig. 4) and B. houstonensis (H1) relative to B. nebulifer (H2). Significant gene flow was also supported between B. houstonensis (H1) and B. americanus (H3; Z = − 76.21; Table 1; Fig. 4) relative to B. woodhousii (H2). We did not find support for significant gene flow between B. houstonensis (H1) and B. nebulifer (H3; Z = − 0.76; Table 1; Fig. 4) or between B. woodhousii (H2) and B. nebulifer (H3; Table 1; Fig. 4).
We saw significantly higher ABBA counts in models with B. nebulifer as the sister group (H1), wherein significant gene flow was indicated between B. houstonensis (H3; Z = 235.78; Table 1; Fig. 4) and B. woodhousii as well as between B. americanus (H3; Z = 184.47; Table 1; Fig. 4) and B. woodhousii.
Discussion
We observed uneven sequencing coverage with heterogeneity in input among samples and variation in locus coverage. Multiple sources such as variable recovery during library preparation, insufficient sequencing coverage, and mutations at enzyme recognition sites have been attributed to missing RADseq data26,27,28,29. Data sets generated using restriction enzymes with recognition sites that are 6-bp long result in a larger number of fragments relative to when using restriction enzymes with recognition sites that are 8-bp in length27. Empirical data suggest that the number of loci shared among taxa in 6-bp cutter data sets are better predicted by sequencing coverage than phylogenetic distance, suggesting such data sets may be consistently under-sequenced27. We acknowledge uneven and low depth of sequencing is a source of missing data for our study. However, we also observed hierarchical data loss in our study, with a strong inverse correlation between number of shared loci and phylogenetic distance. Since our missing data were phylogenetically distributed27, we used computationally tractable, sparse supermatrices of concatenated ddRAD data to potentially increase resolution in our phylogenetic trees30,31 relative to prior taxonomic assessments.
Previous mtDNA based studies that included constituents of the Bufo americanus species group have inferred non-traditional relationships among B. americanus and B. houstonensis. Pauly et al.24 inferred B. woodhousii was sister to a clade that included B. americanus, B. houstonensis, and B. velatus, with monophyly of mitochondrial haplotypes in B. a. charlesmithii, B. houstonensis, and B. velatus to the exclusion of B. a. americanus. Goebel et al.23 inferred similar results with B. woodhousii grouping sister to B. hemiophrys which in turn was sister to a clade that included B. houstonensis and B. americanus. The Bayesian Consensus tree for our entire dataset assigned B. houstonensis and B. americanus to separate groups. Further, lower genetic divergence was observed between B. americanus and B. houstonensis relative to the divergence of each from a clade of B. woodhousii. These results support the identity of B. houstonensis as a taxon that is distinct from, but likely derived from a precursor of, present-day B. americanus. We observed geographically concordant structure within B. houstonensis. Toads from Bastrop and Lee Counties grouped sister to each other, albeit with low (< 70%) support and with five of 16 samples from Bastrop County appearing unresolved within this grouping. Samples from B. houstonensis populations in Austin and Leon grouped distinct from each other and from toads sampled in Bastrop and Lee Counties. Within a clade of B. americanus, the single Eastern American toad (B. americanus americanus) from New York grouped sister to Dwarf American toads (B. americanus charlesmithii) from Missouri.
Additionally, our results also supported genetic structure within the clade of B. woodhousii. Toads from the South-Central Plains in Oklahoma (i.e., Southeast Oklahoma) and Post Oak Savannah region in Texas (i.e., Northeast Texas) formed one group, while B. woodhousii from Blackland Prairie (i.e., North and Central Texas), High Plains (i.e., Northwest Texas), and Central Great Plains in Oklahoma (i.e., North and Central Oklahoma) formed a second group. We lack the data to provide insight into reasons for the observed structure within B. woodhousii. Given that B. woodhousii has a broad distribution32, we suggest prioritizing efforts to characterize genetic variation in the species to identify management units of evolutionary and potential conservation significance. Finally, the greatest genetic divergence in our dataset was between Nearctic taxa and B. nebulifer. However, we observed low support (< 70%) for within group structure in B. nebulifer, which can be attributed to a low number of loci available within constituents of this clade.
Supermatrices of concatenated ddRAD sequence data enable effective tests of evolutionary hypotheses27,31. However, we observed low and uneven sequencing coverage across samples in our dataset. Given such low coverage in our dataset, we sought to also use an approach that avoided basing downstream analysis on raw counts of sequenced bases or called genotypes33. For this reason, we also incorporated a probabilistic approach that used genotype likelihoods to ascertain genetic distinctiveness between B. houstonensis and B. americanus, determine genetic structure within B. houstonensis, and test for historic gene flow among constituent taxa in our study. We observed two B. americanus individuals were clustered in the center of the coordinate space of our PCA plot- indicating these were either different from the remainder of B. americanus (n = 6) in our study or more likely, that these samples clustered in the middle due to a low number of input reads and sequencing coverage. However, admixture analyses clearly corroborate the separation of B. americanus and B. houstonensis into distinct clusters while also indicating population genetic structure within B. houstonensis. Reproductive isolation of B. houstonensis from B. americanus corroborates prior phylogenetic support for the species identity of B. houstonensis.
Using genotypes from ten polymorphic microsatellite markers in 439 individual B. houstonensis from six counties in Texas, McHenry observed two clusters (i.e., North and South) at the uppermost level of hierarchical structure. Both clusters occurred in all counties, except Austin County in which only one cluster (i.e., North) was seen16. Second order analyses indicated at least nine clusters at varying levels of genetic differentiation. Five of these unique clusters were detected in Bastrop County alone and the remaining four were one cluster each from Austin, Milam, Colorado, and Leon Counties16. Sampling bias was a likely influence in McHenry’s study as 95% of all B. houstonensis samples were detected in Bastrop County, where the largest known population occurs16,19. We used genome-wide DNA sequence data with a smaller proportion (33%) of B. houstonensis from Bastrop County. Since we sought comparison with McHenry’s16 characterization of population genetic structure, 40% of our B. houstonensis samples were also used in her study. At the uppermost level of hierarchical structure for B. houstonensis populations in our study, we observed four unique genomic clusters. Toads from Austin, Leon, Lee, and Bastrop Counties were distinct from each other. Additionally, individual toads from Milam County showed mixed ancestry from three and four genomic clusters while the single available sample from Robertson County showed shared ancestry from all four genomic clusters. This is a sampling artefact, since these localities were represented by only one or two individuals.
Despite low sequencing coverage, the genome-wide DNA sequence data used in this study provides considerable power in detecting population genetic structure within B. houstonensis. We recognize four Management Units (MUs) in B. houstonensis and thus, conservation actions for species recovery need to be specific for each MU. Population supplementation to raise juvenile survivorship and habitat restoration to enhance viability of additional subpopulations have been identified as primary measures to reduce extinction risk for B. houstonensis34. Further, an inherently low juvenile survival in B. houstonensis35,36 requires scaled-up investment in captive propagation efforts for population supplementation to be successful34. These conservation actions while expensive are warranted, given the endangered status of this relict species7,8,34. Further, given populations in each county (i.e., Austin, Leon, Lee, and Bastrop) are distinct from each other, we suggest that captive cohorts meant for supplementation need to be of the same provenance as resident populations. The current genetic clustering results are indicative of low levels of gene flow between populations, which contrasts with McHenry’s16 suggestions that B. houstonensis was historically larger and more contiguously distributed with few populations in complete isolation.
McHenry’s16 hypothesis was based on an overall high genetic diversity as evidenced by a considerable number of mitochondrial haplotypes (\(n=14\)) and microsatellite alleles per locus (n = 8–29) across the range of B. houstonensis16. Further, some of these mitochondrial haplotypes and microsatellite alleles were detected range-wide16. Models based on coalescent theory are required to characterize population-level processes (e.g., demographic changes, migration, and isolation) that have resulted in the genetic structure we observed within B. houstonensis37. However, within the context of historic processes, we tested our dataset for signs of historical admixture events. Gene flow between pairs of Nearctic study species was significant. Using Z scores as indices of strength of gene flow, we observed the strongest support for gene flow between B. houstonensis and B. americanus. Second, gene flow between B. houstonensis and B. woodhousii received stronger support relative to gene flow between B. americanus and B. woodhousii. Natural hybridization has been previously documented among members of the Bufo americanus species group and potentially introduced novel genetic variation at the beginning of a radiation of these toads16,20,21,38. Fontenot et al.20 observed high levels of gene flow among pairs in the Bufo americanus species group that occurred in geographic proximity and shared similar male advertisement calls. The substantial signal of ancient gene flow between B. americanus and B. houstonensis may be concordant with a pattern of erstwhile range expansions and secondary contact between species pairs that were geographically proximate and share similar advertisement calls prior to complete reproductive isolation17,20.
Gene flow models did not indicate historic admixture events between B. nebulifer and any Nearctic study species. However, hybridization events between B. houstonensis and B. nebulifer as well as between B. woodhousii and B. nebulifer have been documented16,39. McHenry conducted a range-wide genetic assessment of B. houstonensis for interspecific admixture with B. nebulifer and estimated that nearly 10% of all sampled B. houstonensis were admixed16. Thus, hybridization between B. houstonensis and B. nebulifer can be characterized as an added threat to persistence of the former species. Gene flow models in this study do not support any historic context for hybridization between Nearctic toad taxa and B. nebulifer, possibly due to reproductive isolating mechanisms such as offset breeding times and differences in preferred habitat16,22. Hybridization of Nearctic taxa with B. nebulifer may be more contemporaneous and due to the disruption of ecological barriers and temporal isolation in breeding activity consequent to human-induced land-use and climate change16,22,40. We draw attention to the potential of such hybridization as a contemporaneous driver of extinction in endangered Nearctic toads (e.g., B. houstonensis). We suggest population-level genetic assessments for Nearctic toads that are sympatric with B. nebulifer to determine whether interspecific admixture poses a threat to species persistence.
Historically, the taxonomic status of B. houstonensis has remained uncertain9,16,19. Our study clarified that B. houstonensis is a species distinct from B. americanus via a phylogenetic analysis of a concatenated sequence matrix and an admixture analysis that used genotype likelihoods. An admixture analysis that used genotype likelihoods identified a minimum of four genetically distinct areas or populations (i.e., MUs) within B. houstonensis. These included eastern populations of B. houstonensis from Austin and Leon, as well as western populations from Bastrop and Lee Counties. Given that B. houstonensis is a distinct species with a high risk of extinction, conservation measures such as habitat restoration to increase viability of additional subpopulations and population supplementation to raise juvenile survival are warranted34,35,36. We suggest that captive propagation and population supplementation to raise juvenile survival need to be cognizant of the underlying genetic structure observed within B. houstonensis. Our study strengthens the need and ability for B. houstonensis conservation while re-affirming the evolutionary novelty of an endangered relict.
Methods
Sampling and DNA extraction
We included 48 B. houstonensis sampled from 2001 to 2015 throughout much of their geographic range (Fig. 1, Table S1). We specifically chose B. houstonensis samples from or prior to 2015 since these preceded full-scale population supplementation efforts41. We had a lower number of samples available for B. americanus. We used 8 samples of B. americanus collected from 1996 to 2003, that included 6 samples of Dwarf American toads (B. americanus charlesmithii) and 2 samples of Eastern American toads (B. americanus; Table S1). We included 18 B. woodhousii sampled across much of their range in Texas, USA from 2002 to 2015 and 19 B. nebulifer sampled in Texas, USA and Tamaulipas, Mexico from 2002 to 2014 (Table S1). We specifically included B. nebulifer due to previously documented hybridization with B. houstonensis16,39. All tissue samples were stored in 95% ethanol and afterwards at − 80 °C for long- term storage. Tissue samples were deposited in the Michael R. J. Forstner Frozen Tissue catalog currently held at Texas State University, San Marcos, Texas.
For each putative species we sought to sample the largest spatial extent of species’ range, within the constraints of sample availability. Since we had samples from throughout most of the range of B. houstonensis relative to other study species, the number of samples for the former is considerably larger. However, we sought to exceed a minimum of four samples per taxon. We used a lower number of samples relative to prior studies16 but attempted to offset this with a higher number of markers per sample.
Sample localities for B. woodhousii were both sympatric and allopatric to B. houstonensis and B. americanus, while localities for B. nebulifer were both sympatric and allopatric to B. houstonensis and B. woodhousii (Fig. 1). Sample collection from toads was authorized under Federal Fish and Wildlife Permits (TE039544-0, TE039544-1, TE039544-2, TE004472-0, and TE004472-1), Texas Parks and Wildlife Department Scientific Research Permits (SPR-0102-191, SPR-0290-022). Our experimental protocol was approved by the Institutional Animal Care and Use Committee at Texas State University via permits 5Qrs45_02, HGVMAD_02, 04-0485904A30, 0713_0428_07, 0810_0208_11, and IACUC 201648186. Collection and transport of B. nebulifer samples from Mexico were authorized under CITES Permit Number 05US704066/9 and Costa Rica MINAE Resolucion Numbers 237-98-OFAU and 019-2000-OFAU (Collecting Licenses 0023073 and 0205-00). All procedures were performed in accordance with the relevant guidelines and regulations of each institution and where applicable, in accordance with ARRIVE guidelines (https://arriveguidelines.org/).
Tissue samples included toe-clips or blood tissue from adult toads and tail-clips from tadpoles. All tadpoles used in this study were the only available samples from Lee County. Individual tadpoles were sampled from three different ponds and likely belonged to different cohorts as evidenced by Gosner stage of development. DNA was extracted from samples in ethanol and lysis buffer using Qiagen DNeasy Blood and Tissue kits following manufacturer protocol. High-molecular weight DNA extracted from samples was examined for quality on agarose gels and quantified with a Qubit version 2.0 fluorometer (Thermo Fisher Scientific).
ddRADseq data collection
Genomic libraries were prepared and sequenced at the University of Texas Genomic Sequencing and Analysis Facility (Austin, TX). The ddRADseq data were collected using the protocol described by Peterson et al.42. Samples were normalized to 10 ng/uL gDNA and ~ 100 ng of gDNA were digested with 1 unit each of EcoRI and MspI (at 10,000×) in a single reaction with the manufacturer recommended buffer (New England Biolabs) for 2 h at 37 °C. Samples were purified with AMPure XP beads (Beckman Coulter Life Sciences) prior to ligation of barcoded Illumina adapters onto the fragments. Oligonucleotide sequences used in barcoding and adding Illumina indices to fragments were per Peterson et al.42. Prior to pooling, samples were purified with AMPure XP beads to enrich for a broad size range (150–800 bp). Equimolar amounts of individual libraries were pooled to constitute four libraries. Pooled libraries were size selected over a narrow range (540–660 bp after accounting for adapter length) using a Pippin Prep (Sage Science) size fractionator. PCR amplification of size selected DNA was performed using 1 μL each of 25 μM Illumina dual index primers, 23 μL of size selected DNA, and 25 μL of NEBNext High-Fidelity 2× PCR master mix (New England Biolabs). PCR was performed with an initial denaturation at 98 °C for 30 s, followed by 12 cycles each consisting of denaturing at 98 °C for 10 s, annealing at 65 °C for 30 s, and extension at 72 °C for 30 s, and a final extension step at 72 °C for 5 min. Subsequently, samples were purified using AMPure XP beads (Beckman Coulter Life Sciences) and each library was sequenced, in two replicate runs, on a single lane of an Illumina Miseq platform under a 300 bp paired-end read protocol (2 \(\times\) 300 bp).
De novo assembly and phylogenetic analyses
We processed raw Illumina reads with ipyrad version 0.9.5943. We demultiplexed samples using unique inline barcodes, allowing no mismatches, for each sample and trimmed reads for adapter contamination. Sites with accuracy of base calls under 99% (Phred Quality Score < 20) were assigned ‘N’ characters and reads with ≥ 5 N’s were discarded. Within the ipyrad pipeline, read pairs were merged and clustered with VSEARCH44 and subsequently aligned with MUSCLE45.
Clustering thresholds within the ipyrad pipeline are used to establish homology among reads within samples and identify putative orthologs among samples46. We diagnosed the optimal clustering threshold value by examining the fraction of inferred paralogous clusters, individual heterozygosity, total number of SNPs, and correlation between individual pairwise data-missingness and genetic distance across ten separate runs with clustering thresholds that ranged from 0.50 to 0.99 usually in steps of 0.05 (e.g., 0.50 to 0.55)47. We repeated an examination of clustering thresholds that ranged from 0.90 to 0.99 but in steps of 0.01 while using fraction of inferred paralogs, individual heterozygosity, and total number of SNPs as diagnostic parameters. For runs that increased in steps of ~ 0.05, we used a minimum sequencing depth of 2, based on a relatively low number of raw reads per sample, and a minimum of 7 samples (~ 8%) having data to retain a locus. For ipyrad runs that increased in steps of 0.01, we used a minimum sequencing depth of 2 and a minimum of 15 samples (~ 16%) having data to retain a locus. We used default values to control alignment quality by excluding consensus sequences with ≥ 5% ambiguous and heterozygous bases and excluding loci with ≥ 20% SNPs and ≥ 8 indels. Additionally, we used default values to detect and filter paralogous clusters by allowing a heterozygous site to occur in a maximum of 50% of samples. Among clustering thresholds that increased in broad steps (i.e., ~ 0.05), we determined a clustering threshold of 0.95 as optimal since this minimized the fraction of inferred paralogous clusters, maximized individual heterozygosity and total number of SNPs, and was the threshold value (Figs. S1–S12) at which the correlation between pairwise data-missingness and genetic distance steeply increased47. Similarly, while examining clustering thresholds at narrow consecutive increases (i.e., steps of 0.01) we determined 0.95 as the optimal clustering threshold as this minimized the fraction of inferred paralogous clusters, maximized individual heterozygosity, and the total number of SNPs (Figs. S13–S15).
We generated an output file in custom format (i.e., *.loci) in ipyrad and used Matrix Condenser to visualize the effects of excluding samples with poor coverage and changing the minimum number of samples to retain a locus43,48,49. We excluded two samples with fewer than 10,000 reads and greater than 95% missing data. One of these samples (MF1103, Table S1) was an eastern American toad (B. americanus americanus) from New York State and the other (MF7399, Table S1) was a dwarf American toad (B. americanus charlesmithii) from Oklahoma. We also retained loci that were recovered for a minimum of 41 samples, with 49.8% missing sites in our sequence matrix for 91 samples. Despite a more stringent filtering threshold, we continued to see a systematic pattern to missing data—with few loci overlapping between Middle American and Nearctic species (Fig. S16).
We used AIC and BIC scores in ModelTest-NG50 to select the best-fit DNA substitution model for concatenated ddRAD loci in our final dataset. We evaluated 24 candidate models across three DNA substitution schemes (i.e., JC, HKY, and GTR) and four rate-heterogeneity models (i.e., Uniform, Invariant, Gamma, Gamma + Invariant). Bayesian analyses were conducted in MrBayes 3.2.751 to generate phylogenetic hypotheses from matrices of concatenated loci for 91 samples. We conducted two independent runs of 3 × 106 generations from random starting trees using four Markov chains (one cold, three heated, temperature of 0.01) sampling every 1000 generations. We used the average standard deviation of split frequencies (ASDF) and potential scale reduction factor (PSRF) as convergence diagnostics, with analyses run until ASDF \(=\) 0.10 and PSRF approached 1.0. We discarded 25% of the samples as burn-in and used the remaining samples to compute a majority consensus tree. We used a B. nebulifer sequence as an outgroup to Nearctic taxa included in our study. We visualized the Bayesian consensus tree from this phylogenetic analysis in FIGTREE, version 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree).
Probabilistic genomic analysis, genotype structure, and ancient admixture
We aligned 48 B. houstonensis and 8 B. americanus sequences processed in ipyrad43 to a common toad (Bufo bufo) reference genome (5.04 Gb in 1307 sequence scaffolds, GenBank assembly accession no.GCA_905171765.1) using the local algorithm set to default parameters in Bowtie252. A majority (99.1%) of this congeneric reference genome was assigned to 11 chromosomal-level scaffolds and represents the closest related high quality genomic resource that was available53. We discarded reads with more than one match to the genome and converted alignments to indexed BAM format using SAMtools54. We merged alignments from each sequencing run and subsequently removed duplicates using Picard Tools. We used the probabilistic framework implemented in Analysis of Next Generation Sequencing Data (ANGSD) for population genetic analysis. Given low sequencing coverage in our dataset, we sought to also use an approach that avoided basing downstream analysis on raw counts of sequenced bases or called genotypes33. We generated beagle likelihood files (doGlf 2) in ANGSD while using a minimum mapping quality score (minMapQ) of 20, minimum base quality score (minQ) of 25, SAMtools genotype likelihood model (GL1), and fixed major and minor allele frequencies (doMaf 1). Further, we designated polymorphic sites at Minor Allele Frequency (MAF) > 0.05 and probability of site not being polymorphic < 1 × 10–6. Downstream analyses (e.g., NgsAdmix) estimated composite likelihoods that were robust to non-independence of sites55.
We assessed relatedness among tadpoles collected from Lee County, prior to including these samples in downstream analyses. We used IBSrelate to identify pairs of related individuals without requiring population allele frequencies and generated KING-robust kinship coefficient estimates for every pair56. We observed negative estimates of kinship coefficients across pairs (Fig. S17) and concluded there was no relatedness among pairs, enabling inclusion of these samples for population genetic analyses. We examined the overall quality of discovered SNPs and their reliability in distinguishing between B. houstonensis and B. americanus as well as among populations (sensu McHenry, 2010) of B. houstonensis by conducting a Principal Component Analysis (PCA) of genetic structure in the ngsCovar module of ngsTools57. We also investigated genetic structure between B. houstonensis and B. americanus as well as among populations of B. houstonensis (sensu McHenry, 2010) by calculating individual admixture proportions at different grouping levels (1–10) using NgsAdmix55. For each between and within-species admixture analyses, we used 10 independent runs of 20,000 iterations and determined the most likely grouping level (K) per Evanno et al.58.
We also sought to detect historical gene flow between B. houstonensis, B. americanus, B. woodhousii, and B. nebulifer and performed the ABBA-BABA test of ancient admixture (or wrong tree topology) in ANGSD59. This provides a way to test the correctness of a hypothetical genetic relationship among four groups. We examined distribution of ancestral (‘A’) and derived (‘B’) alleles across the genomes of four groups of individuals that include an ancestral outgroup. Two allelic patterns ‘ABBA’ or ‘BABA’ should occur equally frequently under a scenario with no introgression. An excess of either ‘ABBA’ or ‘BABA’ would indicate gene flow between two taxa59. We aligned all individual sequences in each group to the Bufo bufo reference genome and determined polymorphic sites in ANGSD, as described previously. We assessed the distribution of ancestral (‘A’) and derived (‘B’) alleles within taxa wherein allele states in Bufo bufo were considered ancestral states and all bases at each position were considered (doAbbababa2 1). We evaluated 12 gene flow models for ancient admixture between sister Nearctic representatives as well as between Middle American and Nearctic species. We set B. americanus, B. houstonensis, and B. nebulifer each as the sister group (H1) in 6, 4, and 2 models, respectively. The number of ABBA and BABA counts and Patterson’s D-statistic were calculated without error correction and ancient transition removal. Significance of the D-statistic was assessed with Z values from a block Jackknife procedure implemented in the module59.
Data availability
Nexus files used in phylogenetic analyses, XML files used in BEAST analyses, and individual alignment files (bam files) used in probabilistic genomic analyses are available in the Dryad Digital Repository (https://doi.org/10.5061/dryad.m37pvmd52). All scripts used in phylogenetic and population genetic analysis will be made available by Shashwat Sirsi (s_s477@txstate.edu) on request.
References
Howard, S. D. & Bickford, D. P. Amphibians over the edge: Silent extinction risk of data deficient species. Divers. Distrib. 20, 837–846 (2014).
Stuart, S. N. et al. Status and trends of amphibian declines and extinctions worldwide. Science 306, 1783–1786 (2004).
Kiesecker, J. M., Blaustein, A. R. & Belden, L. K. Complex causes of amphibian population declines. Nature 410, 681–684 (2001).
Lips, K. R., Burrowes, P. A., Mendelson, J. R. III. & Parra-Olea, G. Amphibian declines in Latin America: Widespread population declines, extinctions, and impacts. Biotropica 37, 163–165 (2005).
Sodhi, N. S. et al. Measuring the meltdown: Drivers of global amphibian extinction and decline. PLoS ONE 3, e1636 (2008).
Brown, L. E. The status of the near-extinct Houston toad (Bufo houstonensis) with recommendations for its conservation. Herpetol. Rev. 6, 37–40 (1975).
Gottschalk, J. S. United States list of endangered native fish and wildlife. Fed. Regist. 35, 16047–16048 (1970).
Honegger, R. E. Red Data Book Vol. 3 Amphibia and Reptilia. (International Union for Conservation of Nature and Natural Resources, 1970).
Blair, W. F. Bufo of North and Central America. In Evolution in the Genus Bufo (ed. Blair, W. F.) 93–101 (University of Texas Press, 1972).
Blair, W. F. Evolution in the Genus Bufo (University of Texas Press, 1972).
United States Fish and Wildlife Service. Houston Toad (Bufo houstonensis) 5-Year Review: Summary and Evaluation. https://www.fws.gov/southwest/es/documents/r2es/houstontoad_5-yr_review_nov2011.pdf (Accessed 12 December 2023) (United States Fish and Wildlife Service, 2011).
Brown, L. E. & Mesrobian, A. Houston toads and Texas politics. In Amphibian Declines (ed. Lannoo, M. J.) 150–167 (University of California Press, 2005).
Buzo, D. A GIS Model for Identifying Potential Breeding Habitat for the Houston Toad (Bufo houstonensis). M.S. Thesis, Texas State University, San Marcos (2008).
Kennedy, J. Spawning season and experimental hybridization of the Houston toad, Bufo houstonensis. Herpetologica 17, 239–245 (1962).
Sanders, O. A new species of toad, with a discussion of morphology of the bufonid skull. Herpetologica 9, 25–47 (1953).
McHenry, D. Genetic Variation and Population Structure in the Endangered Houston Toad in Contrast to Its Common Sympatric Relative, the Coastal Plain Toad. Doctoral Dissertation, University of Missouri, Columbia (2010).
Blair, W. F. Evolutionary relationships of North American toads of the genus Bufo: A progress report. Evolution 17, 1–16 (1963).
Brown, L. E. Bufo houstonensis. In Catalogue of American Amphibians and Reptiles (ed. Zweifel, R. G.) 131–133 (University of Texas Press, 1973).
Potter, F. E., Brown, L. E., McClure, W. L., Scott, N. J. & Thomas, R. A. Recovery Plan for the Houston Toad (Bufo houstonensis) (United States Fish and Wildlife Service, 1984).
Fontenot, B. E., Makowsky, R. & Chippindale, P. T. Nuclear-Mitochondrial discordance and gene flow in a recent radiation of toads. Mol. Phylogenet. Evol. 59, 66–80 (2011).
Masta, S. E., Sullivan, B. K., Lamb, T. & Routman, E. J. Molecular systematics, hybridization, and phylogeography of the Bufo americanus complex in Eastern North America. Mol. Phylogenet. Evol. 24, 302–314 (2002).
Vogel, L. S. & Johnson, S. G. Estimation of hybridization and introgression frequency in toads (genus: Bufo) using DNA sequence variation at mitochondrial and nuclear loci. J. Herpetol. 42, 61–75 (2008).
Goebel, A. M., Ranker, T. A., Corn, P. S. & Olmstead, R. G. Mitochondrial DNA evolution in the Anaxyrus boreas species group. Mol. Phylogenet. Evol. 50, 209–225 (2009).
Pauly, G., Hillis, D. M. & Cannatella, D. C. The history of a Nearctic colonization: Molecular phylogenetics and biogeography of the Nearctic toads (Bufo). Evolution 58, 2517–2535 (2004).
Després, L. One, two or more species? Mitonuclear discordance and species delimitation. Mol. Ecol. 28, 3845–3847 (2019).
Cariou, M., Duret, L. & Charlat, S. Is RAD-seq suitable for phylogenetic inference? An in-silico assessment and optimization. Ecol. Evol. 3, 846–852 (2013).
Eaton, D. A., Spriggs, E. L., Park, B. & Donoghue, M. J. Misconceptions on missing data in RAD-seq phylogenetics with a deep-scale example from flowering plants. Syst. Biol. 66, 399–412 (2017).
Escudero, M., Eaton, D. A., Hahn, M. & Hipp, A. L. Genotyping-by-sequencing as a tool to infer phylogeny and ancestral hybridization: A case study in Carex (Cyperaceae). Mol. Phylogenet. Evol. 79, 359–367 (2014).
Huang, H. & Knowles, L. L. Unforeseen consequences of excluding missing data from next-generation sequences: Simulation study of RAD sequences. Syst. Biol. 65, 357–365 (2016).
Leache, A. D. et al. Phylogenomics of phrynosomatid lizards: Conflicting signals from sequence capture versus restriction site associated DNA sequencing. Genome Biol. Evol. 7, 706–719 (2015).
Wagner, C. E. et al. Genome-wide RAD sequence data provide unprecedented resolution of species boundaries and relationships in the Lake Victoria cichlid adaptive radiation. Mol. Ecol. 22, 787–798 (2013).
Stebbins, R. C. A Field Guide to Western Reptiles and Amphibians 3rd edn. (Houghton Mifflin, 2003).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: Analysis of next generation sequencing data. BMC Bioinform. 15, 356 (2014).
Forstner, M. R. J. & Crump, P. Houston toad population supplementation in Texas, USA in global re-introduction perspectives: 2011. In More Case Studies from Around the Globe (ed. Soorae, P. S.) 71–76 (IUCN/SSC Re-introduction Specialist Group & Environment Agency, 2011).
Greuter, K. Survivorship and Growth in the Endangered Houston Toad. Master’s Thesis. Texas State University, San Marcos (2004)
Swannack, T. M., Grant, W. E. & Forstner, M. R. J. Projecting population trends of endangered amphibian species in the face of parametric uncertainty. Ecol. Model. 220, 148–159 (2009).
Sukumaran, J. & Knowles, L. L. Multispecies coalescent delimits structure, not species. Proc. Natl. Acad. Sci. U.S.A. 114, 1607–1612 (2017).
Green, D. M. & Parent, C. Variable and asymmetric introgression in a hybrid zone in the toads, Bufo americanus and Bufo fowleri. Copeia 2003, 34–43 (2003).
Hillis, D. M., Hillis, A. M. & Martin, R. F. Reproductive ecology and hybridization of the endangered Houston toad (Bufo houstonensis). J. Herpetol. 18, 56–72 (1984).
Milko, L. V. Integrating museum and GIS data to identify changes in species distributions driven by a disturbance-induced invasion. Copeia 2012, 307–320 (2012).
Valles, D. et al. Houston Toad Recovery: A Comprehensive Monitoring, Evaluation, and Support Program for Head-Start Success (Texas Parks and Wildlife Department, 2019).
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7, e37135 (2012).
Eaton, D. A. & Overcast, I. Ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics 36, 2592–2594 (2020).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 4, e2584. https://doi.org/10.7717/peerj.2584 (2016).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Eaton, D. A. PyRAD: Assembly of de novo RADseq loci for phylogenetic analyses. Bioinformatics 30, 1844–1849 (2014).
McCartney-Melstad, E., Gidiş, M. & Shaffer, H. B. An empirical pipeline for choosing the optimal clustering threshold in RADseq studies. Mol. Ecol. Resour. 19, 1195–1204 (2019).
De Medeiros, B. A. S. & Farrell, B. D. Whole-genome amplification in double-digest RADseq results in adequate libraries but fewer sequenced loci. PeerJ 6, e5089. https://doi.org/10.7717/peerj.5089 (2018).
de Medeiros, B. A. Matrix Condenser v.1.0. https://github.com/brunoasm/matrix_condenser/ (2019).
Darriba, D. et al. ModelTest-NG: A new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294 (2020).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Streicher, J. W. The genome sequence of the common toad, Bufo bufo (Linnaeus, 1758). Wellcome Open Res. 6, 1. https://doi.org/10.12688/wellcomeopenres.17298.1 (2021).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Skotte, L., Korneliussen, T. S. & Albrechtsen, A. Estimating individual admixture proportions from next generation sequencing data. Genetics 195, 693–702 (2013).
Waples, R. K., Albrechtsen, A. & Moltke, I. Allele frequency‐free inference of close familial relationships from genotypes or low‐depth sequencing data. Mol. Ecol. 28, 35–48 (2019).
Fumagalli, M., Vieira, F. G., Linderoth, T. & Nielsen, R. ngsTools: Methods for population genetics analyses from next-generation sequencing data. Bioinformatics 30, 1486–1487 (2014).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Soraggi, S., Wiuf, C. & Albrechtsen, A. Powerful inference with the D-statistic on low-coverage whole-genome data. G3 Genes Genom. Genet. 8, 551–566 (2018).
Acknowledgements
Many thanks to all those who helped with sample collection and laboratory work: Jim Bell, D.J. Stout, Lauren Schumacher, Heather Diedrick, Jessica Podnar, and Drs. Todd Swannack, Donald Brown, Ivana Mali, and Melissa Jones. The authors are also thankful to the Boy Scouts of America and stakeholders of the Lost Pines Habitat Conservation Plan for facilitating site access and sample collection. Thanks to Dr Chris Nice and Thom Marshall for their tips on analytical pipelines. They thank Saniya Chaplod for assistance with creating figures. They would like to thank the USFWS, particularly Jeff Hill, for enabling our research. This work was supported by the USFWS, Magellan Midstream Partners, L.P., the Bastrop Utilities HCP, and the Alexander Stone Endowment for Genetics.
Author information
Authors and Affiliations
Contributions
S.S., D.R., and M.R.J.F. designed the study; S.S. and D.R. performed laboratory work and M.R.J.F. contributed reagents; S.S. analyzed the data; S.S. wrote the manuscript with contributions and oversight from D.R. and M.R.J.F.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sirsi, S., Rodriguez, D. & Forstner, M.R.J. Using genome-wide data to ascertain taxonomic status and assess population genetic structure for Houston toads (Bufo [= Anaxyrus] houstonensis). Sci Rep 14, 3306 (2024). https://doi.org/10.1038/s41598-024-53705-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-53705-w
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
