
Abstract
Several observational studies have revealed an association between autoimmune diseases (AIDs) and colorectal cancer (CRC), although their causal association remained controversial. Therefore, our study used a two-sample Mendelian randomization (MR) analysis to verify the causal association between AIDs and CRC. We employed three common MR approaches, including inverse variance weighted (IVW), weighted median, and MR-Egger methods, to assess the causal association between type 1 diabetes (T1D), systemic lupus erythematosus, rheumatoid arthritis, psoriasis, multiple sclerosis, juvenile idiopathic arthritis, celiac disease, and primary sclerosing cholangitis (PSC) and CRC. The reverse MR analysis was performed to assess the possibility of reverse causation. To evaluate the validity of the analysis, we also performed sensitivity analysis, such as the heterogeneity test, the horizontal pleiotropy test, and the leave-one-out sensitivity analysis, and validated the results in the validation cohort. Our results showed that genetically predicted T1D was nominally associated with a lower risk of CRC (IVW OR = 0.965, 95% CI = 0.939–0.992, P = 0.012). However, genetic susceptibility to psoriasis nominally increased the risk of CRC (IVW OR = 1.026, 95% CI = 1.002–1.050, P = 0.037). Genetically predicted PSC had a significant causal effect on the increasing risk of CRC (IVW OR = 1.038, 95% CI = 1.016–1.060, P = 5.85 × 10−4). Furthermore, the MR analysis between PSC and the CRC validation cohort indicated consistent results. We found no causal association between genetically predicted other five AIDs and CRC (P > 0.05). The results of reverse MR analysis showed that genetically predicted CRC had no causal effect on T1D, psoriasis, and PSC (P > 0.05). The sensitivity analysis demonstrated that the results of the MR analysis were reliable. Our findings help to understand the causal association between AIDs and CRC, which deserves further investigation.
Similar content being viewed by others
Introduction
Colorectal cancer (CRC) is the third most common cancer and the second leading cause of cancer-related deaths globally1. It is estimated that by 2035, there could be up to 2.5 million new cases of colorectal cancer globally due to the acceleration of population aging2. Therefore, to improve CRC prevention and treatment, it is crucial to explore the risk factors and etiology of CRC3. The etiology of CRC is complicated and includes the accumulation of epigenetic and genetic risk factors, obesity, dietary habits, a sedentary lifestyle with little or no physical activity, alterations in gut microbiota, and others4. An increasing number of studies indicate that the immune system is critical to the occurrence, progression, and treatment of CRC5.
Autoimmune diseases (AIDs) are a group of complicated, chronic illnesses with unidentified causes. It is now generally accepted that AIDs are caused by excessive host immunoreaction due to impaired host immunoregulation6. AIDs can be systemic or can affect specific organs or body systems and affect approximately 10% of the world’s population7. Furthermore, AIDs could result in severe complications in multiple systems, which affects the population’s life expectancy. Some studies indicate that the global incidence of autoimmune diseases is increasing yearly8,9. Therefore, we need to pay more attention to these diseases.
As we all know, the dysfunction of the immune system may lead to the occurrence of AIDs. Besides the protection against pathogens, the immune system is also intensely involved in CRC prevention, development, and defense. Therefore, the immune system plays a crucial role in both AIDs and CRC, which might indicate a specific association between these two diseases. For example, numerous studies demonstrated the significance of the P2X7 receptor (P2X7R) in the pathogenesis of AIDs, such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), multiple sclerosis (MS), and inflammatory bowel diseases (IBD)10. Furthermore, researchers have demonstrated that P2X7R promotes the proliferation and metastasis of CRC cells, and its expression level is negatively related to the overall survival of CRC patients11. Therefore, AIDs and CRC might have a shared genetic susceptibility. Many studies have supported the intimate relationship between AIDs and CRC, but they remain partially controversial. Certainly, IBD, such as Crohn’s disease and ulcerative colitis, increase the probability to develop CRC, which.is because the gut chronic inflammatory state inherent in patients with IBD increases the risk of CRC12,13. Moreover, IBD have been demonstrated as an essential risk factor for the development of CRC14. However, when it comes to the association between other AIDs and the risk of CRC, there are inconsistent findings in patients with type 1 diabetes (T1D), RA, primary sclerosing cholangitis (PSC), and psoriasis15,16,17. Current studies in this area are primarily observational, which might be susceptible to confounding factors such as disease development, treatment, and environmental exposure. Inevitably, observational studies might reverse causality and make it difficult to distinguish the actual causal association. For example, a previous observational study showed that patients with psoriasis had a higher risk of CRC than those without psoriasis15. However, a recent study indicated no significant association between psoriasis and CRC18.
Mendelian randomization (MR) is an analytical method used to evaluate actual causal relationships between diseases, especially when randomized controlled trials are not feasible to examine the causation and observational studies provide biased associations due to confounding or reverse causal relationships19. It uses single nucleotide polymorphisms (SNPs) as instrument variables (IVs) to deduce causal relationships. On the one hand, confounding factors can theoretically be avoided. On the other hand, genetic variation, which explains exposure, occurs before the outcome, thus excluding the interference of reverse causation20,21. However, when the precise biological function of a genetic variant used in MR analysis is unknown, traditional MR analysis is likely to provide erroneous conclusions about the direction of causation. Bidirectional MR can quantify the effect of each variable on causality and minimize the occurrence of such errors22.
In this study, we utilized summary data from large scale genome-wide association studies (GWAS) for the two-sample MR analysis to identify the causal effect of eight major AIDs, including T1D, SLE, RA, psoriasis, MS, juvenile idiopathic arthritis (JIA), celiac disease (CD), and PSC on the risk of CRC. Furthermore, we verified our results in the validation cohort. Finally, a reverse MR analysis was performed to assess the possibility of reverse causation.
Materials and methods
Study design
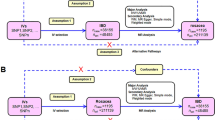
Figure 1 shows the elementary study design of the two-sample MR analysis. In short, for the process of MR analysis, three assumptions must be satisfied: (1) relevance assumption: IVs are closely related to exposure; (2) independence assumption: IVs are not associated with confounders in the exposure-outcome association; (3) exclusivity assumption: IVs directly through exposure influence the outcome instead of through other pathways. Satisfying these assumptions, we are able to confirm that IVs directly influence the outcome through exposure instead of confounders, thus demonstrating the causal association between exposure and outcome. Moreover, we performed a reverse MR analysis to explore the effect of genetically predicted CRC on AIDs.

Flow diagram illustrating the core assumptions and study design of two-sample bidirectional MR analysis. Assumption 1: IVs are closely related to exposure. Assumption 2: IVs are not associated with confounders. Assumption 3: IVs influence the outcome only through the exposure. T1D type 1 diabetes, SLE systemic lupus erythematosus, RA rheumatoid arthritis, MS multiple sclerosis, JIA juvenile idiopathic arthritis, CD celiac disease, PSC primary sclerosing cholangitis, CRC colorectal cancer, AIDs autoimmune diseases.
Data sources
We obtained summary data from the genome-wide association study (GWAS) meta-analysis based on the European population for T1D (N = 520,580), SLE (N = 14,267), RA (N = 97,173), psoriasis (N = 44,161), MS (N = 115,803), JIA (N = 12,501), CD (N = 23,649), and PSC (N = 14,890)23,24,25,26,27,28,29,30. For CRC, we used two GWAS data, one from recently published GWAS data based on the European population (N = 185,616) as primary data for analysis, and the other from the UK Biobank study (N = 377,673) as validation cohort31,32. The detailed data sources were listed in Table S1.
Instrumental variables selection
To satisfy the first assumption, we chose SNPs that were associated with exposure at a level of genome-wide significance (P < 5 × 10−8) and performed linkage disequilibrium (LD) clumping by setting r2 < 0.001 or clump distance > 10,000 kb to ensure the SNPs are independent. In addition, to determine whether there was a weak IV deviation, we calculated the F-statistics (F = R2 (n − k − 1)/k(1 − R2)), where R2 represents the variance of exposure explained by the IV, n is the sample size of the GWAS, and k is the total IVs. The F-statistics > 10 demonstrates a weak IV deviation. To eliminate the influence of confounders, we searched for pleiotropic SNPs of confounders in PhenoScanner (http://www.phenoscanner.medschl.cam.ac.uk/). After removing IVs that showed a strong association (P < 1E−5) with obesity, smoking, alcohol consumption, processed meat consumption, diabetes, inflammatory bowel disease, physical activity, leisure sedentary behaviors, cholesterol, and platelet, we utilized the remaining IVs for MR analysis3,4,33,34.
Reverse Mendelian randomization analysis
We performed a reverse MR analysis using SNPs as IVs, which are associated with CRC, to demonstrate whether CRC has any causal effect on the identified significant association AIDs (i.e., CRC as exposure and the identified significant association AIDs as outcome). The IV selection conditions in reverse MR analysis were the same as in the primary MR analysis.
Statistical analysis
MR analyses were conducted by the “TwoSampleMR” R package (version 0.5.6) and the “MRPRESSO” R package in R software (version 4.2.2). We used three common MR methods, including inverse variance weighted (IVW), weighted median (WM), and MR-Egger methods, to assess the causal association between eight major AIDs and CRC. For causal estimations of multiple SNPs, IVW is the main evaluation method for MR analysis because it has the most significant statistical power and can identify reliable causal estimates without directional pleiotropy35. So, we used IVW as the primary evaluation method. When heterogeneity exists in the analysis, random effects IVW can provide accurate causal estimation. We used the MR-Egger and the WM methods to supplement IVW estimates because they could provide reliable estimates in wider situations but were less efficient. The WM method provides consistent causal estimates when the valid IVs have more than 50% of the weight36. MR-Egger method can detect multiplicity by the intercept. However, its statistical power is impaired37.
Moreover, we carried out integrated sensitivity analyses to detect potential violations of the assumptions in the MR analysis. We used Cochran’s Q test to evaluate the heterogeneity across the individual causal effects (value of P < 0.05 suggesting heterogeneity). The MR-Pleiotropy Residual Sum and Outlier (MR-PRESSO) method was employed to explore outlier IVs and correct horizontal pleiotropy38. We used the MR-Egger intercept test to detect the horizontal and directional pleiotropy of IVs (value of P < 0.05 suggesting pleiotropy)37. Additionally, we conducted the Leave-one-out analysis to assess if a single SNP was responsible for the causal estimates or if it was the cause of bias.
Multiple comparisons were made by Bonferroni correction (P < 0.00625), which was regarded as evidence of statistical significance. However, P < 0.05 was assumed nominally significant of a causal association.
Ethics approval
Ethical approval was waived because this study used the data from publicly available databases.
Results
Instrumental variables selection
First, we selected 81 IVs for T1D, 43 IVs for SLE, 76 IVs for RA, 55 IVs for psoriasis, 71 IVs for MS, 13 IVs for JIA, 37 IVs for CD, and 18 IVs for PSC, respectively. 2 SNPs of T1D, 2 SNPs of RA, 3 SNPs of psoriasis, 5 SNPs of MS, 3 SNPs of JIA, and 1 SNP of CD were excluded because they were not found in the dataset of the outcome. Next, we excluded 14SNPs of T1D, 1 SNP of SLE, 13 SNPs of RA, 6 SNPs of psoriasis, 9 SNPs of MS, 2 SNPs of JIA, 3 SNPs of CD, and 1 SNP of PSC because they were palindromic SNPs. Then, we searched for pleiotropic SNPs of confounders in PhenoScanner and removed them. Finally, we identified 42 SNPs for T1D, 34 SNPs for SLE, 46 SNPs for RA, 32 SNPs for psoriasis, 41 SNPs for MS, 7 SNPs for JIA, 17 SNPs for CD, and 11 SNPs for PSC to conduct MR analysis, respectively. The F-statistics of all IVs were > 10, indicating a slight possibility of a weak IV bias. The details of IVs for MR analysis are shown in Tables S2–S9.
T1D and CRC
The MR analysis results showed no causal association between T1D and CRC (IVW P = 0.080, MR-Egger P = 0.368, WM P = 0.070). However, Cochran’s Q test indicated significant heterogeneity (P < 0.001), and the MR-PRESSO global test showed horizontal pleiotropy (P < 0.001). After 2 outliers were removed (rs12742756 and rs2493411), we conducted MR analysis again. Based on Bonferroni correction, the result of the IVW method showed that genetically predicted T1D nominally decreased the risk of CRC (IVW OR = 0.965, 95% CI = 0.939–0.992, P = 0.012). In addition, the consistent impact direction was shown in other methods (Fig. 2). Although we excluded outliers detected in MR-PRESSO, there was still heterogeneity among SNPs (P = 0.007). We used the random effects IVW method to alleviate this problem. The MR-Egger intercept test showed no horizontal pleiotropy (P = 0.806) (Table 1). The scatter plot showed the nominally causal relationship between T1D and CRC using different MR methods (Fig. 3). Figure S1 shows the forest and funnel plots of all SNPs in MR analysis. The plot of leave-one-out sensitivity analysis determined that no SNP affected the overall results in this analysis (Fig. S1).

Forest plot for results of the Mendelian randomization analysis. T1D type 1 diabetes, SLE systemic lupus erythematosus, RA rheumatoid arthritis, MS multiple sclerosis, JIA juvenile idiopathic arthritis, CD celiac disease, PSC primary sclerosing cholangitis.

Scatter plots of the causal effect of T1D, SLE, RA, psoriasis, MS, JIA, CD, and PSC on CRC in the MR analysis. (A) T1D on CRC. The F-statistics of IVs were from 30.38 to 196.60. (B) SLE on CRC. The F-statistics of IVs were from 29.96 to 374.85. (C) RA on CRC. The F-statistics of IVs were from 29.70 to 321.46. (D) psoriasis on CRC. The F-statistics of IVs were from 30.60 to 728.17. (E) MS on CRC. The F-statistics of IVs were from 26.86 to 525.02. (F) JIA on CRC. The F-statistics of IVs were from 33.50 to 121.17. (G) CD on CRC. The F-statistics of IVs were from 29.83 to 920.79. (H) PSC on CRC. The F-statistics of IVs were from 27.32 to 574.41. T1D type 1 diabetes, SLE systemic lupus erythematosus, RA rheumatoid arthritis, MS multiple sclerosis, JIA juvenile idiopathic arthritis, CD celiac disease, PSC primary sclerosing cholangitis, CRC colorectal cancer.
SLE and CRC
MR analysis showed no causal association between SLE and CRC (IVW P = 0.109, MR-Egger P = 0.407, WM P = 0.311) (Fig. 2). The Cochran’s Q test indicated no heterogeneity (P = 0.243), and the MR-Egger intercept test (P = 0.087) also showed no significant horizontal pleiotropy (Table 1). The MR-PRESSO detected no outlier (P = 0.254), which indicated our results were reliable. The scatter plot showed the causal relationship between SLE and CRC using different MR methods (Fig. 3). Figure S2 shows the forest and funnel plots of all SNPs in MR analysis. The plot of leave-one-out sensitivity analysis determined that no abnormal IV in our analysis affected the overall results (Fig. S2).
RA and CRC
MR analysis showed no causal association between RA and CRC (IVW P = 0.900, MR-Egger P = 0.573, WM P = 0.743). The Cochran’s Q test showed significant heterogeneity (P < 0.001), and the MR-PRESSO global test showed horizontal pleiotropy (P < 0.001). After 3 outliers were removed (rs114435492, rs17534670, and rs3087243), the MR analysis was performed again. The results of MR analysis did not change (IVW P = 0.818, MR-Egger P = 0.982, WM P = 0.775) (Fig. 2). We found no heterogeneity in our MR analysis (P = 0.138), and the MR-Egger intercept test (P = 0.862) also showed no significant horizontal pleiotropy (Table 1). The scatter plot showed the causal relationship between RA and CRC using different MR methods (Fig. 3). The forest and funnel plots of all SNPs in MR analysis were displayed in Fig. S3. The plot of leave-one-out sensitivity analysis indicated that no IV influenced the overall results in this analysis (Fig. S3).
Psoriasis and CRC
MR analysis showed no causal association between psoriasis and CRC (IVW P = 0.803, MR-Egger P = 0.692, WM P = 0.607). The Cochran’s Q test showed significant heterogeneity (P < 0.001), and the MR-PRESSO global test showed horizontal pleiotropy (P < 0.001). After 3 outliers were removed (rs10893885, rs582757, and rs9481169), the MR analysis was performed again. However, the results of MR analysis changed. After Bonferroni correction for multiple tests, we found that genetically predicted psoriasis nominally increased the risk of CRC (IVW OR = 1.026, 95% CI = 1.002–1.050, P = 0.037) (Fig. 2). The heterogeneity in our MR analysis was eliminated (P = 0.095), and the MR-Egger intercept test (P = 0.814) showed no horizontal pleiotropy (Table 1). The scatter plot showed the nominally causal relationship between psoriasis and CRC using different MR methods (Fig. 3). The forest and funnel plots of all SNPs in MR analysis were displayed in Fig. S4. The plot of leave-one-out sensitivity analysis determined that no SNP affected the overall results in this analysis (Fig. S4).
MS and CRC
MR analysis showed no causal association between MS and CRC (IVW P = 0.341, MR-Egger P = 0.904, WM P = 0.821). The Cochran’s Q test showed significant heterogeneity (P < 0.001), and the MR-PRESSO global test showed horizontal pleiotropy (P < 0.001). After 4 outliers were removed (rs4325907, rs57116599, rs7855251, and rs12365699), the MR analysis was performed again. The results of MR analysis did not change (IVW P = 0.480, MR-Egger P = 0.794, WM P = 0.815) (Fig. 2). The Cochran’s Q test still showed heterogeneity (P = 0.015), and we used the random effects IVW method to alleviate this problem. The MR-Egger intercept test showed no horizontal pleiotropy (P = 0.945) (Table 1). The scatter plot showed the causal relationship between MS and CRC using different MR methods (Fig. 3). The forest and funnel plots of all SNPs in MR analysis were displayed in Fig. S5. The plot of leave-one-out sensitivity analysis indicated that no IV influenced the overall results in this analysis (Fig. S5).
JIA and CRC
MR analysis showed no causal association between JIA and CRC (IVW P = 0.590, MR-Egger P = 0.668, WM P = 0.799) (Fig. 2). The Cochran’s Q test indicated no heterogeneity (P = 0.200), and the MR-Egger intercept test (P = 0.797) showed no significant horizontal pleiotropy (Table 1). The MR-PRESSO global test (P = 0.265) also showed a consistent result. The scatter plot showed the causal relationship between JIA and CRC using different MR methods (Fig. 3). Figure S6 shows the forest and funnel plots of all SNPs in MR analysis. The plot of leave-one-out sensitivity analysis demonstrated that no IV influenced the overall results in this analysis (Fig. S6).
CD and CRC
MR analysis showed no causal association between CD and CRC (IVW OR = 1.016, 95% CI = 0.983–1.051, P = 0.343). However, Cochran’s Q test indicated significant heterogeneity (P < 0.001), and the MR-PRESSO global test showed significant horizontal pleiotropy (P < 0.001). After 3 outliers were removed (rs1018326, rs10790269, and rs79758729), we conducted MR analysis again. The results of MR analysis did not change, which indicated no causal association between CD and CRC (IVW P = 0.086, MR-Egger P = 0.505, WM P = 0.065) (Fig. 2). The heterogeneity in our MR analysis was eliminated (P = 0.137), and the MR-Egger intercept test (P = 0.698) showed no horizontal pleiotropy (Table 1). The scatter plot showed the causal relationship between CD and CRC using different MR methods (Fig. 3). The forest and funnel plots of all SNPs in MR analysis were displayed in Fig. S7. The plot of leave-one-out sensitivity analysis demonstrated that no abnormal IV influenced the results in MR analysis (Fig. S7).
PSC and CRC
The result of the WM method showed that genetically predicted PSC increased the risk of CRC (WM OR = 1.033, 95% CI = 1.005–1.061, P = 0.019), but the IVW method showed that there was no causal association between PSC and CRC (P = 0.206). However, Cochran’s Q test indicated significant heterogeneity (P = 0.001), and the MR-PRESSO global test showed horizontal pleiotropy (P = 0.003). After 2 outliers were removed (rs41316239 and rs72837826), we conducted MR analysis again. After Bonferroni correction for multiple tests, PSC and CRC showed a significant causal association. We found that genetically predicted PSC could increase the risk of CRC using the IVW method (IVW OR = 1.038, 95% CI = 1.016–1.060, P = 5.85 × 10−4). Additionally, the consistent impact direction was shown in other methods (Fig. 2). The heterogeneity in our MR analysis was eliminated (P = 0.408), and the MR-Egger intercept test (P = 0.986) showed no horizontal pleiotropy (Table 1). The scatter plot showed the causal relationship between PSC and CRC using different MR methods (Fig. 3). The forest and funnel plots of all SNPs in MR analysis were displayed in Fig. S8. The plot of leave-one-out sensitivity analysis determined that no IV affected the overall results in MR analysis (Fig. S8). Furthermore, the same analysis was performed between PSC and the CRC validation cohort, and the results were consistent (IVW OR = 1.0005, 95% CI = 1.0001–1.0010, P = 0.025). The detailed results were showed in Table S10.
Reverse MR analysis
Finally, we performed a reverse MR analysis between CRC and T1D, psoriasis, and PSC, but we did not find reverse causal relationships between them in all MR methods (Table 2). The details of IVs for CRC were shown in Table S11. The Cochran’s Q test showed partial heterogeneity in the reverse MR analysis, and the MR-Egger intercept test showed no horizontal pleiotropy in the reverse MR analysis (Table S12). The plots of other sensitivity analyses were showed in Figs. S9–S11.
Discussion
We used the bidirectional two-sample MR analysis for the first time to systematically reveal the causal association between eight major AIDs and CRC. After Bonferroni correction for multiple tests, our study showed that genetically predicted T1D nominally decreased the risk of CRC (IVW OR = 0.965, 95% CI = 0.939–0.992, P = 0.012). However, genetically predicted psoriasis was nominally associated with an increased risk of CRC (IVW OR = 1.026, 95% CI = 1.002–1.050, P = 0.037). PSC and CRC showed a significant causal association. We found that genetically predicted PSC was significantly associated with an increased risk of CRC (IVW OR = 1.038, 95% CI = 1.016–1.060, P = 5.85 × 10−4). Furthermore, the MR analysis between PSC and the CRC validation cohort indicated consistent results (IVW OR = 1.0005, 95% CI = 1.0001–1.0010, P = 0.025). However, we found no causal association between five other AIDs (SLE, RA, MS, JIA, and CD) and CRC. The results of reverse MR analysis demonstrated that genetically predicted CRC had no causal effect on T1D, psoriasis, and PSC (P > 0.05). As far as we know, our study is the first to ascertain causal associations between AIDs and CRC using MR analysis.
Studies of the association of T1D with cancers were limited. In a retrospective study in Australia, T1D was positively correlated with CRC in females but found no association in males39. Conversely, a meta-analysis of observational studies showed no significant association between T1D and the risk of CRC (RR = 0.90, 95% CI = 0.61–1.31, I2 = 20.9)16. However, our study demonstrated that genetic vulnerabilities to T1D nominally decreased the risk of CRC using MR analysis. The discrepancies between our results and previous observational findings might be caused by various measurement errors, underlying biases, and confounding factors (e.g., use of insulin and incidence of complications) in the observational studies. These factors might reverse causality and make it challenging to clarify the real causal relationship. Based on previous studies, the nominal association between T1D and a decreased risk of CRC might involve the following aspects. First, mast cells have been discovered to have a protective anti-tumor role in the development of intestinal tumors40. There were very few mast cells in a healthy pancreas but a rise in pancreatic mast cell density appeared among individuals with T1D41. Second, T1D could result in severe complications in multiple systems, which might affect the population’s life expectancy. However, CRC is an age-related disease, and early mortality from T1D could reduce CRC prevalence. Third, T1D was genetically heterogeneous, and for the most common forms of T1D, alleles of human leukocyte antigen (HLA)-DR were an essential determinant of T1D42. Nevertheless, some research indicated that HLA-DR was a favorable prognostic indicator in CRC43. Forth, an MR study showed a positive correlation between the Bifidobacterium genus and the incidence of T1D44. On the other hand, an increased abundance of Bifidobacterium could enhance antitumor immunity and inhibit the development of CRC45.
Our study indicated that psoriasis nominally increased the risk of CRC, which was consistent with a previous meta-analysis of observational studies15. The possible mechanisms may be due to the following reasons. First, chemokine CCL27 played an essential role in psoriasis. Meanwhile, it was also involved in the cellular proliferation and migration of tumor cells46. Second, IL-17 and IL-22 are related to the etiology of autoimmune diseases such as psoriasis and numerous inflammatory-associated cancers, including CRC47. Third, Chang et al. found that several cancer metabolic pathways, including those of tryptophan metabolism and lipid biosynthesis proteins, were significantly enhanced in the blood microbiome in patients with psoriasis based on 16S rRNA metagenome sequences48. At the same time, tryptophan metabolism and lipid biosynthesis proteins were significantly associated with the development of CRC49,50.
Generally, PSC was significantly associated with susceptibility to colorectal cancer, consistent with a previous meta-analysis of observational studies17. The explanation for this phenomenon may involve several complicated factors. First, PSC was closely linked to IBD, and more than two-thirds of PSC patients were accompanied by IBD51. IBD has been demonstrated to be a risk factor for CRC. In addition, compared to patients with IBD alone, those with IBD and PSC had a fivefold increased risk of developing CRC52. These evidences indicated that PSC could directly increase the risk of CRC or indirectly promote CRC through IBD. Second, recent research found that the number of plasma cells that secreted immunoglobulin G and FOXP3+ CD4+ T cells that produced the cytokine IL-17 was increased in PSC patients’ right colonic tissue53. This indicated that the burden of intestinal inflammation was heavy in PSC patients, which might cause CRC. Third, Nakamoto et al. found K. pneumoniae, Proteus mirabilis, and Enterococcus gallinarum were prevalently detected in patients with PSC and demonstrated that PSC-derived K. pneumoniae could damage the intestinal epithelium in animal experiments54. The disturbance of intestinal flora could damage the intestinal barrier and result in bacterial translocation, which increased the risk of CRC. The recent discovery of alteration in intestinal fungi composition also reinforced the role of gut dysbiosis in PSC promoting CRC55. Fourth, Gao et al. used multidrug resistance protein 2 knockout (Mdr2−/−) mice to mirror human PSC and found that Mdr2−/− mice appeared to have alterations in fecal bile acid composition and enhanced colitis susceptibility. In Mdr2−/− mice, ursodeoxycholic acid treatment attenuated colitis susceptibility, which demonstrated that bile acid metabolism might be involved in promoting colitis and CRC by PSC56.
This study demonstrated no causal association between five other AIDs (SLE, RA, MS, JIA, and CD) and CRC, which was consistent with previous partial observational studies57,58,59. However, there were also partial studies inconsistent with our results. A Meta-analysis reported that patients with RA had a decreased risk of CRC60. A cohort study showed the risk of CRC was higher in MS patients than in healthy people61. Above all, the previously observed association between RA and MS and CRC could be mediated by hitherto unknown confounding factors. Therefore, further research is required to clarify the association between these AIDs and CRC using robust designs to eliminate confounding and survival bias. It is necessary to conduct cohort studies with large sample size adjusting for potential confounders. Current observational studies inevitably have confounding factors or reverse causation bias. Thus, animal experiments are equally necessary to eliminate these influences and clarify the underlying mechanisms between AIDs and CRC. In addition, we first reported that genetically predicted CRC had no effect on T1D, psoriasis, and PSC, which demonstrated the associations of these diseases were unidirectional.
Our study has several major strengths. First, the most primary strength was the MR design, which avoided confounding factors and reverse causality in previous observational studies. Second, we performed a variety of sensitivity analyses and explained the potential mechanisms involved in these associations combined with previous studies, which made our conclusions more convincing. Third, we demonstrated that T1D, psoriasis, and PSC had causal effects on CRC. Although the odds ratio was not high, these described associations might have clinical implications. For example, we suggested that it was necessary for us to strengthen the prevention and screening of CRC in patients with AIDs, especially those with the use of immunosuppressive treatments. Fourth, we indicated that there were causal associations between AIDs and CRC. The elucidation of the mechanisms between AIDs and CRC might allow us to exploit these mechanisms to monitor and treat these two diseases in the future.
There are several limitations to our study. First, we selected the IVs and GWAS data based on the European population to construct MR analysis in this study. It means whether our results are consistent in other ethnic populations remains to be verified. Second, although the GWAS data of AIDs and CRC came from different samples in our study, it is inevitable that there may be a sample overlap. However, sample overlap in the two-sample MR analysis may lead to an overestimation of the results. Third, we confirmed no causal association between five other AIDs (SLE, RA, MS, JIA, and CD) and CRC. However, we cannot exclude the possibility that this association was not detected due to insufficient sample size at present. With larger sample sizes of GWAS in the future, the association between AIDs and CRC might become apparent. Fourth, we could not rule out the possibility that the association between AIDs and CRC may be non-linear. Current MR methods assume that the exposure–outcome correlation is linear. Furthermore, the causal association and underlying mechanisms between AIDs and CRC must be further confirmed in animal experiments or cohort studies with large sample size.
In summary, our study demonstrated that genetic vulnerabilities to psoriasis and PSC could increase the risk of CRC using MR analysis. Genetically predicted T1D was associated with a decreased risk of CRC. However, there was no evidence to show a causal association between five other AIDs (SLE, RA, MS, JIA, and CD) and CRC. The results of reverse MR analysis demonstrated that genetically predicted CRC had no causal effect on T1D, psoriasis, and PSC. Our findings help to comprehend the causal association between AIDs and CRC, which deserves further investigation.
Data availability
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
References
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer. J. Clin. 73, 17–48 (2023).
Dekker, E., Tanis, P. J., Vleugels, J. L. A., Kasi, P. M. & Wallace, M. B. Colorectal cancer. Lancet 394, 1467–1480 (2019).
Baidoun, F. et al. Colorectal cancer epidemiology: Recent trends and impact on outcomes. Curr. Drug Targets 22, 998–1009 (2021).
Kumar, R., Harilal, S., Carradori, S. & Mathew, B. A. Comprehensive overview of colon cancer—A grim reaper of the 21st century. Curr. Med. Chem. 28, 2657–2696 (2021).
Sieminska, I. & Baran, J. Myeloid-derived suppressor cells in colorectal cancer. Front. Immunol. 11, 1526 (2020).
Yasunaga, M. Antibody therapeutics and immunoregulation in cancer and autoimmune disease. Semin. Cancer Biol. 64, 1–12 (2020).
Gawałko, M. et al. Cardiac arrhythmias in autoimmune diseases. Circ. J. 84(5), 685–694 (2020).
Glover, K., Mishra, D. & Singh, T. R. R. Epidemiology of ocular manifestations in autoimmune disease. Front. Immunol. 12, 744396 (2021).
Rose, N. R. Prediction and prevention of autoimmune disease in the 21st century: A review and preview. Am. J. Epidemiol. 183, 403–406 (2016).
Cao, F. et al. P2X7 receptor: A potential therapeutic target for autoimmune diseases. Autoimmun. Rev. 18, 767–777 (2019).
Bai, X. et al. Progress in the relationship between P2X7R and colorectal cancer. Mol. Biol. Rep. 50, 1687–1699 (2023).
Olén, O. et al. Colorectal cancer in ulcerative colitis: A Scandinavian population-based cohort study. Lancet 395, 123–131 (2020).
Olén, O. et al. Colorectal cancer in Crohn’s disease: A Scandinavian population-based cohort study. Lancet Gastroenterol. Hepatol. 5, 475–484 (2020).
Terzić, J., Grivennikov, S., Karin, E. & Karin, M. Inflammation and colon cancer. Gastroenterology 138, 2101-2114.e5 (2010).
Fu, Y., Lee, C. H. & Chi, C. C. Association of psoriasis with colorectal cancer. J. Am. Acad. Dermatol. 85, 1429–1436 (2021).
Sona, M. F., Myung, S. K., Park, K. & Jargalsaikhan, G. Type 1 diabetes mellitus and risk of cancer: A meta-analysis of observational studies. Jpn. J. Clin. Oncol. 48, 426–433 (2018).
Båve, A. L. et al. Increased risk of cancer in patients with primary sclerosing cholangitis. Hepatol. Int. 15, 1174–1182 (2021).
Watanabe, T. et al. Risks of malignancies among patients with psoriasis: A cohort study of 360 patients. J. Dermatol. 50, 615–621 (2023).
Sekula, P., Del Greco, M. F., Pattaro, C. & Köttgen, A. Mendelian randomization as an approach to assess causality using observational data. J. Am. Soc. Nephrol. 27, 3253–3265 (2016).
Burgess, S., Small, D. S. & Thompson, S. G. A review of instrumental variable estimators for Mendelian randomization. Stat. Methods Med. Res. 26, 2333–2355 (2017).
Bowden, J. & Holmes, M. V. Meta-analysis and Mendelian randomization: A review. Res. Synth. Methods 10, 486–496 (2019).
Evans, D. M. & Smith, G. D. Mendelian randomization: New applications in the coming age of hypothesis-free causality. Annu. Rev. Genom. Hum. Genet. 16, 327–350 (2015).
Chiou, J. et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 594, 398–402 (2021).
Bentham, J. et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47, 1457–1464 (2015).
Ishigaki, K. et al. Multi-ancestry genome-wide association analyses identify novel genetic mechanisms in rheumatoid arthritis. Nat. Genet. 54, 1640–1651 (2022).
Stuart, P. E. et al. Transethnic analysis of psoriasis susceptibility in South Asians and Europeans enhances fine-mapping in the MHC and genomewide. HGG Adv. 3, 100069 (2022).
International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365, eaav7188 (2019).
López-Isac, E. et al. Combined genetic analysis of juvenile idiopathic arthritis clinical subtypes identifies novel risk loci, target genes and key regulatory mechanisms. Ann. Rheum. Dis. 80, 321–328 (2021).
Trynka, G. et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 43, 1193–1201 (2011).
Ji, S. G. et al. Genome-wide association study of primary sclerosing cholangitis identifies new risk loci and quantifies the genetic relationship with inflammatory bowel disease. Nat. Genet. 49, 269–273 (2017).
Fernandez-Rozadilla, C. et al. Deciphering colorectal cancer genetics through multi-omic analysis of 100,204 cases and 154,587 controls of European and east Asian ancestries. Nat. Genet. 55, 89–99 (2023).
Conroy, M. C. et al. UK Biobank: A globally important resource for cancer research. Br. J. Cancer 128, 519–527 (2023).
Zhu, X. et al. Evaluation of platelet indices as diagnostic biomarkers for colorectal cancer. Sci. Rep. 8, 11814 (2018).
Jacobs, R. J., Voorneveld, P. W., Kodach, L. L. & Hardwick, J. C. Cholesterol metabolism and colorectal cancers. Curr. Opin. Pharmacol. 12, 690–695 (2012).
Burgess, S. et al. Using published data in Mendelian randomization: A blueprint for efficient identification of causal risk factors. Eur. J. Epidemiol. 30, 543–552 (2015).
Bowden, J., Smith, G. D., Haycock, P. C. & Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Bowden, J., Smith, G. D. & Burgess, S. Mendelian randomization with invalid instruments: Effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Harding, J. L., Shaw, J. E., Peeters, A., Cartensen, B. & Magliano, D. J. Cancer risk among people with type 1 and type 2 diabetes: Disentangling true associations, detection bias, and reverse causation. Diabetes Care 38, 264–270 (2015).
Sinnamon, M. J. et al. A protective role of mast cells in intestinal tumorigenesis. Carcinogenesis 29, 880–886 (2008).
Martino, L. et al. Mast cells infiltrate pancreatic islets in human type 1 diabetes. Diabetologia 58, 2554–2562 (2015).
Melanitou, E., Fain, P. & Eisenbarth, G. S. Genetics of type 1A (immune mediated) diabetes. J. Autoimmun. 21, 93–98 (2003).
Dunne, M. R. et al. Characterising the prognostic potential of HLA-DR during colorectal cancer development. Cancer Immunol. Immunother. 69, 1577–1588 (2020).
Xu, Q. et al. Causal relationship between gut microbiota and autoimmune diseases: A two-sample mendelian randomization study. Front. Immunol. 12, 746998 (2022).
Yang, X. et al. Probiotic powder ameliorates colorectal cancer by regulating Bifidobacterium animalis, Clostridium cocleatum, and immune cell composition. PLoS One 18, e0277155 (2023).
Martínez-Rodríguez, M. & Monteagudo, C. CCL27 signaling in the tumor microenvironment. Adv. Exp. Med. Biol. 1302, 113–132 (2021).
Blake, S. J. & Teng, M. W. Role of IL-17 and IL-22 in autoimmunity and cancer. Actas. Dermosifiliogr. 105, 41–50 (2014).
Chang, C. J. et al. Compositional features of distinct microbiota base on serum extracellular vesicle metagenomics analysis in moderate to severe psoriasis patients. Cells 10, 2349 (2021).
Ala, M. Tryptophan metabolites modulate inflammatory bowel disease and colorectal cancer by affecting immune system. Int. Rev. Immunol. 41, 326–345 (2022).
You, X. et al. Loss of mitochondrial aconitase promotes colorectal cancer progression via SCD1-mediated lipid remodeling. Mol. Metab. 48, 101203 (2021).
Boonstra, K. et al. Primary sclerosing cholangitis is associated with a distinct phenotype of inflammatory bowel disease. Inflamm. Bowel Dis. 18, 2270–2276 (2012).
Loftus, E. V. Jr. et al. PSC-IBD: A unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut 54, 91–96 (2005).
Shaw, D. G. et al. Antigen-driven colonic inflammation is associated with development of dysplasia in primary sclerosing cholangitis. Nat. Med. 29, 1520–1529 (2023).
Nakamoto, N. et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat. Microbiol. 4, 492–503 (2019).
Lemoinne, S. et al. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut 69, 92–102 (2020).
Gao, R. Y. et al. Bile acids modulate colonic MAdCAM-1 expression in a murine model of combined cholestasis and colitis. Mucosal Immunol. 14, 479–490 (2021).
Song, L. et al. The risks of cancer development in systemic lupus erythematosus (SLE) patients: A systematic review and meta-analysis. Arthritis Res. Ther. 20, 270 (2018).
Lasa, J., Rausch, A. & Zubiaurre, I. Risk of colorectal adenomas in patients with celiac disease: A systematic review and meta-analysis. Rev. Gastroenterol. Mex. (Engl Ed) 83, 91–97 (2018).
Marrie, R. A. et al. Cancer incidence and mortality rates in multiple sclerosis: A matched cohort study. Neurology 96, e501–e512 (2021).
Simon, T. A., Thompson, A., Gandhi, K. K., Hochberg, M. C. & Suissa, S. Incidence of malignancy in adult patients with rheumatoid arthritis: A meta-analysis. Arthritis Res. Ther. 17, 212 (2015).
Bosco-Lévy, P. et al. Incidence and risk of cancer among multiple sclerosis patients: A matched population-based cohort study. Eur. J. Neurol. 29, 1091–1099 (2022).
Acknowledgements
We are grateful to the IMSGC and related institutions for making the GWAS summary data publicly available, and grateful to all the researchers and participants who contributed to those studies.
Author information
Authors and Affiliations
Contributions
Conceptualization, L.C. and B.C.; methodology, L.C. and F.W.; validation, formal analysis, and investigation, L.C. and B.C.; writing—original draft and visualization, L.C., F.W., H.Z. and B.C.; writing—review and editing, L.C., F.W., H.Z. and B.C.; supervision, project administration, and funding acquisition, B.C. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, L., Wang, F., Zhang, H. et al. Exploring potential causal associations between autoimmune diseases and colorectal cancer using bidirectional Mendelian randomization. Sci Rep 14, 1557 (2024). https://doi.org/10.1038/s41598-024-51903-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-51903-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
