
Abstract
Calorie restriction (CR), which is a reduction in calorie intake without malnutrition, usually extends lifespan and improves tissue integrity. This report focuses on the relationship between nuclear genomic instability and dietary-restriction and its effect on cell survival. We demonstrate that the cell survival rates of the genomic instability yeast mutant rrm3 change under metabolic restricted conditions. Rrm3 is a DNA helicase, chromosomal replication slows (and potentially stalls) in its absence with increased rates at over 1400 natural pause sites including sites within ribosomal DNA and tRNA genes. Whereas rrm3 mutant cells have lower cell death rates compared to wild type (WT) in growth medium containing normal glucose levels (i.e., 2%), under CR growth conditions cell death rates increase in the rrm3 mutant to levels, which are higher than WT. The silent-information-regulatory (Sir) protein complex and mitochondrial oxidative stress are required for the increase in cell death rates in the rrm3 mutant when cells are transferred from growth medium containing 2% glucose to CR-medium. The Rad53 checkpoint protein is highly phosphorylated in the rrm3 mutant in response to genomic instability in growth medium containing 2% glucose. Under CR, Rad53 phosphorylation is largely reduced in the rrm3 mutant in a Sir-complex dependent manner. Since CR is an adjuvant treatment during chemotherapy, which may target genomic instability in cancer cells, our studies may gain further insight into how these therapy strategies can be improved.
Similar content being viewed by others


Introduction
Calorie restriction (CR), which is a well-known metabolic intervention, is a reduction in calorie intake without malnutrition. CR usually extends the lifespan in many model organisms from yeast to primates and improves tissue integrity in higher eukaryotes1,2,3,4,5,6. CR has beneficial physiological effects on genome stability (e.g., preventing the shortening of telomeres or instability in ribosomal DNA (rDNA)); CR reduces also the effects of numerous diseases including obesity or predisposition and progression of cancer3,5,7.
However, under certain conditions and/or genotype settings CR may have adverse effects on cell survival. Some translational studies demonstrated that CR in the form of intermittent fasting or CR-mimetic drugs as adjuvant therapies enhance the efficacy of chemotherapy and reduce metastasis5,8,9,10,11,12,13,14,15,16,17,18,19,20. The combined action of the chemotherapeutic agents and the CR conditions lead to increased DNA breakage in cancer cells as it has been determined by the comet assay15,21. CR-mediated longevity has been shown to be genotype dependent in numerous organisms including yeast, flies and mice5,22. The underlying mechanisms of how maintenance of genome stability and metabolic state of a cell interact are still not well understood.
In this report we used the rrm3 yeast mutant as a model system for nuclear genomic instability. Rrm3 is a DNA helicase23. In its absence, replication pauses (sometimes stalls) with increased rates at over 1400 distinct chromosomal sites (also called: replication-pause-sites (RPS))23,24,25,26,27. RPS include tRNA genes, centromeres, telomeres, silent replication origins and sites within the ribosomal DNA (rDNA)23,26,27,28,29,30,31,32,33. Usually these RPS are occupied by non-histone protein complexes, which may cause replication slowing/stalling24,26,29,34. Some of these sites were identified in chromosome translocations and numerous of these sites even superimpose with the locations of gamma-H2AX phosphorylation events which indicate the presence of single-stranded DNA leading potentially to double-strand DNA (dsDNA) breaks23,26,27,31,33,35,36,37,38,39. Although the exact function of Rrm3 is not known, it is proposed that during DNA replication Rrm3 transiently displaces the non-histone protein complexes, which are tightly bound to the RPS24,28,40,41,42,43. Rrm3 may also have a role in facilitating replication past highly-transcribed regions and in terminating DNA replication at natural RPS24,25,44. Although it appears that Rrm3 is acting only at specific chromosomal locations, this helicase migrates with the replication complex and affects replication of all yeast chromosomes (even in regions lacking RPS)24,25,45. The functions of Rrm3 are conserved in the fission yeast S. pombe, which is evolutionary closer to mammalian systems, and they overlap also with some functions of its yeast homolog Pif146,47,48,49,50,51. Rrm3/Pif1 has a homolog protein in humans (human Pif1 (hPif1)) which affects telomeres52,53. There are reports that hPif1 is involved in tumor cell proliferation and it is proposed to be a cancer therapy target54,55,56,57. Thus, the Rrm3/Pif1 yeast studies may have relevance to understanding tumor initiation and progression.
Though rrm3 mutant cells are viable, survival of the rrm3 mutant cells is dependent on multiple checkpoint activities26,58,59. Examples are the intra S-phase checkpoint which includes Sgs1, the MRX complex, Srs2, Top3, Mec1, Mrc1 and Rad53. Rad53 is the mammalian CHK2 homolog and an essential serine/threonine kinase regulating checkpoint responses throughout the cell cycle. Rad53 is hyperphosphorylated in response to DNA damage60,61. Rad53 phosphorylation in rrm3 mutant cells indicates DNA damage such as collapsed forks and/or DNA breakage. Rrm3 itself is also phosphorylated by Rad53 in response to replication fork stalling (induced by hydroxy urea, HU)62. Rrm3 is proposed to be required for the survival after replication fork breakage and for efficient sister chromatid exchange63. Rrm3p enriches at inducible DNA breaks (i.e., induced by an HO endonuclease) in a replication-dependent manner63. In a recent report, helicase activities of Rrm3 and Rad5 were demonstrated to interact genetically to prevent recombinogenic DNA lesions40.
There is a relationship between glucose restriction, repressive chromatin, and presence/activity of Rad5364. The Sir (Silent Information Regulatory protein) complex plays a role in this relationship. The Sir complex consists of the Sir2/Sir3/Sir4 proteins65. It is mainly present in subtelomeric and telomeric regions, at the silent mating type loci HML and HMR, and at the rDNA locus (particularly Sir2) and mediates silencing of gene transcription at those locations. Sir2 is a NAD-dependent histone deacetylase which is required for heterochromatin formation at telomeres, the silent mating type loci HML and HMR, and at the rDNA locus66,67. The Sir complex is also recruited to dsDNA breaks and in addition the Sir2/Sir3 proteins can be present directly at euchromatic DNA replication origins68,69,70,71. CR-restricted growth conditions can promote rDNA repeat stability and extension of lifespan by Sir2-dependent and -independent pathways3,72,73,74,75,76. For example, CR greatly decreases RNA–DNA hybrid levels in the intergenic spacers of rDNA (DNA location between the 5S and 35S rDNA genes) in WT as well as sir2 mutants75.
Here, we found that compared to wild-type (WT) cells, chromosomal DNA breakage is lower genome-wide in rrm3 mutant cells, which are cultured in medium containing 2% glucose. However, DNA breakage rates in rrm3 mutant cells increase in all these regions in growth conditions of CR (compared to rrm3 mutant cells grown in 2% glucose). CR-rrm3 mutant cells have increased cell death rates compared to rrm3 cells grown in medium containing 2% glucose. This increase in cell death rates in the CR-rrm3 cell is dependent on members of the Sir-complex (Sir2/3/4) as well as on mitochondrial reactive oxygen species (ROS). In addition, the Rad53 checkpoint protein is less (or not at all) phosphorylated in CR-rrm3 mutant. In the absence of the Sir-complex, Rad53 phosphorylation is fully restored in the CR-rrm3 mutant. Since cancer cells have genomic instability (like rrm3 mutant cells), our studies should give further insight into how CR, which is a cancer-adjuvant therapy strategy, may help to battle cancer.
Results
In rrm3 mutant cells genome-wide DNA breakage rates are lower than wild type under normal growth conditions (2% glucose), but increase after transfer to caloric-restricted (CR) growth medium
Previously we demonstrated that in the absence of the DNA helicase Rrm3, replication pauses/stalls at multiple sites within ribosomal DNA (rDNA) and at other regions in the yeast genome23,26,27. Because the increased pausing/stalling of replication forks may result in DNA breakage, we determined the natural chromosomal DNA breakage rates in these (and other) regions using the standard terminal deoxynucleotidyl transferase (TdT) assay combined with PCR (see “Methods” section; Supplement Fig. 1)77. To study the interaction between genome stability and metabolic state of a cell we applied caloric-restricted (CR) growth conditions.
We measured DNA breakage rates in wild-type (WT) and rrm3 mutant yeast cells, which were grown either in regular, rich growth medium (YEPD, containing 2% glucose) or in CR growth medium (YEPD, containing 0.05% glucose) to an early-logarithmic growth phase (OD about 0.1; see also “Methods” section for growth conditions). Initially we determined DNA breakage rates in rDNA, which consists of a repeated chromosomal array (100–150 identical 9.1 kbp repeats on chromosome XII), but also extrachromosomal rDNA circles (ERCs) (Supplement Fig. 1C)27,78,79. Each rDNA repeat can be split into two about equal-sized 4.5 kbp DNA fragments by digestion of the rDNA with the restriction enzyme BglII (called Bgl A and B fragments; Supplement Fig. 1C)78. Whereas the Bgl A fragment does not contain any sites, which perturb rDNA replication, the Bgl B fragment contains the site for the replication fork barrier (RFB), which blocks rDNA replication unidirectional (Supplement Fig. 1C)78,80. In addition, the Bgl B fragment contains the 5S rDNA gene and a site, where rDNA replication can be initiated (called ARS, autonomous replicative sequence). Replication forks may pause at both the 5S rDNA genes and inactive replication origins, when these sites are replicated passively by forks moving through this region (Supplement Fig. 1C)27,81.
Using the TdT assay, we demonstrate that natural DNA breakage rates are consistently three–fourfold lower in both rDNA fragments (Bgl A and B) of the rrm3 mutant compared to WT cells (both grown in 2% glucose) (Fig. 1, rDNA Bgl A and B). WT cells experienced about 15% higher DNA breakage rates in medium containing 0.05% glucose (CR) compared to medium containing 2% glucose. DNA breakage rates in both rDNA fragments (Bgl A and B) of CR rrm3 mutant cells increased on average 5.7-fold compared to rrm3 mutant cells grown in 2% glucose. DNA breakage rates in rDNA of the CR-rrm3 mutant cells are 40–80% higher than DNA breakage rates of WT-cells (2% glucose) in both Bgl A and B fragments.

Genome-wide DNA breakage rates of rrm3 mutant cells are low (compared to WT) under normal growth conditions (2% glucose) and increase after transferring to caloric-restricted (CR) growth conditions. (A) WT and rrm3 mutant cells were grown in medium containing 2% or 0.05% (CR) glucose to an early logarithmic phase (OD600 ~ 0.1). Total genomic DNA was prepared, and DNA breakage rates were determined by using the terminal deoxynucleotidyl transferase (TdT) assay combined with PCR. Agarose gels display the PCR products for ribosomal DNA (rDNA) fragments #1 and #2 (see also Supplement Fig. 1C) and 11 indicated non-rDNA chromosomal locations. (−) and (+) biotin-dCTP refer to “no addition” or “addition” of biotin-dCTP to the TdT reaction. In the positive-control experiment, we demonstrate that the TdT assay can readily detect dsDNA breakage events, since genomic DNA which has been digested with a restriction enzyme prior to carrying out the TdT assay, results into higher DNA breakage rates (Supplement Fig. 1B). The full-sized images (pellet) are shown as Supplement Figures. (B) The intensity of the bands in the gels shown in panel A was determined by using the program ImageLab (Bio-Rad™ Laboratories). Each pellet value was first normalized by the input value, then the (−)-biotin-dCTP value (i.e., background) was subtracted from the (+)-biotin-dCTP value. The DNA breakage value for WT (2% glucose) was set at the arbitrary number 1. Experiments were carried out three times. Standard errors are shown.
To investigate whether in the rrm3 mutant the low DNA breakage rates (compared to WT) and their increase after the switch from regular growth (2% glucose) to CR-growth conditions are specific for rDNA, we determined DNA breakage rates of several non-rDNA chromosomal regions. Some of these regions contain known (or putative) RPS (e.g., tRNA genes and silent replication origins). As described above, we determined DNA breakage rates by using the TdT assay (see Supplement Fig. 1). DNA was digested with the restriction enzyme BglII prior to the capture of the broken DNA fragments (having biotinylated extensions) with avidin-coated beads. DNA breakage rates were determined for the following eleven regions; the names of the probes (gene names) are listed as well as the sizes of the (unbroken) BglII fragments are indicated in parenthesis: PHO87 (~ 23 kbp), MET10/SMC2 (~ 2.1 kbp), YFH7 (~ 6.2 kbp), DCV1 (~ 9.2 kbp), HIS2 (~ 5.3 kbp), STE12 (~ 6 kbp), NUC1 (~ 3 kbp), MHR1 (~ 5 kbp), KSS1 (~ 3 kbp), CCE1 (~ 20 kbp) and SOD1 (~ 10 kbp) (Fig. 1). Some of the investigated DNA fragments contain known (or putative) RPS. These fragments contain known (or putative) silent replication origins (in parenthesis): PHO87 (ARS313/314), MET10/SMC2 (ARS608), KSS1 (ARS723) and CCE1 (ARS1113)26,28. These fragments contain tRNA genes (in parenthesis): YFH7 (tRNAF), DCV1 (tRNAY), HIS2 (tRNAA) and SOD1 (tRNAL)26.
In summary, the natural nuclear DNA breakage rates follow similar trends as observed for rDNA, though there are a few differences: (i) in average, about tenfold reduced DNA breakage rates in the rrm3 mutant compared to WT cells (both 2% glucose), the presence of natural RPS (e.g., tRNA genes and silent replication origins) does not appear to have an effect on the changes in the DNA breakage rates; (ii) an in average 5.4-fold increase in DNA breakage rates in the rrm3 mutant when cells are switched from regular growth medium (2% glucose) to CR-growth medium (0.05% glucose), however (unlike in rDNA) DNA breakage rates do not reach WT-levels (except for the DNA fragment containing CCE1); (iii) the breakage rates in chromosomal DNA of CR-WT cells are in average 5% increased or decreased compared to WT cells grown in medium containing 2% glucose (Fig. 1).
In caloric-restricted (CR) rrm3 mutant cells replication pausing/stalling is decreased at selective sites in ribosomal DNA, at tRNA genes and silent replication origins, but high-molecular weight DNA structures increase in ribosomal DNA
Rrm3 is a DNA helicase involved in DNA replication23,25,26,27. We investigated therefore whether replication pausing/stalling is altered particularly at natural RPS (e.g., the inactive RFB, 5S rDNA genes, and silent rDNA replication origins (ARS)) (Fig. 2). To analyze rDNA replication intermediates by neutral–neutral DNA 2D agarose gel electrophoresis, we digested the DNA with the restriction enzyme ClaI which generates mainly an about 6 kb fragment from the 9.1 kb rDNA repeat unit (Fig. 2A)27,79,81.

Replication pausing/stalling is decreased at sites in ribosomal DNA, at tRNA genes and silent replication origins of caloric-restricted rrm3 mutant cells, but high-molecular weight DNA structures increase in ribosomal DNA under CR-growth conditions. (A) rDNA replication is initiated in a subset of the rDNA repeats. The pattern of replication intermediates for any rDNA fragment containing a replication origin (ARS: autonomous replicative sequence) is a composite of intermediates generated from repeats with an active ARS and those without an active ARS27,78. For repeats with an active ARS, initiation begins and proceeds bi-directionally from the ARS (bubble structures, BU). They are converted to simple Y structures when the rightward-moving fork reaches the end of the fragment. The leftward-moving fork stops at the site-specific replication fork barrier (RFB) creating increased hybridization on the arc of simple Y-shaped intermediates. The remaining part of ClaI fragments with active ARSs is replicated by forks converging on the fork stalled at the RFB. These forks generate intermediates that emanate from the RFB and terminate with a mass of close to 2N (X). Repeats without an active origin are replicated uni-directionally, creating a full arc of simple Y-shaped intermediates (passive replication movement). Schema of rDNA replication intermediates (ClaI DNA fragment): BU bubble arc (i.e., replication initiation); RFB replication fork barrier; X termination of rDNA replication; RF replication forks; RI, recombination intermediates; CF converging forks. Pauses of replication forks at the natural RPS in rDNA are indicated (asterisks). A StuI-BglII fragment within the analyzed ClaI-rDNA fragment served as a probe81. 2D gels of ClaI-digested DNA of the indicated strains and growth conditions are displayed. Probe: rDNA (StuI-BglII). Histogram shows quantifications by phosphor-imager analysis. Pauses of replication forks at the natural RPS in rDNA are indicated by asterisks (*, **, ***). RFB refers to the in-active replication fork barrier (*); 5S means 5S rDNA gene (**); ARS refers to the silent replication origin (***). The amount of each replication intermediate is expressed as ratios (i.e., specific intermediate versus total replication structures (i.e., sum of all replication forks (including pauses and RFB), RI/CF, and BU). In this way, we normalized for different amounts of total replication structures. Experiments were carried out two times. Standard errors are displayed. (B) 2D gels of EcoRV-digested (top, left 2 panels; probe: ARS313/ARS314), BglII-digested (bottom, 2 panels; probe: MET10/SMC2 (ARS608)) and EcoRV–digested (top, right 2 panels; probe: tRNAY) DNA of the indicated strains and growth conditions. Replication pausing at the ARS313/ARS314 locations is about three-fold, at the ARS608 locations about four-fold, and at the tRNAY gene about seven-fold reduced in the CR-rrm3 cells. Experiments were carried out two times. (C) Separation of circular from linear DNA structures by 2D gels (ethidium-bromide in both dimensions) of the indicated strains (WT, rrm3, rad52, rrm3 rad52) grown in medium containing 2% glucose or 0.05% glucose (CR). Structure A and B are indicated in the cartoon and appear only in CR-grown cells. (D) 2D gels of ClaI-digested DNA of the indicated strains and growth conditions. Probe: rDNA (StuI-BglII). Pauses of replication forks at the natural RPS in rDNA are indicated by asterisks (*, **, ***; see also A).
As expected, and previously demonstrated, replication pausing/stalling at the inactive RFB, 5S rDNA genes and silent rDNA replication ARS was increased in the rrm3 mutant compared to WT, both strains were grown in medium containing 2% glucose (Fig. 2A)27. Under conditions of CR, replication pausing/stalling at the inactive RFB and silent rDNA replication ARS was also increased in the rrm3 mutant compared to WT, in contrast replication pausing/stalling did not increase at 5S rDNA genes in the CR-rrm3 mutant cells (Fig. 2A). The rDNA replication pattern of WT cells grown in regular medium (2% glucose) and CR-medium were indistinguishable (P < 0.05; Fig. 2A).
Previously we demonstrated that replication pausing at the tRNAA gene is reduced in CR-rrm3 mutant cells81. Here we demonstrate that replication pausing/stalling at the silent replication origins ARS313, ARS314 as well as ARS608 and the tRNAY gene is also reduced in CR-rrm3 mutant cells (Fig. 2B).
When we separated intact, undigested total genomic DNA in two dimensions in the presence of ethidium-bromide (in both dimensions27), and probed the membrane after the Southern-transfer with radioactive-labeled rDNA, we observed slower migrating DNA species (containing rDNA) particularly in CR cells (both WT and rrm3 mutant cells; marked as structures A and B) (Fig. 2C). We also observed these DNA structures in the absence of the recombination protein Rad52 (rad52 mutant background) (Fig. 2C). The amounts of ERCs (presence is dependent on Rad52) were similar in rrm3 mutant cells grown in medium containing 2% glucose or 0.05% glucose (CR) (2% glucose: eightfold enrichment of ERCs in rrm3 mutant over WT; CR: 8.2-fold enrichment of ERCs in rrm3 mutant over WT; Fig. 2C). Replication patterns of rDNA in the rad52 and rrm3 rad52 strains display the same patterns observed in WT and rrm3 strains, respectively (compare Fig. 2A,D).
In summary, these results suggest that in rrm3 mutant cells, which were transferred from growth medium containing 2% glucose to CR-growth medium, replication pausing/stalling is in fact reduced likely genome-wide at numerous natural RPS (including tRNA and 5S rDNA genes as well as selective silent replication origins (ARSs)) to various extents. However, high-molecular weight structures in rDNA increase under CR-growth conditions in a Rad52-independent manner.
Numbers of rrm3 mutant cells stained with propidium iodide (PI) are low under normal growth conditions (2% glucose) and increased under caloric restriction (CR)
DNA breakage occurs in dying or dead cells because of the digestion of chromosomal DNA during the cell death process (e.g., apoptosis or necrosis)82,83. To determine the number of dying and dead cells, we added the dye propidium iodide (PI) to growing yeast cultures. PI can enter dying/dead cells and binds to DNA, because these cells have disrupted cell membranes83. Whereas fewer than 1% of the WT cells were PI-positive in YEPD medium containing 2% glucose, we discovered that rrm3 mutant cells (2% glucose) had even ten times fewer PI-positive cells (0.07%; Fig. 3A; Supplement Fig. 2). The percentage of PI-positive rrm3 mutant cells is greatly increased in CR-growth medium containing 0.05% glucose (i.e., from 0.07% (2% glucose) to 4.9% (CR); Fig. 3A; Supplement Fig. 2). This phenotype can be rescued by expression of the functional RRM3 gene, whereas expression of mutant versions of rrm3 (K260A, K260R), which affect the binding of ATP to Rrm3 (K260A) and the hydrolysis of ATP (K260R), resemble the same effect as a complete lack of Rrm3 (Fig. 3A; Supplement Fig. 2)27. The mutant rrm3 versions (K260A, K260R) are expressed at WT levels as previously demonstrated27.

Numbers of propidium iodide (PI)-stained rrm3 mutant cells (indicating dying or dead cells) growing in 2% glucose-medium are low and increased under caloric-restricted (CR) growth conditions. (A) The histogram displays the percentage of PI-stained cells. Standard errors are shown. Cells of the indicated strains were grown in medium containing 2% or 0.05% glucose to an early logarithmic phase (OD600 ~ 0.1). These are: WT, pRS315; rrm3, pRS315; rrm3, pRS315-RRM3; rrm3, pRS315-rrm3 (K260A); rrm3, pRS315-rrm3 (K260R). Representative microscopic images (PI-stained cells and the corresponding brightfield images) are displayed in Supplement Fig. 2. The two alleles rrm3 (K260A) and rrm3 (K260R) are properly expressed as previously demonstrated27. Experiments were carried out three times. (B) Microscopic images of rrm3 mutant cells grown in medium containing 2% or 0.05% (CR) glucose. Cells were stained with fluorescein diacetate (FDA) and PI. In addition, the corresponding brightfield images are shown. Bar: 25 µm. (C) A population of cells containing PI-stained cells (CR rrm3) were sorted resulting in a fraction of cells enriched with stained cells (PI-positive). The other fraction contained non-stained cells (PI-negative). Brightfield images (superimposed with the PI-staining) are displayed below. Bar: 25 µm. Equal numbers of cells were plated on YEPD plates (2% glucose). Colonies were counted. The histogram shows percentage survival expressed as colonies on the PI-positive plate versus colonies on the PI-negative plate. Experiments were carried out three times. Standard errors are shown. DNA breakage in rDNA of cells stained or not stained with PI was determined by using the terminal deoxynucleotidyl transferase (TdT) assay combined with PCR as described in Fig. 1. Agarose gels display the PCR products for the rDNA fragment #1 (see also Supplement Fig. 1C). (−) and (+) biotin-dCTP refer to “no addition” or “addition” of biotin-dCTP to the TdT reaction. The histogram displays the quantification of the agarose gels on the left side as described in Fig. 1. (D) WT and rrm3 mutant cells were plated on YEPD medium containing 2% or 0.05% (CR) glucose. Images display colonies after 2, 3, and 4 days of growth at 30 °C. Growth curves at 30 °C of WT and rrm3 mutant cells grown in YEPD medium containing 2% or 0.05% (CR) glucose. (E) The histogram displays the percentage of PI-stained cells. Standard errors are shown. Cells of the indicated strains were grown in medium containing 2% or 0.05% glucose to an early logarithmic phase (OD600 ~ 0.1). These are WT, rrm3, pif1-m2, pif1-m2 rrm3, rad52, rad52 rrm3, hdf1, hdf1 rrm3, hxk2, hxk2 rrm3. Representative microscopic images (PI-stained cells and the brightfield images) are displayed in Supplement Fig. 3.
Whether PI-stained cells are indeed dead/dying cells, we applied two approaches. We were co-staining rrm3 mutant cells grown in medium containing 2% or 0.05% (CR) glucose with fluorescein diacetate (FDA) and PI (Fig. 3B)84. Living cells are stained in green (FDA), whereas dead cells (or undergoing cell death) display a red/orange color (PI). We noticed that some of the CR-rrm3 cells were stained both in green and red/orange (indicated with arrows; Fig. 3B). We propose that these cells are in a transition-state to cell death. In the second approach, we enriched PI-stained cells by cell sorting (Fig. 3C). Whereas one fraction contained cells (here: CR-rrm3 mutant cells) with 25–50% PI-positive cells, the other fraction contained cells, which were not stained with PI (PI-negative cells; Fig. 3C). Equal numbers of cells of these two fractions were spread on YEPD plates. About 70% of the fraction containing PI-stained cells formed viable colonies (Fig. 3C). An aliquot of these fractions (stained or not stained with PI) was also used to purify intact genomic DNA and was used to determine DNA breakage by PCR as shown in Fig. 1. Broken DNA (rDNA) is mainly detected in the fraction with the PI-stained cells (Fig. 3C). Regarding the general growth behavior, colonies of WT and rrm3 mutant cells on plates containing medium with 2% or 0.05% glucose were of similar size, respectively (Fig. 3D). CR cells (both WT and rrm3 mutant background) had a 5–10% slower growth curve (Fig. 3D).
We were analyzing loss-of-function mutations of several genes (i.e., deletion mutations) to address whether they affect (e.g., promote, suppress) the increase of the rates of PI-stained cells, when rrm3 deletion mutant cells are switched from normal glucose levels (i.e., 2% glucose) to CR-restricted growth conditions (0.05% glucose). For example, the nuclear proteins Pif1, Rad52 and Hdf1, which are all involved in DNA repair/maintenance mechanisms, may promote/affect the formation of potential toxic DNA intermediates under CR growth conditions.
We tested mutants lacking the homolog of Rrm3, the DNA helicase Pif185,86. Since Pif1 is present in both the mitochondria and the nucleus, we used mutants only lacking the nuclear form of Pif1 (pif1-m2)85. The pif1-m2 mutant strain does not have a mitochondrial defect (i.e., petite growth phenotype). The double mutant strain rrm3 pif1-m2 shows the same trend as the single mutant strain rrm3. We observed that the number of PI-stained cells increases in rrm3 pif1-m2 from 0.2% (in 2% glucose) to 6.7% (in 0.05% glucose), and in the rrm3 mutant from 0.13% (in 2% glucose) to 5.1% (in 0.05% glucose) (Fig. 3E; Supplement Fig. 3), the increase in the double mutant is about 30% larger. The number of PI-stained cells remains low in the pif1-m2 single mutant in both under CR (0.6%) and non-CR (0.48%) growth conditions and are lower compared to WT (Fig. 3E; Supplement Fig. 3).
We investigated whether lack of the repair genes Rad52 and Hdf1, which are involved in homologous and non-homologous DNA repair, have an influence on the percentage of PI-stained cells in rrm3 mutant cells87,88,89. Neither lack of Rad52 nor Hdf1 has any influence on the increase in the number of PI-positive cells, when rrm3 mutant cells are transferred from growth medium containing 2% glucose to CR-growth medium (Fig. 3E; Supplement Fig. 3). However, an increase in the number of PI-stained cells can be observed in the single mutants (rad52, hdf1), when these cells are transferred from medium containing 2% glucose to CR-growth medium (Fig. 3E; Supplement Fig. 3).
Hxk2 encodes one of three hexokinases involved in converting glucose into glucose-6-phosphate, thus introducing glucose into the glycolysis pathway90. Absence of the non-essential Hxk2 protein has similar effects on cells compared to CR-growth conditions in some genetic backgrounds1,91,92. We determined the frequency of the appearance of PI-stained cells of hxk2 and rrm3 hxk2 mutant strains grown in medium containing 2% glucose and CR (Fig. 3E). Interestingly, whereas under CR-growth conditions in all other combination (WT versus rrm3, pif1-m2 versus pif1-m2 rrm3, rad52 versus rad52 rrm3, hdf1 versus hdf1 rrm3) the number of PI-stained cells increases, there is a decrease in these numbers, when hxk2 is compared to rrm3 hxk2 suggesting the increase in cell death in the CR-rrm3 cells is partially independent of the glucose concentration (Fig. 3E; Supplement Fig. 3).
In summary, (i) though the DNA breakage rates (Fig. 1) and appearance of PI-stained cells (Fig. 3) follow similar trends to some extent, there are differences. Whereas the increase in DNA breakage rates of rrm3 cells after transferring from medium containing 2% glucose to CR medium does not reach WT-levels for most of the tested DNA fragments (except for rDNA), numbers of PI-stained (and presumably dying or dead) cells are fivefold higher in CR-rrm3 cells compared to WT cells (Fig. 1). (ii) The absence of Pif1, Rad52 or Hdf1 has no influence on the increase in the number of PI-stained cells of rrm3 mutant cells (comparison of WT with the rrm3 mutant, and single mutants (pif1-m2, rad52, hdf1) with rrm3 double mutants) under CR-growth conditions. (iii) In contrast, when hxk2 and rrm3 hxk2 mutant strains are compared under CR-growth conditions, the number of PI-stained cells decrease in the rrm3 hxk2 mutant strain.
The checkpoint protein Rad53 is less (or not at all) phosphorylated in the rrm3 single and other rrm3-combination mutants under CR
The rrm3 mutant has a delay in the cell cycle when cells pass through the G2/M phases23,25. We determined by FACS analysis whether the cell cycle profiles of WT and rrm3 mutant cells alter when cells are switched to CR-growth conditions (Fig. 4A, FACS). There are 22.8% of the WT cells in S-phase in medium containing 2% glucose, and 16.5% S-phase cells in CR-medium (1.4-fold decrease). There are 62.0% of the WT-cells in G2/M-phase in medium containing 2% glucose, and 64.2% of the cells in G2/M-phase in CR-medium. There are 15.2% of the WT-cells in G1-phase in medium containing 2% glucose, and 19.3% of the cells in G1-phase in CR-medium, which is a 1.3-fold increase. In contrast, whereas 83.9% of rrm3 cells grown in medium containing 2% glucose reside or pass slowly through the G2/M phases, the number of cells in these phases drop to 70.1% in CR-growth medium. The number of rrm3 cells in S-phase increases from 15.8% to 26.6% (1.7-fold increase), and in G1-phase from 0.24% to 3.2% (13.3-fold increase) when cells are switched to CR-growth medium.

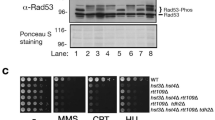
Increased numbers of cells in S-phase when rrm3 mutant cells are grown under caloric-restricted (CR) growth conditions and lower levels of Rad53-phosphorylation in CR-rrm3 cells and several CR-rrm3 double mutations. (A) Cell cycle distribution of WT and rrm3 mutant cells, grown in medium containing 2% or 0.05% (CR) glucose, determined by FACS analysis. The distribution of cells in G1, S and G2/M were modelled according to the ModFit LT program. Percentages of cells with 1N, 2N and intermediate DNA content are displayed as a histogram. Standard errors are shown. (B) Western-blots probed for Rad53p of the indicated strains and grown in YEPD containing 2% (CR: −) or 0.05% (CR: +) glucose. (C) Western-blots probed for Rad53p of the indicated strains and grown in YEPD containing 2% (CR: −) or 0.05% (CR: +) glucose in the presence of 0.2 M hydroxy urea (HU) for one hour. Percentage-distributions are displayed below. (D) Time course experiment with WT and rrm3 mutant cells grown in medium containing 2% or 0.05% (CR) glucose and containing 0.2 M HU. Samples were taken at the indicated times and probed for Rad53p by western-blotting. Asterisks indicate Rad53 protein species which were still non-phosphorylated under CR-growth conditions. Experiments in (A–D) were carried out three times. Images of the western-blots in full size are displayed in supplementary figures.
Since DNA damage occurs in rrm3 mutant cells which activates the intra S-phase checkpoint26,58,59, we determined the status of the Rad53 protein phosphorylation in WT, various single mutant strains (i.e., rrm3, pif1-m2, rad52, hdf1) and rrm3 double mutant strains (i.e., with pif1-m2, rad52, hdf1) in growth medium containing 2% or 0.05% (CR) glucose (Fig. 4B). As previously demonstrated, as an indication for DNA damage Rad53 is phosphorylated in rrm3 cells, which were grown in medium containing 2% glucose (Fig. 4B)26,58,59. However, Rad53 is phosphorylated at largely reduced levels in CR-rrm3 cells (Fig. 4B). Similar phenotypes appear also in the pif1-m2 rrm3, rad52 rrm3 and hdf1 rrm3 double mutants, i. e. intense phosphorylation of Rad53 in cells grown in 2% glucose and reduced/missing phosphorylation in the corresponding CR-cells (Fig. 4B). Rad53 protein exhibits limited phosphorylation in rad52 cells grown in medium containing 2% glucose which is markedly reduced in CR-rad52 cells (Fig. 4B). No phosphorylation of Rad53 occurs in WT, pif1-m2 and hdf1 mutant cells (grown in medium containing 2% glucose or grown under CR-conditions) (Fig. 4B).
Since no phosphorylation of Rad53 is observed in CR-rrm3 cells, we investigated whether the checkpoint can still be activated. We determined the status of Rad53 phosphorylation in cells experiencing induced DNA damage and growing at normal glucose levels (i.e., 2%) or under CR (i.e., 0.05%). We treated WT, various single mutants (i.e., rrm3, pif1-m2, rad52, hdf1) and rrm3 double mutants (i.e., with pif1-m2, rad52, hdf1) with hydroxy-urea (HU; 0.2 M, final concentration) for 1 h (Fig. 4C). Rad53 protein in WT and pif1-m2 cells grown in 2% glucose and in the presence of HU is highly phosphorylated; in contrast, levels of Rad53 protein phosphorylation are markedly reduced in the corresponding CR-cells (Fig. 4C). Although in the single mutants rrm3, rad52, hdf1, and the rrm3 double mutants (i.e., with pif1-m2, rad52, hdf1) Rad53 protein is phosphorylated in cells grown both in 2% glucose and in CR-medium, the non-phosphorylated form is more abundant under CR-growth conditions (except in the case of hdf1) (Fig. 4C, histogram). Further, in a time course experiment we determined that in WT cells grown in medium containing 2% glucose and HU, Rad53 is activated (by phosphorylation) faster compared to WT cells grown under CR growth conditions or in rrm3 mutant cells grown in HU-medium containing 2% glucose or 0.05% (CR) (Fig. 4D; asterisks indicate remaining non-phosphorylated form in CR-cells).
In summary, although Rad53 does not appear to be phosphorylated in CR-rrm3 cells in unchallenged conditions (in contrast to rrm3 cells grown in medium containing 2% glucose), CR-rrm3 cells under induced DNA damage conditions does result into phosphorylation of Rad53, thus the damage response mechanisms in the CR-rrm3 mutant cells appear to be functional.
The Sir2/Sir3/Sir4 complex is needed for the increase of the number of PI-stained cells of CR-rrm3 mutant cells
Given there is a relationship between glucose restriction, repressive chromatin, and presence/activity of Rad5364, we investigated whether absence of any member of the Sir-complex affects the increase in the number of PI-stained cells when rrm3 cells are transferred from normal growth medium (2% glucose) to CR-growth medium (0.05%). In all single deletion mutants of the Sir-complex (sir2, sir3 and sir4) the percentage of PI-stained cells is elevated compared to WT in medium containing 2% glucose (WT: 1.1%; sir2: 2.6%; sir3: 3.8%; sir4: 2.7%; Fig. 5A; Supplement Fig. 4). Under CR growth conditions in both single (sir2, sir3 and sir4) and double mutants (sir2 rrm3, sir3 rrm3 and sir4 rrm3), the percentage of PI-stained cells is either only minimally increased (sir2: 2.6% (2% glucose) and 3% (CR); sir2 rrm3: 0.61% (2% glucose) and 1.3% (CR)) or even decreased (sir3: 3.8% (2% glucose) and 0.78% (CR); sir4: 2.7% (2% glucose) and 1.5% (CR); sir3 rrm3: 1.4% (2% glucose) and 0.43% (CR); sir4 rrm3: 0.86% (2% glucose) and 0.39% (CR)) (Fig. 5A; Supplement Fig. 4).

The Sir2/3/4 proteins are needed for the increase in cell death rates in the rrm3 mutant after switch to CR-restricted growth conditions. (A) The histogram displays percentage of propidium iodide (PI)-stained cells. Standard errors are shown. Cells of the indicated strains were grown in medium containing 2% or 0.05% glucose to an early logarithmic phase (OD600 ~ 0.1). These are WT, rrm3, sir2, sir2 rrm3, sir3, sir3 rrm3, sir4, sir4 rrm3. Representative microscopic images (PI-stained cells and the brightfield images) are displayed in Supplement Fig. 4. (B) rrm3 mutant cells were grown under CR conditions in the absence or presence of the Sir2-inhibitor sirtinol (20 µM final concentration). Microscopic images display PI-stained cells (orange) and the corresponding brightfield images (PI-staining superimposed). Bar: 50 µm. The histogram displays the percentage of PI-stained cells. Experiments were carried out three times. Standard errors are shown. (C) Western-blots probed for Rad53p of the indicated strains and grown in YEPD containing 2% (CR: −) or 0.05% (CR: +) glucose. The image on the left side (WT, rrm3; 2% and 0.05% glucose) is taken from Fig. 4B for comparison. Experiments were carried out two times. (D) Western-blots probed for Rad53p of the indicated strains and grown in YEPD containing 2% (CR: −) or 0.05% (CR: +) glucose in the presence of 0.2 M hydroxy urea (HU) for one hour. Asterisks indicate the fully phosphorylated Rad53 protein. The image on the left side (WT, rrm3; 2% and 0.05% glucose) is taken from Fig. 4C for comparison. Percentage-distributions are displayed below. Experiments were carried out two times. Images of the western-blots in full size (C,D) are displayed in supplementary figures.
The Sir2-inhibitor sirtinol reduces the enzymatic activity of the Sir complex (Sir2/3/4)93. Interestingly, culturing of rrm3 mutant cells in CR-medium in the presence of sirtinol causes an increase in the number of PI-stained cells (Fig. 5B).
In summary, absence of the Sir2/Sir3/Sir4 complex decreases the numbers of PI-stained cells in rrm3 mutant strains under CR-growth conditions (Fig. 5A; Supplement Fig. 4) though the Sir2 protein may have a separate/additional role in this process (e.g., Sir2, but not Sir3/4, is present in the nucleolus).
The checkpoint protein Rad53 is phosphorylated at about the same levels in rrm3 sir2, rrm3 sir3, or rrm3 sir4 mutants grown in medium containing 2% glucose or in CR-medium
We determined the status of the Rad53 protein phosphorylation in both single (sir2, sir3 and sir4) and double mutants (sir2 rrm3, sir3 rrm3 and sir4 rrm3) in growth medium containing 2% or 0.05% (CR) glucose (Fig. 5C). Rad53p is phosphorylated equally in the double mutant rrm3 (with sir2, sir3 and sir4) cells grown in medium containing 2% or 0.05% glucose (Fig. 5C). Also, here we investigated the status of Rad53 phosphorylation in cells experiencing induced DNA damage and growing at normal glucose levels (i.e., 2%) or under CR (i.e., 0.05%). We treated WT, the single mutants (rrm3, sir2, sir3 and sir4) and rrm3 double mutants (sir2 rrm3, sir3 rrm3 and sir4 rrm3) with hydroxy-urea (HU; 0.2 M, final concentration) for 1 h (Fig. 5D). Whereas in WT and rrm3 cells grown in 2% glucose Rad53 protein is highly phosphorylated and at a reduced level in the corresponding CR-cells, Rad53 is phosphorylated at similarly high levels in all the sir-mutants in cells grown both in 2% glucose and in CR-medium (Fig. 5D, histogram).
Mitochondrial oxidative stress contributes to the increase in PI-stained cells of the rrm3 mutant under CR growth conditions
Cell death including apoptosis may involve and be initiated by the opening of the mitochondrial structure (e.g., opening of the MTP (mitochondrial permeability transition pore), rupture of the outer mitochondrial membrane with release of cytochrome c) which may lead to an increase in oxidative stress94,95. Using fluorescence microscopy, we visualized which cells exhibit excessive oxidative stress and undergo cell death. Cells with excessive numbers of reactive oxygen species (ROS) were visualized by using the Singlet Oxygen Sensor Green (SOSG, Molecular Probes) reagent, and cells potentially undergoing cell death were detected with PI as described above (Supplement Fig. 5)96. Whereas almost all PI-stained cells had clear overlapping SOSG signals in WT and rrm3 mutant cells (both with medium containing 2% glucose and CR-medium), there were additional cells with excessive ROS (compared to the number of PI-positive cells) under normal growth conditions (i.e., 2% glucose) both in WT and rrm3 mutant cells, which however did not appear (yet) to undergo cell death (PI-positive) (WT: ~ 50% increase; rrm3: ~ 100% increase)(SOSG-positive/PI-negative cells labeled with asterisk; Supplement Fig. 5A). In contrast, almost all the WT and rrm3 mutant cells grown in CR-medium, which showed excessive oxidative stress (i.e., green staining by SOSG) were also PI-positive (i.e., likely undergoing cell death), there were almost no SOSG-positive cells, which were not PI-positive.
Calorie restriction increases respiration activity74. We were co-staining cells with SOSG and MitoTracker Red CMXRos, which is a red-fluorescent dye that stains mitochondria in live cells and which reflects the intensity of membrane potential (i.e., respiration activity; Supplement Fig. 5B). In general, cells grown under CR conditions (both WT and rrm3 mutant cells) exhibit stronger staining with MitoTracker Red CMXRos reflecting higher respiration activity.
To investigate whether an increase in mitochondrial oxidative stress is a prerequisite for the increase in the numbers of PI-positive cells in the CR-grown rrm3 mutant cells, we treated CR-rrm3 mutant cells with the compound Mito-TEMPO (SigmaAldrich, St. Louis, MO) which reduces oxidative stress in mitochondria97. Treating CR-grown rrm3 mutant cells in the presence of 160 µM Mito-TEMPO reduces the number of cells with PI-positive signals to about 50% of untreated CR-rrm3 mutant cells (Supplement Fig. 5C).
In summary, minimizing mitochondrial oxidative stress decreases the cell death rates in CR-rrm3 mutant cells.
Discussion
Growth conditions of calorie restriction (CR) usually induce lifespan extension, improvement of tissue quality, and an increase in genome stability5. We discovered that the chromosomal DNA breakage as well as survival rates of the rrm3 DNA helicase mutant are impacted by CR-growth conditions. By using the terminal deoxynucleotidyl transferase (TdT) assay we observed that despite the presence of about 1400 chromosomal replication pause sites (RPS) DNA breakage rates of rrm3 mutant cells were lower compared to WT in normal growth medium (i.e. 2% glucose) (Fig. 1). However, after transferring the cells to CR-growth conditions (i.e. 0.05% glucose) DNA breakage rates increased in the rrm3 mutant. Except for rDNA, where DNA breakage rates were higher in the CR-rrm3 mutant compared to WT, DNA breakage rates in the CR-rrm3 mutant did not reach WT levels at numerous other (non-rDNA) genomic loci (Fig. 1). The numbers of PI-stained cells, which reflect cell death rates, mirrored the DNA breakage rates to some degree, however there were a few differences (Fig. 3). Though the cell death in WT and rrm3 mutant cells may have different causes, cell death rates are lower in the rrm3 mutant compared to WT when grown in medium containing 2% glucose. One possibility to interpret this is that rrm3 mutant cells (in 2% glucose medium) have numerous replication defects which activate the S-phase checkpoint25,26,58,59. These conditions lead to a delay in the cell cycle (around G2/M phases) to allow repair of any defects, thus cell death rates are reduced. However, under CR-growth conditions cell death rates increase in the rrm3 mutant to fivefold higher levels (compared to CR-WT cells), which contrasts with the changes in DNA breakage rates (for most of the tested genomic loci) in the CR-rrm3 mutant. Though it is unknown what the primary cause of the increase in cell death rates in the rrm3 mutant is after transfer of the cells from 2% glucose medium to CR-growth medium, the cell death events (in the rrm3 mutant) might be the consequence of single deleterious events in individual cells. For example, loss of a single telomere may lead to the loss of the affected chromosome and consequently to cell death98. In individual cells, different chromosome parts might be prone to severe damage/loss, which may lead to cell-death as an overlapping result in all affected cells. DNA breakage events may occur in individual cells at different genomic locations, this might be a possible explanation why the DNA breakage rates are lower at individual sites compared to the overall cell death rates. rDNA might be an exception because it is a repeated sequence. Further, there are differences in the cell cycle-distribution in rrm3 cells grown in 2% glucose versus CR-growth conditions (Fig. 4), which may also contribute to a difference in the breakage rates. In addition, the measured DNA breakage events might result from different events. Breakage events may occur during deleterious replication/recombination processes in chromosomal DNA, but may also be the result of cells undergoing cell death as a consequence of a deleterious chromosomal damage event that cannot be repaired (i.e., degradation of chromosomal DNA). There is however an indication that the majority of the detected breakage events originate from the dead cells (see sorting experiment in Fig. 3C). Determining the locations of DNA breaks on potentially single-cell level is a direction to address where DNA breaks initially before cells may enter the cell death pathway99,100,101.
Interestingly, the Rad53 checkpoint protein is phosphorylated at largely reduced levels in CR-rrm3 mutant cells (Fig. 4). The Rad53 checkpoint protein normally ensures that cells (transiently) arrest, when DNA is damaged (e.g., broken DNA as a possible consequence of stalled replication forks) until the damage is repaired102,103. We demonstrate that replication fork pausing/stalling at tRNA and 5S rDNA genes as well as at selective silent replication origins is in fact reduced/missing in the CR-rrm3 cells compared to rrm3 cells propagated in medium containing 2% glucose (Fig. 2). There are about 270 tRNA genes, 100–150 5S rDNA genes and 50–100 (or more) silent replication origins in the haploid yeast genome. Conditions of CR lead to a down-regulation of protein synthesis activities which include translation regulation, ribosome synthesis and assembly as well as mRNA export from the nucleus104,105,106. Recently we also found less binding of Tfc1 (part of the pre-initiation complex) to 5S rDNA and tRNA genes in CR-cells, which may explain the largely reduced replication pausing at 5S rDNA and tRNA genes in the CR-rrm3 mutant cells81. Alternatively, the reduced replication pausing/stalling phenotype may also be the result of the higher expression of Rrm3p-like helicases in CR-rrm3 mutant cells.
The reduction in pausing/stalling at tRNA and 5S rDNA genes as well as at selective silent replication origins would fit to the data with the reduced phosphorylation rates of Rad53 in CR-rrm3 cells assuming that paused forks may also stall and collapse leading to Rad53 activation by phosphorylation. Since about 5% of the CR-rrm3 cells are undergoing cell death (stained with PI; Fig. 3), we speculate that the non-phosphorylated form of Rad53 and the reduced pausing/stalling at the RPS originates (likely mainly) from the 95% remaining living rrm3 cells. Western-blotting and DNA 2D gels are not sensitive enough to detect protein-modifications and DNA structures, respectively, only occurring in the 5% cell population which represent the dying cell population. However, Rad53 protein can be activated by induced DNA damage in CR-growth medium (by exposing to HU treatment; Fig. 4), thus if DNA damage (such as broken replication forks or long single-stranded DNA tracts) is present, Rad53 can be activated by phosphorylation. We propose therefore that Rad53 protein residing in the 95% cell population of the living CR-rrm3 cells is (almost) not activated under non-challenging conditions, thus DNA breakage may occur only at an extremely low frequency (e.g., in a few single cells).
Another possibility to explain the elevated levels of non-phosphorylated Rad53 in the CR-rrm3 mutant is that more cells of this mutant are in S-phase as demonstrated by FACS analysis (Fig. 4A). Schleker et al. demonstrated that Rad53 exhibits a small phosphorylation-dependent electrophoretic mobility shift in G2, M and G1 phases of the cell cycle that is missing in S-phase and that occurs independently of nuclear DNA damage107. The G2, M and G1 phases-dependent phosphorylation of Rad53 appears to be accomplished by the Cdc5 and Cdc28 protein kinases, but the loss of phosphorylation during S-phase does not depend on the protein phosphatases Ptc2 and Ptc3, which are normally involved in checkpoint adaptation107. However, under CR-growth conditions other protein phosphatase may dephosphorylate Rad53 efficiently such as Irc21, which acts together with other protein phosphatases including PP2A108. A proposed reason for the fluctuation in Rad53 phosphorylation is that the checkpoint response is attenuated in S-phase to allow for naturally occurring single-strand DNA at the replication forks107,109,110. In unchallenged cells temporarily activation of Rad53 at the beginning of S-phase has also the purpose of raising the amount of the dNTP pool which is needed during a faithful DNA replication process111.
What could be the reason why rrm3 mutant cells undergo increased cell death when transferred from 2% glucose medium to CR-medium (Fig. 6)? We demonstrate here that the increase in cell death rates in the rrm3 mutant after shifting cells from 2% glucose medium to CR-growth conditions is dependent on the Sir-complex (Sir2/3/4), but appears to be independent of Pif1, Rad52 or Hdf1 (Figs. 3 and 5). Whereas there is still a small increase in PI-positive cells in the sir2 rrm3 mutant after transferring cells from 2% glucose-medium to CR-growth medium, there is clearly a reduction in PI-positive cells when sir3 rrm3 or sir4 rrm3 mutant cells are transferred to CR-growth medium (Fig. 5). Whereas Sir2 alone can be found localized to the rDNA locus, the Sir complex consisting of the Sir2/Sir3/Sir4 proteins binds to telomeres and sub-telomeric regions as well as to the silent mating type loci HML and HMR (and surrounding area)65. Besides the well-known function of the Sir complex in silencing of genes at telomeres, the silent mating type loci or the rDNA locus, the Sir proteins also affect genome stability. For example, the Sir complex (Sir3 and Sir4) may promote non-homologous-end-joining (NHEJ), but there are also reports that Sir4 inhibits NHEJ, which may induce telomere end fusions112,113,114. Previously we described that in the absence of Rrm3 replication forks pause (or potentially stall) within telomeric DNA and the silent mating type loci HML and HMR, and that this pausing/stalling is still occurring in the absence of the Sir-complex (Sir2/Sir3/Sir4)26. Interestingly, we found that in the CR-sir3 rrm3 or sir4 rrm3 double mutant cells Rad53 protein is activated (phosphorylated; two bands visible) in contrast to the CR-rrm3 single mutant in which Rad53 does not appear to be activated (not phosphorylated; only one band visible on the western blot) (Fig. 5). The Sir-complex has known structural and enzymatic roles at telomeres and the silent mating type sites65. Under CR growth conditions, Rrm3 may have an important (essential) function as a helicase when the replication fork passes through these Sir-protein-complex covered regions (or at other Sir-dependent regions). In this scenario when Rrm3 is absent, telomeres which are just being replicated may be prone to higher breakage rates because of unfinished replication leading possibly to a chromosome without a telomere. In addition, CR-rrm3 mutant cells treated with the Sir2 inhibitor sirtinol have an increased cell death rate (Fig. 5B) which suggests that an enzymatic activity (e.g., deacetylation) linked to the Sir complex may also be involved in causing the high cell death rates of the CR-rrm3 sir2 cells. Numerous mammalian and yeast studies demonstrate also that specific histone modifications (e.g., acetylation, methylation, ubiquitination) are important features of the cell death program115. For example, the gamma-H2AX phosphorylation-modification, which occurs during the formation of dsDNA breaks, is also present during apoptosis as well as phosphorylation of histone H2B at serine-14 is associated with chromatin condensation and DNA fragmentation (references in115). Future studies will address why Rad53 is phosphorylated in the CR-sir3 rrm3 or sir4 rrm3 double mutant cells but not in the CR-rrm3 single mutant cells as well as we will address the possible role of chromatin modifications in the increased cell death rates in the CR-rrm3 cells.

The Sir protein complex is needed for the increase in cell death rates in the rrm3 mutant after switch to CR-restricted growth conditions. In CR rrm3 mutant cells cell death rates are increased compared to cells grown in regular growth medium (2% glucose), this increase in cell death rates is prevented in the absence of the Sir complex (Sir2/Sir3/Sir4). The Sir-complex is present at telomeres and the silent mating type loci HML and HMR. Whereas the Rad53 checkpoint is activated (i.e., phosphorylated) in rrm3 cells grown in regular growth medium (2% glucose), Rad53 is not activated in CR-rrm3 cells. Rad53 phosphorylation is restored in CR-rrm3 sir2/3/4 cells.
Whereas rrm3 mutant cells grown in 2% glucose medium pass with a delay through the G2/M phases, the number of cells in S-phase increases when rrm3 mutant cells are transferred to CR-growth conditions (Fig. 4). The cell-cycle arrest/delay during the DNA damage checkpoint in the G2/M phases is mainly regulated by the chaperone protein Pds1 (securin in mammals) and its binding partner, the cohesin protease Esp1 (separase)116,117. Pds1 binds Esp1 in undamaged cells and inhibits its protease activity118. When cells transit from metaphase to anaphase, Pds1 is ubiquitinated by the E3 ubiquitin ligase APC-Cdc20119. It is then degraded by the proteasome. Degradation of Pds1 releases Esp1 to cleave cohesin rings. This process allows sister chromatids to segregate. When DNA is damaged, Pds1 is phosphorylated by Chk1, blocking its interaction with APC-Cdc20116. Cdc20 is phosphorylated by Rad53, which prevents APC-Cdc20 from ubiquitinating Pds1120. Together, both Pds1 and Cdc20 phosphorylation inhibit mitosis progression by keeping Esp1 inactive. Deletion of Pds1 reduces the duration of G2/M arrest by 60% while arrest is completely abrogated in pds1 rad53 mutants121,122. It will be interesting to test whether Cdc20 is not phosphorylated by Rad53 in CR-rrm3 mutant cells thereby releasing cells into M-phase before DNA damage is repaired.
Numerous proteins interact with Rrm3 on a genetic and/or functional level to affect telomeric or replicative processes40,58,59,123,124,125. Examples are the fork protection complex Tof1/Csm3 and Mrc1, proteins that are present at telomeres including the Mre11/Rad50/Xrs2 complex, or the cullin Rtt101. Viability of rtt101 mutants depends on Rrm3123. Further, the increased cell death rates in the CR-rrm3 mutant are also induced by mitochondrial reactive oxygen species (ROS) since treatment with the drug Mito-TEMPO, which reduces mitochondrial oxidative stress, lowers the cell death rates in the CR-rrm3 mutant cells (Supplement Fig. 5C). It remains to be established how these genetic and metabolic factors interact to elucidate why rrm3 mutant cells in CR growth medium experience higher cell death rates compared to rrm3 mutant cells grown in medium 2% glucose. Though the genomic-instability mutant rrm3 behaves worse under CR-conditions, dietary restriction for example in progeroid DNA repair-deficient mice (DNA repair-deficient Ercc1∆/- mice) dramatically extend health span and tripled life span126. Thus, future studies are needed to explore how nuclear genomic instability, which is a prominent feature associated with cancer, affects cell survival under caloric/dietary restriction.
Methods
Experimental model and subject detail
We used haploid WT and mutant S. cerevisiae strains (rrm3, sir2, sir3, sir4, pif1-m2, rad52, hdf1, hxk2, rrm3 sir2, rrm3 sir3, rrm3 sir4, rrm3 pif1-m2, rrm3 rad52, rrm3 hdf1, rrm3 hxk2) (a list of used yeast strains is provided in the Supplemental Experimental Procedures as Table S1)23,26,27,85. HXK2 was knocked out by LEU2 as described previously127.
Method details
Yeast methods, cell growth, incubation with drugs
Yeast strains were thawed from frozen stocks, streaked out on YEPD (yeast extract, pepton, dextrose) or selection plates containing 2% glucose and grown at 30 °C 2–3 days until small colonies were visible. Colonies were suspended in YEPD growth medium containing 2% glucose and grown for 6 h. Optical density (OD at 600 nm) was determined, cells were grown in YEPD growth medium containing 2% (regular level) or 0.05% (caloric restriction, CR) glucose to an early-logarithmic growth phase (about 1 × 107 cells/ml). This means, usually cells were diluted to an OD600 of 0.001, grown to an OD600 of 0.1 (12–15 h) and then resuspended in fresh growth medium (containing 2% or 0.05% glucose) for about two hours to avoid growth limitations. Strains harboring the plasmids pRS315 (empty plasmid (LEU2)) or pIA20 (pRS315-RRM3, pRS315-rrm3 (K260A), pRS315-rrm3 (K260R)) were maintained in synthetic growth medium lacking the amino acid leucine27. Sirtinol (Sigma-Aldrich, St. Louis, MO) was dissolved in DMSO at a concentration of 5 mM and used in the experiment at a final concentration of 20 µM. Mito-TEMPO (Sigma-Aldrich, St. Louis, MO) was dissolved in DMSO at a concentration of 20 mM and used in the experiment at a final concentration of 160 µM.
Determination of the DNA breakage rates using the terminal deoxynucleotidyl transferase (TdT) assay combined with PCR
The TdT assay is used to map the locations of double-strand DNA (dsDNA) breaks on a genome-wide scale by end-labeling of the DNA ends77. We purified highly intact whole genomic DNA and performed the TdT assay reaction using biotinylated dCTP. DNA was digested with a restriction enzyme (i.e., BglII) and the DNA fragments with the extended biotinylated tags (indicating DNA breakage) were recovered using avidin-coated magnetic beads (called: pellet; Supplement Fig. 1A,B). PCR was used to determine the abundance of specific DNA fragments in the sample. As a control and to normalize all samples, the same DNA sequences were amplified from samples taken before the broken DNA fragments (i.e., tagged with the biotinylated nucleotide) were recovered using the avidin-coated beads (called: input; Supplement Fig. 1A,B).
DNA of 5 × 108 cells in an early-logarithmic growth phase (about 1 × 107 cells/ml) was purified with the MasterPure™ purification kit (Epicentre, Madison, WI). The TdT reaction using terminal transferase (NEB, Ipswich, MA) was carried out in the presence or absence (negative control) of Biotin-14-dCTP (Invitrogen, Carlsbad, CA). DNA was precipitated and then digested in a 50 µl volume with the restriction enzyme BglII (NEB, Ipswich, MA). A small aliquot (3 µl) was removed, which was diluted 1:100 in 1xPBS and served for the “Input” PCR. The rest was diluted with 1 ml 1xPBS and 10 µl avidin-coated magnetic beads (NEB, Ipswich, MA; washed with 1xPBS) were added, which removed the broken DNA fragments with the biotinylated nucleotide extension. The DNA solution was incubated with the beads for 2 h at 4 °C. To remove nonspecifically bound DNA fragments, the beads were washed with a buffer containing 5 mM Tris–HCl (pH 8.0), 0.5 mM EDTA (pH 8.0), 1 M NaCl, and 0.05% TWEEN 20 at 50 °C for 2 h. Afterwards the beads were washed with 1 ml 1xPBS three times and resuspended in 100 µl 1xPBS (called “Pellet”). PCR at the indicated locations (see list of used primer-sets in the Supplemental Experimental Procedures) were carried out with “Pellet” and “Input” DNA. The PCR condition was 23 cycles (for rDNA) and 32 cycles (for non-rDNA locations) of denaturation at 94 °C for 30 s, annealing at 60 °C for 45 s (for rDNA) and 50 °C for 45 s (for non-rDNA locations), and elongation at 68 °C for 165 s. PCR fragments were analyzed on an 1% agarose gel and DNA bands were visualized on a Bio-Rad™ Laboratories imaging system and the intensity of the bands was determined by using the program ImageLab (Bio-Rad™ Laboratories, Hercules, CA).
Analysis of DNA replication intermediates and determination of ERCs
DNA for visualizing DNA replication intermediates in yeast was prepared essentially as described previously79. Cells were grown to an early-logarithmic growth phase (about 1 × 107 cells/ml). After harvesting the cells and washing with water, the cells were resuspended in nuclear isolation buffer (NIB: 50 mM MOPS free acid; 150 mM KOAc; 2 mM MgCl2; 150 µM Spermine; 500 µM Spermidine; 17% (v/v) Glycerol; pH 7.2) and lysed using glass beads (about 10 times for 30 s on a vortex in the cold room; cooling in between on ice) as described previously79. Glass beads were removed and the nuclei, broken and unbroken cells were harvested, the pellet resuspended in 4.2 ml TEN buffer (50 mM Tris–HCl, pH 8.0; 50 mM EDTA; 100 mM NaCl), Proteinase K (75 µl of a 20 mg/ml solution) and N-lauroylsarcosine (0.8 ml of a 7.5% solution). Samples were incubated at 37 °C for one hour. Non-lysed cells were removed by centrifugation in a microcentrifuge (10 min; 14,000 rpm; 4 °C). Supernatant liquids were combined and CsCl (final concentration: 1.05 g/ml) and Hoechst Dye #33258 (0.025 volumes of 5 mg/ml) were added. DNA was prepared by CsCl-gradient centrifugation (VTi65.2 rotor; 16–20 h, 55,000 rpm, 17 °C). Highly intact whole genomic DNA was digested for about 5 h at 37 °C with the appropriate restriction enzymes (BglII: rDNA; EcoRV: ARS313/314; BglII: ARS608; EcoRV: tRNAY; NEB, Ipswich, MA)26,27,28. The structure of DNA replication intermediates were determined by neutral–neutral DNA 2D agarose gel electrophoresis and Southern technique using radioactive-labeled probes26,27,79. Typically, the first dimension was performed in 0.35% (w/v) 1xTBE (Tris–borate-EDTA) agarose gels at 0.6–0.7 V/cm for 45–48 h at room temperature. The second dimension was performed in 0.9% (w/v) 1xTBE agarose gels in the presence of 0.3 μg/ml ethidium bromide at 2.6–3 V/cm for 18–24 h at 4 °C. Primer sequences for the PCR amplifications of DNA probes are listed under Supplemental Information. The amounts of specific replication intermediates were determined by phosphor imager analysis.
The amount of extrachromosomal rDNA circles (ERCs) was determined by 2D gels (0.5 μg/ml ethidium bromide in both dimensions)27.
Staining procedures
Yeast cells were grown to an early-logarithmic growth phase (about 1 × 107 cells/ml) in the appropriate medium containing 2% or 0.05% glucose. Dead yeast cells were stained with propidium iodide (PI; final concentration: 0.3 µg/ml) in the growth medium for ~ 20 min at room temperature. PI-stained cells were visualized by epifluorescence microscopy (excitation: 493 nm). Living cells were detected with fluorescein diacetate (FDA; 5 mg/ml in acetone) as previously described84. FDA-stained cells were visualized by epifluorescence microscopy (excitation: 488 nm).
Staining with Singlet Oxygen Sensor Green (SOSG, Invitrogen/Molecular Probes; dilution 1:1000) was carried out on live cells together with PI. SOSG-stained cells were visualized by epifluorescence microscopy (excitation: 488 nm). Staining with MitoTracker® Red CMXRos (Invitrogen/Molecular Probes, Eugene, OR) (excitation: 493 nm) was carried out on live cells together with SOSG.
The numbers of cells on the images were determined using the software NIH imageJ128.
Cell sorting
Yeast cells were grown to an early logarithmic growth phase, PI was added to a final concentration of 0.3 µg/ml. PI-stained cells were separated from unstained cells using a BD FACSAria II cell sorter with excitation off the 561 nm laser and acquisition with the PI filter set in the low 600 nm region. PI-stained and non-stained cells were collected. To determine cell viability, equal cell numbers were plated on YEPD plates (2% glucose) and incubated at 30 °C for 3 days. An aliquot of the cells was used to determine DNA breakage as described above for the TdT assay.
Fluorescent activated cell sorting (FACS)
Yeast cells were fixed with formaldehyde (final concentration: 3.7%; early logarithmic growth phase (about 1 × 107 cells/ml)) for one hour at room temperature. Cells were washed two times with 50 mM sodium citrate buffer. Cells were stained by adding PI (final concentration: 25 µg/ml) for one hour at room temperature after digestion of RNA with RNase A (final concentration: 5 µg/ml) overnight at 37 °C. The distribution of cells in G1, S and G2/M were modelled according to the ModFit LT program.
Rad53 western blotting
Yeast cells were grown to an early-logarithmic growth phase (about 1 × 107 cells/ml) in the appropriate medium containing 2% or 0.05% glucose. About 2 × 107 cells were harvested, resuspended in 150 µl lysis-solution (1.85 M NaOH, 2% beta-mercapto-ethanol) and incubated on ice for 10 min. 150 µl of 50% Tri-chlor-acid were added, the solution mixed and incubated on ice for 30 min. Proteins were recovered by centrifugation (10 min, 14,000 rpm, 4 °C). Pellet was resuspended in 50 µl of protein sample buffer (containing 1 M Tris–HCl, pH8.0). Samples were heated at 70 °C for 5 min. 10 µl of each sample were loaded on an 6% SDS-poly-acryl-amide-gel and proteins transferred to nitrocellulose. Rad53 protein was detected with a rabbit polyclonal anti-Rad53 antibody (1: 2000; abcam, Cambridge, MA) followed by a horse-radish-peroxidase-conjugated secondary antibody (1:3000; Bio-Rad™ Laboratories, Hercules, CA).
Statistics
Experiments were carried out two to three times as indicated. Standard errors were calculated.
Data availability
Further information and requests for resources and reagents as well as details about data should be directed to and will be fulfilled by the Lead Contact, Andreas Ivessa (ivessaan@njms.rutgers.edu). All unique strains and reagents generated in this study are available from the Lead Contact without restriction. Requests for strains and reagents donated by other laboratories should be directed to the specific laboratory from which they were received.
References
McCormick, M. A. et al. A comprehensive analysis of replicative lifespan in 4,698 single-gene deletion strains uncovers conserved mechanisms of aging. Cell Metab. 22, 895–906. https://doi.org/10.1016/j.cmet.2015.09.008 (2015).
Longo, V. D. & Anderson, R. M. Nutrition, longevity and disease: From molecular mechanisms to interventions. Cell 185, 1455–1470. https://doi.org/10.1016/j.cell.2022.04.002 (2022).
Abraham, K. J., Ostrowski, L. A. & Mekhail, K. Non-coding RNA molecules connect calorie restriction and lifespan. J. Mol. Biol. 429, 3196–3214. https://doi.org/10.1016/j.jmb.2016.08.020 (2017).
Colman, R. J. et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 325, 201–204. https://doi.org/10.1126/science.1173635 (2009).
Wilson, K. A. et al. Evaluating the beneficial effects of dietary restrictions: A framework for precision nutrigeroscience. Cell Metab. 33, 2142–2173. https://doi.org/10.1016/j.cmet.2021.08.018 (2021).
Novak, J. S. S., Baksh, S. C. & Fuchs, E. Dietary interventions as regulators of stem cell behavior in homeostasis and disease. Genes Dev. 35, 199–211. https://doi.org/10.1101/gad.346973.120 (2021).
Vaquero, A. & Reinberg, D. Calorie restriction and the exercise of chromatin. Genes Dev. 23, 1849–1869. https://doi.org/10.1101/gad.1807009 (2009).
Mercier, B. D. et al. Dietary interventions in cancer treatment and response: A comprehensive review. Cancers https://doi.org/10.3390/cancers14205149 (2022).
Montegut, L., de Cabo, R., Zitvogel, L. & Kroemer, G. Science-driven nutritional interventions for the prevention and treatment of cancer. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-22-0504 (2022).
Isaac-Lam, M. F. & DeMichael, K. M. Calorie restriction and breast cancer treatment: A mini-review. J. Mol. Med. https://doi.org/10.1007/s00109-022-02226-y (2022).
Caffa, I. et al. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 583, 620–624. https://doi.org/10.1038/s41586-020-2502-7 (2020).
Salvadori, G. et al. Fasting-mimicking diet blocks triple-negative breast cancer and cancer stem cell escape. Cell Metab. 33, 2247–2259. https://doi.org/10.1016/j.cmet.2021.10.008 (2021).
Deligiorgi, M. V., Liapi, C. & Trafalis, D. T. How far are we from prescribing fasting as anticancer medicine?. Int. J. Mol. Sci. 21, 9175. https://doi.org/10.3390/ijms21239175 (2020).
Vidoni, C. et al. Calorie restriction for cancer prevention and therapy: Mechanisms, expectations, and efficacy. J. Cancer Prev. 26, 224–236. https://doi.org/10.15430/JCP.2021.26.4.224 (2021).
Brandhorst, S., Harputlugil, E., Mitchell, J. R. & Longo, V. D. Protective effects of short-term dietary restriction in surgical stress and chemotherapy. Ageing Res. Rev. 39, 68–77. https://doi.org/10.1016/j.arr.2017.02.001 (2017).
O’Flanagan, C. H., Smith, L. A., McDonell, S. B. & Hursting, S. D. When less may be more: Calorie restriction and response to cancer therapy. Bmc Med. https://doi.org/10.1186/S12916-017-0873-X (2017).
Caccialanza, R. et al. To fast, or not to fast before chemotherapy, that is the question. BMC Cancer 18, 337. https://doi.org/10.1186/s12885-018-4245-5 (2018).
Castejon, M. et al. Energy restriction and colorectal cancer: A call for additional research. Nutrients https://doi.org/10.3390/nu12010114 (2020).
Shingler, E. et al. Dietary restriction during the treatment of cancer: Results of a systematic scoping review. BMC Cancer 19, 811. https://doi.org/10.1186/s12885-019-5931-7 (2019).
Levesque, S. et al. Trial watch: Dietary interventions for cancer therapy. Oncoimmunology 8, 1591878. https://doi.org/10.1080/2162402X.2019.1591878 (2019).
Ferguson, L. R. et al. Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin. Cancer Biol. 35(Suppl), S5–S24. https://doi.org/10.1016/j.semcancer.2015.03.005 (2015).
Szafranski, K. & Mekhail, K. The fine line between lifespan extension and shortening in response to caloric restriction. Nucleus 5, 56–65. https://doi.org/10.4161/nucl.27929 (2014).
Ivessa, A. S., Zhou, J.-Q., Schulz, V. P., Monson, E. M. & Zakian, V. A. Saccharomyces Rrm3p, a 5’ to 3’ DNA helicase that promotes replication fork progression through telomeric and sub-telomeric DNA. Genes Dev. 16, 1383–1396 (2002).
Claussin, C., Vazquez, J. & Whitehouse, I. Single-molecule mapping of replisome progression. Mol. Cell 82, 1372–1382. https://doi.org/10.1016/j.molcel.2022.02.010 (2022).
Azvolinsky, A. et al. cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev. 20, 3104–3116 (2006).
Ivessa, A. S. et al. The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol. Cell 12, 1525–1536 (2003).
Ivessa, A. S., Zhou, J.-Q. & Zakian, V. A. The Saccharomyces Pif1p DNA helicase and the highly related Rrm3p have opposite effects on replication fork progression in ribosomal DNA. Cell 100, 479–489 (2000).
Azvolinsky, A., Giresi, P. G., Lieb, J. D. & Zakian, V. A. Highly transcribed RNA polymerase II genes are impediments to replication fork progression in Saccharomyces cerevisiae. Mol. Cell 34, 722–734. https://doi.org/10.1016/j.molcel.2009.05.022 (2009).
Deshpande, A. M. & Newlon, C. S. DNA replication fork pause sites dependent on transcription. Science 272, 1030–1033 (1996).
Greenfeder, S. A. & Newlon, C. S. Replication forks pause at yeast centromeres. Mol. Cell Biol. 12, 4056–4066 (1992).
Lemoine, F. J., Degtyareva, N. P., Lobachev, K. & Petes, T. D. Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120, 587–598 (2005).
Makovets, S., Herskowitz, I. & Blackburn, E. H. Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. Mol. Cell Biol. 24, 4019–4031 (2004).
Szilard, R. K. et al. Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nat. Struct. Mol. Biol. 17, 299–305. https://doi.org/10.1038/nsmb.1754 (2010).
Hizume, K. & Araki, H. Replication fork pausing at protein barriers on chromosomes. FEBS Lett. https://doi.org/10.1002/1873-3468.13481 (2019).
Di Rienzi, S. C., Collingwood, D., Raghuraman, M. K. & Brewer, B. J. Fragile genomic sites are associated with origins of replication. Genome Biol. Evol. 2009, 350–363 (2009).
Hu, Z. et al. Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging. Genes Dev. 28, 396–408. https://doi.org/10.1101/gad.233221.113 (2014).
Lindstrom, D. L., Leverich, C. K., Henderson, K. A. & Gottschling, D. E. Replicative age induces mitotic recombination in the ribosomal RNA gene cluster of Saccharomyces cerevisiae. PLoS Genet. 7, e1002015. https://doi.org/10.1371/journal.pgen.1002015 (2011).
Raveendranathan, M. et al. Genome-wide replication profiles of S-phase checkpoint mutants reveal fragile sites in yeast. EMBO J. 25, 3627–3639. https://doi.org/10.1038/sj.emboj.7601251 (2006).
Song, W., Dominska, M., Greenwell, P. W. & Petes, T. D. Genome-wide high-resolution mapping of chromosome fragile sites in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 111, E2210-2218. https://doi.org/10.1073/pnas.1406847111 (2014).
Muellner, J. & Schmidt, K. H. Helicase activities of Rad5 and Rrm3 genetically interact in the prevention of recombinogenic DNA lesions in Saccharomyces cerevisiae. DNA Repair. 126, 103488. https://doi.org/10.1016/j.dnarep.2023.103488 (2023).
Prado, F. & Aguilera, A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. Embo J. 24, 1267–1276 (2005).
Osmundson, J. S., Kumar, J., Yeung, R. & Smith, D. J. Pif1-family helicases cooperatively suppress widespread replication-fork arrest at tRNA genes. Nat. Struct. Mol. Biol. 24, 162–170. https://doi.org/10.1038/nsmb.3342 (2017).
Fachinetti, D. et al. Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol. Cell 39, 595–605. https://doi.org/10.1016/j.molcel.2010.07.024 (2010).
Choudhary, R. et al. Sen1 and Rrm3 ensure permissive topological conditions for replication termination. Cell Rep. 42, 112747. https://doi.org/10.1016/j.celrep.2023.112747 (2023).
Syed, S., Desler, C., Rasmussen, L. J. & Schmidt, K. H. A novel Rrm3 function in restricting DNA replication via an Orc5-binding domain is genetically separable from Rrm3 function as an ATPase/helicase in facilitating fork progression. PLoS Genet. 12, e1006451. https://doi.org/10.1371/journal.pgen.1006451 (2016).
McDonald, K. R. et al. Pfh1 is an accessory replicative helicase that interacts with the replisome to facilitate fork progression and preserve genome integrity. PLoS Genet. 12, e1006238. https://doi.org/10.1371/journal.pgen.1006238 (2016).
McDonald, K. R., Sabouri, N., Webb, C. J. & Zakian, V. A. The Pif1 family helicase Pfh1 facilitates telomere replication and has an RPA-dependent role during telomere lengthening. DNA Repair. 24, 80–86. https://doi.org/10.1016/j.dnarep.2014.09.008 (2014).
Sabouri, N., Capra, J. A. & Zakian, V. A. The essential Schizosaccharomyces pombe Pfh1 DNA helicase promotes fork movement past G-quadruplex motifs to prevent DNA damage. BMC Biol. 12, 101. https://doi.org/10.1186/s12915-014-0101-5 (2014).
Sabouri, N., McDonald, K. R., Webb, C. J., Cristea, I. M. & Zakian, V. A. DNA replication through hard-to-replicate sites, including both highly transcribed RNA Pol II and Pol III genes, requires the S. pombe Pfh1 helicase. Genes Dev. 26, 581–593. https://doi.org/10.1101/gad.184697.111 (2012).
Zhou, J. Q. et al. Schizosaccharomyces pombe pfh1+ encodes an essential 5’ to 3’ DNA helicase that is a member of the PIF1 subfamily of DNA helicases. Mol Biol. Cell 13, 2180–2191. https://doi.org/10.1091/mbc.02-02-0021 (2002).
Tran, P. L. T. et al. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nat. Commun. 8, 15025. https://doi.org/10.1038/Ncomms15025 (2017).
Mateyak, M. K. & Zakian, V. A. Human PIF helicase is cell cycle regulated and associates with telomerase. Cell Cycle 5, 2796–2804 (2006).
Zhang, D. H., Zhou, B., Huang, Y., Xu, L. X. & Zhou, J. Q. The human Pif1 helicase, a potential Escherichia coli RecD homologue, inhibits telomerase activity. Nucleic Acids Res. 34, 1393–1404. https://doi.org/10.1093/nar/gkl029 (2006).
Li, C. W. & Chen, B. S. Investigating core genetic-and-epigenetic cell cycle networks for stemness and carcinogenic mechanisms, and cancer drug design using big database mining and genome-wide next-generation sequencing data. Cell Cycle 15, 2593–2607. https://doi.org/10.1080/15384101.2016.1198862 (2016).
Gagou, M. E. et al. Suppression of apoptosis by PIF1 helicase in human tumor cells. Cancer Res. 71, 4998–5008. https://doi.org/10.1158/0008-5472.CAN-10-4404 (2011).
Dehghani-Tafti, S., Levdikov, V., Antson, A. A., Bax, B. & Sanders, C. M. Structural and functional analysis of the nucleotide and DNA binding activities of the human PIF1 helicase. Nucleic Acids Res. 47, 3208–3222. https://doi.org/10.1093/nar/gkz028 (2019).
Wang, K. et al. Petite Integration Factor 1 knockdown enhances gemcitabine sensitivity in pancreatic cancer cells via increasing DNA damage. J. Appl. Toxicol. https://doi.org/10.1002/jat.4494 (2023).
Schmidt, K. H. & Kolodner, R. D. Requirement of Rrm3 helicase for repair of spontaneous DNA lesions in cells lacking Srs2 or Sgs1 helicase. Mol. Cell. Biol. 24, 3213–3226 (2004).
Torres, J. Z., Schnakenberg, S. L. & Zakian, V. A. The Saccharomyces cerevisiae Rrm3p DNA helicase promotes genome integrity by preventing replication fork stalling: Viability of rrm3 cells requires the intra S phase checkpoint and fork restart activities. Mol. Cell. Biol. 24, 3198–3212 (2004).
Stern, D. F., Zheng, P., Beidler, D. R. & Zerillo, C. Spk1, a new kinase from Saccharomyces cerevisiae, phosphorylates proteins on serine, threonine, and tyrosine. Mol. Cell Biol. 11, 987–1001. https://doi.org/10.1128/mcb.11.2.987-1001.1991 (1991).
Zheng, P. et al. SPK1 is an essential S-phase-specific gene of Saccharomyces cerevisiae that encodes a nuclear serine/threonine/tyrosine kinase. Mol. Cell Biol. 13, 5829–5842. https://doi.org/10.1128/mcb.13.9.5829-5842.1993 (1993).
Rossi, S. E., Ajazi, A., Carotenuto, W., Foiani, M. & Giannattasio, M. Rad53-mediated regulation of Rrm3 and Pif1 DNA helicases contributes to prevention of aberrant fork transitions under replication stress. Cell Rep. 13, 80–92. https://doi.org/10.1016/j.celrep.2015.08.073 (2015).
Munoz-Galvan, S. et al. A new role for Rrm3 in repair of replication-born DNA breakage by sister chromatid recombination. PLoS Genet. 13, e1006781. https://doi.org/10.1371/journal.pgen.1006781 (2017).
Bruhn, C. et al. The Rad53(CHK1/CHK2)-Spt21(NPAT) and Tel1(ATM) axes couple glucose tolerance to histone dosage and subtelomeric silencing. Nat. Commun. 11, 4154. https://doi.org/10.1038/s41467-020-17961-4 (2020).
Gartenberg, M. R. & Smith, J. S. The nuts and bolts of transcriptionally silent chromatin in Saccharomyces cerevisiae. Genetics 203, 1563–1599. https://doi.org/10.1534/genetics.112.145243 (2016).
Moazed, D., Kistler, A., Axelrod, A., Rine, J. & Johnson, A. D. Silent information regulator protein complexes in Saccharomyces cerevisiae: A SIR2/SIR4 complex and evidence for a regulatory domain in SIR4 that inhibits its interaction with SIR3. Proc. Natl. Acad. Sci. USA 94, 2186–2191 (1997).
Straight, A. F. et al. Net1, a Sir2-associated nucleolar protein required for rDNA silencing and nucleolar integrity. Cell 97, 245–256 (1999).
Tamburini, B. A. & Tyler, J. K. Localized histone acetylation and deacetylation triggered by the homologous recombination pathway of double-strand DNA repair. Mol. Cell Biol. 25, 4903–4913. https://doi.org/10.1128/MCB.25.12.4903-4913.2005 (2005).
Martin, S. G., Laroche, T., Suka, N., Grunstein, M. & Gasser, S. M. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell 97, 621–633 (1999).
Mills, K. D., Sinclair, D. A. & Guarente, L. MEC1-dependent redistribution of the Sir3 silencing protein from telomeres to DNA double-strand breaks. Cell 97, 609–620 (1999).
Hoggard, T. A. et al. Yeast heterochromatin regulators Sir2 and Sir3 act directly at euchromatic DNA replication origins. PLoS Genet. 14, e1007418. https://doi.org/10.1371/journal.pgen.1007418 (2018).
Kaeberlein, M., Kirkland, K. T., Fields, S. & Kennedy, B. K. Sir2-independent life span extension by calorie restriction in yeast. PLoS Biol. 2, E296. https://doi.org/10.1371/journal.pbio.0020296 (2004).
Kaeberlein, M., McVey, M. & Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 13, 2570–2580 (1999).
Lin, S. J. et al. Calorie restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418, 344–348. https://doi.org/10.1038/nature00829 (2002).
Salvi, J. S. et al. Enforcement of a lifespan-sustaining distribution of Sir2 between telomeres, mating-type loci, and rDNA repeats by Rif1. Aging Cell 12, 67–75. https://doi.org/10.1111/acel.12020 (2013).
Kobayashi, T. & Ganley, A. R. D. Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science 309, 1581–1584. https://doi.org/10.1126/science.1116102 (2005).
Leduc, F. et al. Genome-wide mapping of DNA strand breaks. PLoS ONE 6, e17353. https://doi.org/10.1371/journal.pone.0017353 (2011).
Brewer, B. J. & Fangman, W. L. A replication fork barrier at the 3’ end of yeast ribosomal RNA genes. Cell 55, 637–643 (1988).
Ivessa, A. S. Analysis of DNA structures from eukaryotic cells by two-dimensional native-native DNA agarose gel electrophoresis. Methods Mol. Biol. 1054, 63–81. https://doi.org/10.1007/978-1-62703-565-1_4 (2013).
Brewer, B. J., Lockshon, D. & Fangman, W. L. The arrest of replication forks in the rDNA of yeast occurs independently of transcription. Cell 71, 267–276 (1992).
Cabral, M., Cheng, X., Singh, S. & Ivessa, A. S. Absence of non-histone protein complexes at natural chromosomal pause sites results in reduced replication pausing in aging yeast cells. Cell Rep. 17, 1747–1754 (2016).
Madeo, F., Frohlich, E. & Frohlich, K. U. A yeast mutant showing diagnostic markers of early and late apoptosis. J. Cell Biol. 139, 729–734 (1997).
Carmona-Gutierrez, D. et al. Guidelines and recommendations on yeast cell death nomenclature. Microb. Cell 5, 4–31. https://doi.org/10.15698/mic2018.01.607 (2018).
Cao, G. et al. Effects of X-ray and carbon ion beam irradiation on membrane permeability and integrity in Saccharomyces cerevisiae cells. J. Radiat. Res. 56, 294–304. https://doi.org/10.1093/jrr/rru114 (2015).
Schulz, V. P. & Zakian, V. A. The Saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell 76, 145–155 (1994).
Pohl, T. J. & Zakian, V. A. Pif1 family DNA helicases: A helpmate to RNase H?. DNA Repair 84, 102633. https://doi.org/10.1016/j.dnarep.2019.06.004 (2019).
Bonetti, D., Colombo, C. V., Clerici, M. & Longhese, M. P. Processing of DNA ends in the maintenance of genome stability. Front. Genet. 9, 390. https://doi.org/10.3389/fgene.2018.00390 (2018).
Wu, X. Replication stress response links RAD52 to protecting common fragile sites. Cancers 11, 1467. https://doi.org/10.3390/cancers11101467 (2019).
Bhargava, R., Onyango, D. O. & Stark, J. M. Regulation of single-strand annealing and its role in genome maintenance. Trends Genet. 32, 566–575. https://doi.org/10.1016/j.tig.2016.06.007 (2016).
Walsh, R. B., Kawasaki, G. & Fraenkel, D. G. Cloning of genes that complement yeast hexokinase and glucokinase mutants. J. Bacteriol. 154, 1002–1004. https://doi.org/10.1128/jb.154.2.1002-1004.1983 (1983).
Lin, S. J., Defossez, P. A. & Guarente, L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128 (2000).
Yokoyama, M., Sasaki, M. & Kobayashi, T. Spt4 promotes cellular senescence by activating non-coding RNA transcription in ribosomal RNA gene clusters. Cell Rep. 42, 111944. https://doi.org/10.1016/j.celrep.2022.111944 (2023).
Puddu, F. et al. Chromatin determinants impart camptothecin sensitivity. EMBO Rep. 18, 1000–1012. https://doi.org/10.15252/embr.201643560 (2017).
Strich, R. Programmed cell death initiation and execution in budding yeast. Genetics 200, 1003–1014. https://doi.org/10.1534/genetics.115.179150 (2015).
Madeo, F. et al. Oxygen stress: A regulator of apoptosis in yeast. J. Cell Biol. 145, 757–767 (1999).
Driever, S. M., Fryer, M. J., Mullineaux, P. M. & Baker, N. R. Imaging of reactive oxygen species in vivo. Methods Mol. Biol. 479, 109–116. https://doi.org/10.1007/978-1-59745-289-2_7 (2009).
Jiang, J. et al. A mitochondria-targeted triphenylphosphonium-conjugated nitroxide functions as a radioprotector/mitigator. Radiat. Res. 172, 706–717. https://doi.org/10.1667/RR1729.1 (2009).
Sandell, L. L. & Zakian, V. A. Loss of a yeast telomere: Arrest, recovery and chromosome loss. Cell 75, 729–739 (1993).
Hoffman, E. A., McCulley, A., Haarer, B., Arnak, R. & Feng, W. Break-seq reveals hydroxyurea-induced chromosome fragility as a result of unscheduled conflict between DNA replication and transcription. Genome Res. 25, 402–412. https://doi.org/10.1101/gr.180497.114 (2015).
Tubbs, A. et al. Dual roles of Poly(dA:dT) tracts in replication initiation and fork collapse. Cell 174, 1127–1142. https://doi.org/10.1016/j.cell.2018.07.011 (2018).
Saayman, X., Graham, E., Nathan, W. J., Nussenzweig, A. & Esashi, F. Centromeres as universal hotspots of DNA breakage, driving RAD51-mediated recombination during quiescence. Mol. Cell 83, 523–538. https://doi.org/10.1016/j.molcel.2023.01.004 (2023).
Pellicioli, A. et al. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J. 18, 6561–6572 (1999).
Waterman, D. P., Haber, J. E. & Smolka, M. B. Checkpoint responses to DNA double-strand breaks. Annu. Rev. Biochem. 89, 103–133. https://doi.org/10.1146/annurev-biochem-011520-104722 (2020).
Sun, Y., Yu, R., Guo, H. B., Qin, H. & Dang, W. A quantitative yeast aging proteomics analysis reveals novel aging regulators. Geroscience 43, 2573–2593. https://doi.org/10.1007/s11357-021-00412-3 (2021).
Kaeberlein, M. et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310, 1193–1196 (2005).
Steffen, K. K. et al. Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell 133, 292–302. https://doi.org/10.1016/j.cell.2008.02.037 (2008).
Schleker, T., Shimada, K., Sack, R., Pike, B. L. & Gasser, S. M. Cell cycle-dependent phosphorylation of Rad53 kinase by Cdc5 and Cdc28 modulates checkpoint adaptation. Cell Cycle 9, 350–363. https://doi.org/10.4161/cc.9.2.10448 (2010).
Ferrari, E. et al. PP2A controls genome integrity by integrating nutrient-sensing and metabolic pathways with the DNA damage response. Mol. Cell 67, 266–281. https://doi.org/10.1016/j.molcel.2017.05.027 (2017).
Shimada, K., Pasero, P. & Gasser, S. M. ORC and the intra-S-phase checkpoint: A threshold regulates Rad53p activation in S phase. Genes Dev. 16, 3236–3252 (2002).
Tercero, J. A., Longhese, M. P. & Diffley, J. F. A central role for DNA replication forks in checkpoint activation and response. Mol. Cell. 11, 1323–1336 (2003).
Forey, R. et al. Mec1 is activated at the onset of normal S phase by low-dNTP pools impeding DNA Replication. Mol. Cell 78, 396–410. https://doi.org/10.1016/j.molcel.2020.02.021 (2020).
Marcand, S., Pardo, B., Gratias, A., Cahun, S. & Callebaut, I. Multiple pathways inhibit NHEJ at telomeres. Genes Dev. 22, 1153–1158. https://doi.org/10.1101/gad.455108 (2008).
Astrom, S. U., Okamura, S. M. & Rine, J. Yeast cell-type regulation of DNA repair. Nature 397, 310. https://doi.org/10.1038/16833 (1999).
Bordelet, H. et al. Sir3 heterochromatin protein promotes non-homologous end joining by direct inhibition of Sae2. EMBO J. 41, e108813. https://doi.org/10.15252/embj.2021108813 (2022).
Fahrenkrog, B. Histone modifications as regulators of life and death in Saccharomyces cerevisiae. Microb. Cell 3, 1–13. https://doi.org/10.15698/mic2016.01.472 (2015).
Cohen-Fix, O. & Koshland, D. The anaphase inhibitor of Saccharomyces cerevisiae Pds1p is a target of the DNA damage checkpoint pathway. Proc. Natl. Acad. Sci. USA 94, 14361–14366 (1997).
Yamamoto, A., Guacci, V. & Koshland, D. Pds1p, an inhibitor of anaphase in budding yeast, plays a critical role in the APC and checkpoint pathway(s). J. Cell Biol. 133, 99–110 (1996).
Ciosk, R. et al. An ESP1/PDS1 complex regulates loss of sister chromatid cohesion at the metaphase to anaphase transition in yeast. Cell 93, 1067–1076. https://doi.org/10.1016/s0092-8674(00)81211-8 (1998).
Shirayama, M., Toth, A., Galova, M. & Nasmyth, K. APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature 402, 203–207. https://doi.org/10.1038/46080 (1999).
Agarwal, R., Tang, Z., Yu, H. & Cohen-Fix, O. Two distinct pathways for inhibiting pds1 ubiquitination in response to DNA damage. J. Biol. Chem. 278, 45027–45033. https://doi.org/10.1074/jbc.M306783200 (2003).
Dotiwala, F., Harrison, J. C., Jain, S., Sugawara, N. & Haber, J. E. Mad2 prolongs DNA damage checkpoint arrest caused by a double-strand break via a centromere-dependent mechanism. Curr. Biol. 20, 328–332. https://doi.org/10.1016/j.cub.2009.12.033 (2010).
Gardner, R., Putnam, C. W. & Weinert, T. RAD53, DUN1 and PDS1 define two parallel G2/M checkpoint pathways in budding yeast. EMBO J. 18, 3173–3185 (1999).
Luke, B. et al. The cullin Rtt101p promotes replication fork progression through damaged DNA and natural pause sites. Curr. Biol. 16, 786–792. https://doi.org/10.1016/j.cub.2006.02.071 (2006).
Bairwa, N. K., Mohanty, B. K., Stamenova, R., Curcio, M. J. & Bastia, D. The intra-S phase checkpoint protein Tof1 collaborates with the helicase Rrm3 and the F-box protein Dia2 to maintain genome stability in Saccharomyces cerevisiae. J. Biol. Chem. 286, 2445–2454. https://doi.org/10.1074/jbc.M110.189456 (2011).
Mohanty, B. K., Bairwa, N. K. & Bastia, D. The Tof1p-Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 103, 897–902. https://doi.org/10.1073/pnas.0506540103 (2006).
Vermeij, W. P. et al. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 537, 427–431. https://doi.org/10.1038/nature19329 (2016).
Longtine, M. et al. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961 (1998).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. https://doi.org/10.1038/nmeth.2089 (2012).
Acknowledgements
We thank the two anonymous reviewers for valuable suggestions and Weiwei Dang and David Stern for discussions. This work was supported by National Institutes of Health grant R21AG047423 (to A.S.I.) and National Institutes of Health SIG grant 1S0OD018103-01 (Rutgers NJMS Flow Cytometry and Immunology Core Laboratory) (to S.S.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
S.S. carried out the FACS and cell sorting experiments; A.S.I. designed the experiments, carried out the rest of the experiments, analyzed the data, and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ivessa, A.S., Singh, S. The increase in cell death rates in caloric restricted cells of the yeast helicase mutant rrm3 is Sir complex dependent. Sci Rep 13, 17832 (2023). https://doi.org/10.1038/s41598-023-45125-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-45125-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
