
Abstract
Composition of pulmonary microbiome of patients with severe pneumonia is poorly known. The aim of this work was to analyse the lung microbiome of patients admitted to the intensive care unit (ICU) with severe community acquired pneumonia (CAP) between 2019 and 2021 in comparison with a control group of 6 patients undergoing digestive surgery. As a second objective, the diagnostic capabilities of metagenomics was also studied in a small group of selected patients. The lung microbiome of patients with viral (5 with Influenza A and 8 with SARS-CoV-2) pneumonia at admission showed a similar diversity as the control group (p = 0.140 and p = 0.213 respectively). Contrarily, the group of 12 patients with pneumococcal pneumonia showed a significant lower Simpson´s index (p = 0.002). In the control group (n = 6) Proteobacteria (36.6%), Firmicutes (24.2%) and Actinobacteria (23.0%) were the predominant phyla. In SARS-CoV-2 patients (n = 8), there was a predominance of Proteobacteria (mean 41.6%) (Moraxella and Pelomonas at the genus level), Actinobacteria (24.6%) (Microbacterium) and Firmicutes (22.8%) mainly Streptococcus, Staphylococcus and Veillonella. In patients with Influenza A pneumonia (n = 5) there was a predominance of Firmicutes (35.1%) mainly Streptococcus followed by Proteobacteria (29.2%) (Moraxella, Acinetobacter and Pelomonas). In the group of pneumococcal pneumonia (n = 12) two phyla predominated: Firmicutes (53.1%) (Streptococcus) and Proteobacteria (36.5%) (Haemophilus). In the 7 patients with non-pneumococcal bacterial pneumonia Haemophilus influenzae (n = 2), Legionella pneumophila (n = 2), Klebsiella pneumoniae, Streptococcus pyogenes and Leptospira were detected by metagenomics, confirming the diagnosis done using conventional microbiological techniques. The diversity of the respiratory microbiome in patients with severe viral pneumonia at ICU admission was similar to that of the control group. Contrarily, patients with pneumococcal pneumonia showed a lower grade of diversity. At initial stages of SARS-CoV-2 infection, no important alterations in the pulmonary microbiome were observed. The analysis of bacterial microbiome showed promising results as a diagnostic tool.
Similar content being viewed by others

Introduction
One of the most frequent community acquired infections requiring hospitalization is pneumonia, which in severe cases requires admission into the intensive care units (ICU) and mechanical ventilation1. Despite early antimicrobial treatment and support measures, mortality due to severe pneumonia is still very high and new approaches in respiratory therapy are being sought to try to improve their outcomes2. Among the new research in pneumonia, the study of the role of the lung microbiome has been pointed out as a new key therapeutic target for the prevention and treatment of critical respiratory illness3. Although the lung microbiome is composed of bacterial, viral and fungal populations, most studies are based on the analysis of the bacteriome, in which all the bacterial 16S rRNA genes present in the sample are amplified and sequenced using next-generation sequencing (NGS). This technology has also been used with diagnostic purposes, as a high number of pneumonias remain without etiologic diagnosis despite efforts in detecting the responsible pathogen1,4,5
The lung microbiota in healthy individuals is conceptualized not as a static entity but as a dynamic balance between microbial immigration, clearance and replication under the strict alveolar conditions6,7. It has been demonstrated that bacterial communities of the healthy lung are quite similar to those found in the mouth but at lower concentrations, lower diversity and different composition8. In fact, the lung microbiome of each person seems to be unique9. In this model, pneumonia causative agents will alter the previous homeostasis producing decrease in biodiversity, increase in the microbial biomass and inflammation in the host7. However, and mainly due to the lack of samples from the lower-respiratory tract of patients before developing pneumonia, it is quite difficult to determine whether differences in the microbiota are a cause or a consequence of pulmonary infections10.
Many studies on respiratory microbiome have been done, most of them in patients with chronic pulmonary diseases as asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis or receiving mechanical ventilation11,12. However, the number of studies focused on the lung microbiome in community acquired pneumonia (CAP) are scant, probably due to the great number of challenges that the study of the lung microbiome poses. Among them, the invasiveness of obtaining samples via bronchoalveolar lavages (BAL), the frequent use of antibiotics before sampling which affects the bacterial composition, or the role of contamination of reagents used in amplification and sequencing techniques are the most important13.
In this work we analyse the lung microbiome of 31 patients admitted to the ICU due to severe bacterial and/or viral pneumonia and compare it with the lung microbiome of 6 control patients submitted to digestive surgery. All samples were taken by protected mini-BAL in the first 24 h since admission, and the use of antibiotics was limited to 48 h to avoid antibiotic treatment interferences in the microbiome composition. As no samples previous to the development of pneumonia were available, only description of the microbiome found at admission was analysed. As a secondary aim, the utility of metagenomic NGS (mNGS) as a microbiological diagnostic method for bacterial pneumonia was studied in the non-protected BAL of a subset of 7 selected patients with non-pneumococcal bacterial pneumonia.
Methods
Subjects
Between January 2019 and May 2021, 31 adult patients with severe CAP admitted to the ICU were prospectively enrolled in the study (Table 1). The including criteria were: (1) Adult patients admitted with clinical and radiological criteria of severe CAP according to IDSA/ATS guidelines14, (2) BAL samples collected in the first 24 h since admission to the ICU, (3) No antibiotic treatment for more than 48 h. Of the 31 patients of whom respiratory microbiome was investigated, 7 have not received antibiotics before obtaining the mini-BAL sample (6 of them with SARS-CoV-2 pneumonia), 9 had received antibiotics during the previous 24 h, and 15 had received antibiotics in the previous 48 h (Table 1). As control group, 6 postsurgical intubated patients due to gastrointestinal disease were also analysed. Pulmonary samples were collected using a protected blind mini-BAL (Combicath™ Plastimed, France) and immediately taken to the microbiology department.
Apart from the study of the lung microbiome, the non-protected BAL of 7 patients diagnosed with non-pneumococcal bacterial pneumonia were arbitrarily selected, based on the pathogen detected by microbiological culture or other conventional methods, to study the possible utility of mNGS as a diagnostic method for bacterial pneumonia.
Microbiological procedures
Microbiological analysis for the etiological diagnosis of pneumonia included blood-culture (BD BACTEC™ blood culture systems), urine antigen test for Streptococcus pneumoniae and Legionella pneumophila detection (Sofia FIA® Quidel Corporation, San Diego, CA, USA), and qRT-PCR targeting viral respiratory pathogens (Allplex™ Respiratory Panels 1, 2 and 3, Seegene, South Korea). For other less frequent bacterial pathogens causing pneumonia (Mycoplasma pneumoniae and Chlamydophila pneumoniae) a PCR of a pharyngeal swab was performed according to manufactures instructions (CerTest Biotec SL, Spain).
In patients with diagnosis of SARS-CoV-2 pneumonia, diagnosis of SARS-CoV-2 had been commonly done some days before admission using commercial RT-PCR (Allplex SARS-CoV-2 assay, Seegene, South Korea) on nasopharyngeal swabs. Alpha (lineage B.1.1.7) was the predominant SARS-CoV-2 variant present when samples of these patients were collected.
Microbiome analysis
Nucleic acids from 1 mL of the mini-BAL were extracted using the NUCLISENS easyMAG platform (bioMérieux) according to manufacturer´s recommendations and eluted in a final volume of 100 µl of elution buffer.
Microbiome was determined using the Ion 16S Metagenomics Kit (Thermo Fisher Scientific) that amplifies 7 hypervariable regions (V2, 3, 4, 6–7, 8, 9) of the 16S rRNA gene in two amplification reactions (primer sets V2-4-8 and V3-6,7–9, respectively) with the following PCR conditions: 95 °C for 10 min, 30 cycles at 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 20 s, followed by 72 °C for 7 min. After amplification, samples were run in an agarose gel and those not showing amplification were subjected to a nested PCR using the same primers and PCR conditions.
The nested PCR was performed on 6 samples that had amplified in the initial PCR, and the sequencing results of the first (standard) and nested PCRs were compared to verify if there were differences in the composition of the microbiome between both PCRs as nested PCR was performed on BAL samples with low bacterial load.
Resulting PCR products of each sample were mixed and cleaned up using the Sera-Mag Select magnetic beads (Cytiva, Sheffield, UK). Purified amplicons were quantified using the Qubit Fluorometer kit (Invitrogen, Carlsbad, CA, USA). Libraries were prepared using the Ion Plus fragment Library Kit. Briefly, amplicons were end repaired and purified before adapters and barcodes were ligated and nick repaired using DNA Ligase and nick repair polymerases. Libraries were cleaned using the Sera-Mag Select Reagent and quantified by qRT-PCR. Finally, samples were combined in equimolar concentrations and sequenced with the commercially available ION PGM™ Sequencing 400 kit on an ION PGM™ System (Thermo Fisher Scientific, Waltham, MA, USA), using an Ion 318™ Chip v2, with a maximum of 12 samples per chip.
Data was analysed with the Ion Reporter software 5.18.2 using the consensus data of all variable regions. Alpha diversity results were produced using the QIIME’s open-source bioinformatics pipeline. Alpha diversity was calculated using the Simpson and Shannon diversity indexes.
Utility of metagenomic Next-Generation Sequencing (mNGS) for etiological diagnosis of patients with CAP
The same mNGS technique used in the study of the microbiome was applied to the BAL of 7 patients with radiological and clinical criteria of severe CAP14. These patients were arbitrarily selected according to the etiologic agent found in any of their samples using conventional microbiological diagnostic techniques. In these patients, differing from the protected mini-BAL used in the microbiome analysis, the rest of the BAL used for routine microbiological diagnosis was used for mNGS.
Patients with pneumococcal pneumonia were intentionally excluded from this part of the study as they had been already included in the study of the microbiome of patients with pneumococcal NAC.
Statistics
Continuous variables were compared using the unpaired t test using the Graph pad Prism 5 software. A p value of < 0.05 was considered as statistically significant.
Ethics
The study was approved by the regional (Gipuzkoa) Ethics Committee, reference MOZ-NBI-2017–01 and all patients or their legal representatives gave written informed consent to be included in the study. All experiments were performed in accordance with relevant guidelines and regulations.
Results
Validation of the nested-PCR
The pulmonary microbiome of six different patients was studied both on direct DNA extraction (standard PCR) and after performing a nested PCR of the first amplified product using the same sets of primers and PCR and sequencing conditions to check possible differences in the results. The total number of reads was bigger in five of the six samples in the standard PCR compared to the nested PCR (Table 2). Almost no differences were observed in the distribution and richness of the most frequent genus in each sample (Supplementary Table S1).
Microbiome analysis
Lung microbiome in the control group
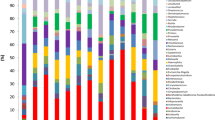
The lung microbiome was studied in the BAL samples of 6 patients undergoing mechanical ventilation before digestive surgery (Table 3). The mini-BAL samples were obtained when the patients were intubated, just before antibiotic prophylaxis was administered and surgery began. On mean, Proteobacteria (36.6%) was the dominant phylum followed by Firmicutes (24.2%) and Actinobacteria (23%) (Table 4). The number of different phyla in each sample varied between 6 and 13.
At the genus level and considering those genera that had more than 1% of the assigned reads in each sample, only the genus Microbacterium was detected in the mini-BAL of all the controls, which represented 3.2% of the mean of all the reads (Supplementary Table S2). The genus Propionibacterium, Corynebacterium, Acinetobacter, Streptococcus and Staphylococcus were found in 5 of the 6 controls (5.3%, 4.5%, 4.4%, 4.3% and 2.8% of the mean of all reads, respectively). The genus Enterococcus and Enhydrobacter were found in 4 of the 6 controls (5.9% and 3.4% of the mean of all reads, respectively). All the other genera with more than 1% of reads were found in the microbiome of 3 or less of the 6 controls.
The Shannon index for diversity in the microbiome of controls ranged between 3.423 and 4.847 (mean 4.214 ± 0.581) and the Simpson index ranged between 0.840 and 0.947 (mean 0.902 ± 0.038).
Lung microbiome of patients with viral pneumonia (non-SARS-CoV-2)
In the lung microbiome of the 5 patients admitted to the ICU with severe pneumonia caused by the 2009 pandemic influenza A virus subtype H1N1 there was a predominance of Firmicutes (mean 35.1%) mainly Streptococcus, Veillonella and Staphylococcus at the genus level followed by Proteobacteria (mean 29.2%) mainly Moraxella, Acinetobacter and Pelomonas at the genus level (Table 5 and Supplementary Table S3). The other two highly abundant phyla were Bacteroidetes (mainly Prevotella and Porphyromonas) and Actinobacteria (mainly Microbacterium and Propionibacterium) with 17.6% and 15.2% mean abundance, respectively.
The Shannon diversity index of the lung microbiome from patients with Influenza virus pneumonia ranged between 2.097 and 4.335 (mean 3.333 ± 0.902). The Simpson diversity index ranged between 0.651 and 0.929 (mean 0.825 ± 0.110) (Fig. 1).

Simpson index showing the microbiome diversity of mini-BAL samples of patients with pneumonia caused by Influenza A virus, SARS-COV-2, Rhinovirus and RSV, and pneumococcal and of control patients without respiratory symptoms.
There were also 4 patients in which Rhinovirus (3 patients) or RSV were the only respiratory pathogen detected in any sample. Proteobacteria (mean 62.1%), Firmicutes (22.4%) and Actinobacteria (9.5%) were the most abundant phyla (Supplementary Table S4). At the genus level, Streptococcus (17.2%), Haemophilus (27.2%) and Microbacterium (4.4%) were the most abundant and were found in 4, 3 and 3 of these patients, respectively. The mean Shannon and Simpson diversity indexes of the microbiome of these 4 patients was 3.114 ± 1.171 and 0.752 ± 0.175.
Pulmonary microbiome of patients with SARS-CoV-2 pneumonia
In total, we studied 8 patients with SARS-CoV-2 severe pneumonia admitted to the ICU between March 2020 and April 2021, this is, during the first year of the pandemic. The diagnosis of SARS-CoV-2 had been commonly done some days before admission (on mean 3.75 days, range 0–10 days) using a commercial RT-qPCR on nasopharyngeal swabs.
In terms of mean organisms´ abundance in the microbiome of SARS-CoV-2 patients, there was a predominance of Proteobacteria (mean of all samples 41.6%) with Acinetobacter, Sphingomonas and Pelomonas as predominant genus followed by Actinobacteria (24.6%) mostly Microbacterium and Propionibacterium and by Firmicutes (22.8%) mainly Streptococcus and Staphylococcus (Table 6 and Supplementary Table S5).
The diversity of the lung microbiome of patients with SARS-CoV-2 pneumonia at admission was very similar (not statistically significant difference) to the diversity of the microbiome of the patients of the control group: the mean Simpson index was 0.880 ± 0.023 for SARS-CoV-2 patients and 0.902 ± 0.038 for controls (p = 0.213) and the Shannon index was 3.881 ± 0.301 for SARS-CoV-2 patients and 4.213 ± 0.580 for controls (p = 0.187).
Pulmonary microbiome of patients with pneumococcal pneumonia
There were 12 patients with pneumococcal pneumonia included in the study. Five had a positive blood culture (four of them also had a positive urine antigen test). Another six had a positive urine antigen test and the last patient was diagnosed by the growth of the pneumococcus on a tracheal aspirate culture.
Four had a mixed pneumonia caused by Influenza virus AH1 and S. pneumoniae, and one each with Human Metapneumovirus (hMPV) and Respiratory Syncytial Virus (RSV). In the other 6 patients, S. pneumoniae was the unique pathogen detected. Because the diversity indexes of the lung microbiome of patients with simple S. pneumoniae pneumonia or coinfected with a virus were similar, patients with a pneumococcal pneumonia were studied as a unique group.
The composition of the microbiome of patients with pneumococcal pneumonia showed that Firmicutes (53.1%) and Proteobacteria (36.5%) comprised the majority of all phyla (Table 7). They were mainly represented by the genus Streptococcus (43.7%) and Haemophilus (18.4%) (Supplementary Table S6). Haemophilus was the predominant genus in two of the four patients that had a mixed Influenza AH1 and S. pneumoniae pneumonia (69.4% and 66.9%) and in one of the patients with pure pneumococcal pneumonia (83.4%).
The diversity of the lung microbiome of patients with pneumococcal pneumonia at admission was lower than in the control group and in patients with viral pneumonia. The mean Simpson index was 0.369 (range 0.022–0.920) compared to 0.902 (range 0.840–0.947) of controls (p = 0.002) or 0.798 (range 0.651–0.929) for patients with pure influenza pneumonia (p = 0.009) and 0.843 (0.532–0.909) of patients with SARS-CoV-2 pneumonia (p < 0.001).
The Shannon index was 1.417 (range 0.1–4.185) for patients with pneumococcal pneumonia, statically lower the diversity Shannon index for of the patients of the control group (4.214, p < 0.001) and for patients with influenza (3.126, p = 0.027) or SARS-CoV-2 pneumonia (3.882, p < 0.001).
Utility of mNGS for etiological diagnosis of non-pneumococcal bacterial pneumonia
The BAL of 7 patients admitted to the ICU with radiologically confirmed pneumonia in whom bacterial pneumonia had been diagnosed using conventional methods (antigen detection, culture, PCR detection) were selected according to their bacterial aetiology to study the possibilities of using the mNGS with diagnostic purposes. Unlike the rest of the patients of this work, conventional BALs were collected in these patients using a non-protected catheter.
Two of them had a positive Legionella pneumophila urinary antigen test. One of them also had a positive PCR on a tracheal aspirate in which later grew a serogroup 1 L. pneumophila. The microbiome of the two patients was almost entirely composed of the pathogenic bacteria: 92.3%, and 89.5% at the species level and 95.4%, and 99.9% at the genus level (Supplementary Table S7).
In the BAL of another patient grew a Streptococcus pyogenes in pure culture. In the same BAL an Influenza AH3 virus was detected by RT-qPCR. The microbiome of this patient was also nearly fully composed of the pathogenic bacteria: 79.1% and 99.9%, at the species and genus level, respectively.
Another two patients had a Haemophilus influenzae pneumonia. In the BAL and in a tracheal aspirate of the first case, a 66 year old man, a H. influenzae grew in pure culture. He also had a RSV detected by RT-qPCR in a pharyngeal exudate. The other, a 60 year old woman, had an H. influenzae cultured from a tracheal aspirate. In both patients, the blood-cultures, the Legionella and S. pneumoniae urine antigens and the qPCR to detect other respiratory viruses and atypical bacterial pneumonia were negative. In the first case, Haemophilus represented 90.4% of the genus detected (H. influenzae 7.8%, Haemophilus aegyptius 48.2% at the species level) and in the second, Haemophilus represented 89.2% of the genus detected (H. influenzae 7.7%, Haemophilus aegyptius 50.8% at the species level).
A 41 years old man was admitted in the ICU due to Leptospirosis, diagnosed by PCR in the urine and blood, and a positive serology. In the BAL of this patient, the genus Leptospira represented 26% of the mapped reads. At the species level, Leptospira interrogans represented 6.5% of the reads. It was the only respiratory sample in which a Leptospira was found.
Finally, a patient with two days of purulent expectoration was admitted to the ICU after a respiratory arrest in the emergency room. Influenza A virus was detected in a pharyngeal swab collected at admission but in the BAL obtained they next day Klebsiella pneumoniae grew in significant number (5,000 cfu/mL). At the phylum level, the microbiome showed a 100% of Protobacteria, 42% of Klebsiella at the genus level (19.8% K. pneumoniae and 21.2% Klebsiella variicola at the species level).
Discussion
Since the existence of a pulmonary microbiome was demonstrated, numerous works have analysed and reviewed different aspects of its composition in relation to respiratory diseases6,11. Most of these works have focused on chronic pulmonary diseases15, such as COPD16, cystic fibrosis17, asthma18, or bronchiectasis19. However, the role of the microbiome in acute pulmonary diseases such as CAP is less well understood, probably due to the difficulty of finding non-antibiotic treated intubated patients in whom a lower respiratory tract sample can be more easily obtained. It is well known the effect of antibiotics in pulmonary microbiota17,20.
First, we validated the use of a nested-PCR to detect the lung microbiome due to the low bacterial load in the lungs of some patients, especially when collected with protected catheter to avoid contamination with microbiota of the upper-respiratory tract21. Our results showed that there were nearly no differences in the distribution and richness of the most frequent genus within the same sample, showing that nested PCR can improve the results of the number of samples being processed. Due to the difficulty in selecting patients for studies of the lung microbiome, considering the need of intubation, absence of antibiotic treatment, etc., nested PCR can improve the number of patients studied without biasing the composition of the lung microbiome16,22.
The pulmonary microbiome of the control group corresponded to that of patients who had undergone elective digestive surgery. Given the impossibility of obtaining mini-BAL samples in healthy people for ethical reasons, we used as control this group of patients that, although not healthy, did not had respiratory pathology. Even so, we could not rule out that the respiratory microbiome of some of them was altered due to the microaspirations of an altered oropharyngeal microbiota8.
As in other studies23, the pulmonary microbiome of the controls showed a high alpha-diversity, with a predominance of Proteobacteria, Firmicutes and Actinobacteria at the phylum level. Twenty-one genera had between 1 and 8% of all assigned reads, demonstrating the great diversity of the lung microbiome in absence of respiratory pathology. Typical genus of the lung as Streptococcus, Prevotella and to a lesser extent Veillonella were included among these genus, as showed in other studies6,11.
In patients with influenza A virus pneumonia alpha-diversity values were similar to those of the microbiome of the control group. This was also observed in patients with other viral infections (Rhinovirus and RSV). As summarized in a recent work, no consistent changes were demonstrated in the diversity of the upper respiratory tract microbiome in virally infected patients and healthy controls in cross-sectional studies24. Also, in a mouse model of influenza virus infection, it was shown that the lower respiratory tract is very stable against viral infection, which produces only minor qualitative changes in the composition of its microbiota25. The fact that these patients were at early stages of pure Influenza AH1 virus pneumonia (no bacterial coinfection) could be the reason why the changes in their lung bacteriome was nearly inapparent.
Patients with SARS-CoV-2 infection also did not show a differentiate lung microbiome at ICU admission. Microbacterium, Streptococcus, Acinetobacter and Propionibacterium were the most frequent genera found. The changes of microbial diversity in patients with SARS-CoV-2 pneumonia is controversial: some studies found a higher diversity (Shannon index) among SARS-CoV-2 patients compared to controls although microbial composition was different26. In the upper respiratory tract of patients with SARS-CoV-2 pneumonia it has also been observed a decrease diversity in the microbiome with increasing length of stay in ICU27. Other studies, however, have found dysbiosis in the lungs of patients with SARS-CoV-2 pneumonia although in these patients the use of antibiotics was not ruled out28.
The lung microbiome of patients with pneumococcal pneumonia showed a lower diversity than the microbiome of controls and of patients with viral pneumonia, with a clear decrease in Actinobacteria and the predominance of two phyla, Firmicutes and Proteobacteria, mainly represented by the genera Streptococcus and Haemophilus, respectively. The lower diversity observed in the microbiome of patients with pneumococcal pneumonia seems to be due to an increase in the number of pathogenic bacteria, which could also compete with commensal microbiota. The mini-BAL of two patients with pneumonia of unknown origin was also studied. All microbiological test (blood cultures, Legionella and pneumococcal urine antigens, viral and atypical bacterial PCRs, etc.) were negative. Both patients had a predominance of Firmicutes (68.9% and 72.1%) mainly Streptococcus (59.6% and 51.2%, respectively) (Supplementary Table S8). However, the aetiology of S. pneumoniae as responsible of the pneumonia could not be proved.
The number of studies on the lung microbiome in bacterial CAP are scarce, mainly focused in sputum samples that can be contaminated with upper respiratory tract microbiota hindering the interpretation of the results. In an study performed on canine bacterial pneumonia it was observed, as well as in ours, that the relative diversity of bacterial communities was decreased in dogs with bacterial pneumonia29. In critically ill patients it was observed a worse clinical outcome (ventilator free days) associated with increased lung bacterial burden and lung enrichment with gut-associated bacteria30. Moreover, half of the pneumococcal pneumonias of this study were concomitant to viral infections, specially to influenza AH1 virus, which makes it even more difficult to establish pathogen-microbiome associations. The finding of three patients with pneumococcal pneumonia and a predominance of Haemophilus in the metagenomic analysis is not surprising, as mixed CAP are not infrequent, specially the combination of S. pneumoniae and H. influenzae31. Both bacteria habit the same respiratory mucosal environment although the cooperative or competitive nature of this relationship has yet to be determined32.
The utility of mNGS to determine the aetiology of bacterial CAP was not specifically the main objective of this study. However, the availability of samples from patients with severe pneumonia caused by different bacterial species prompted us to do so. In the BAL of all of these patients, the pathologic agent was detected, including two cases of L. pneumophila and one of Leptospira culture-negative respiratory samples. Some studies have found discrepancies between bacterial culture and molecular testing results in patients with pneumonia33,34. Molecular techniques appear to be more sensitive than culture for bacterial detection5,34, however more studies are needed to show the real potential of metagenomics in the diagnosis of pneumonia.
This study has several limitations. The number of patients included in each type of pneumonia (viral or bacterial) was low, which limits the representativeness of the results. Some samples showed a very low bacterial load and had to be studied after a nested PCR. Although we proved in several samples to have similar results in standard and nested PCRs, small alterations in the microbiome composition after two amplification processes could not be completely ruled out. Another limitation was that only the bacterial microbiome (bacteriome) was studied. The lower alpha diversity observed in bacterial pneumonias was a consequence of the increase in the etiologic pathogen that unbalanced the resident microbiota. In viral pneumonias we could not rule out a decrease in the virome diversity and the fungome (mycobiome) was not studied in any sample. Techniques capable of studying the microbiome as a whole are needed to determine alterations in all the microbes of the lung microbiome. Finally, although a limit of 48 h of antibiotic treatment was admitted as not to cause detectable alterations in the metagenomics study of the lung microbiome, some of the patients had already been treated, what could have altered the microbiome composition. Other studies have demonstrated that bacterial DNA can be detected unaltered by molecular methods even after completed antibiotic treatment35.
In conclusion, most of the aetiologic diagnosis of patients admitted to ICU with severe CAP were done applying extensive microbiological diagnostic methods (culture, antigen detection, molecular testing). In patients with viral pneumonia, lung microbiome diversity and composition was similar to that of the microbiome of controls. On the other hand, patients with pneumococcal pneumonia showed a lower microbiome diversity. Further studies including more patients are needed to assess the utility of metagenomic analysis as a complementary tool in the diagnosis of bacterial pneumonia.
Data availability
All raw sequencing data is available as supplementary material.
Change history
01 February 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41598-024-53030-2
References
Jain, S. et al. Community-acquired pneumonia requiring hospitalization among U.S. adults. N. Engl. J. Med. 373, 415–427 (2015).
Prina, E., Ranzani, O. T. & Torres, A. Community-acquired pneumonia. Lancet 386, 1097–1108 (2015).
Dickson, R. P. The microbiome and critical illness. Lancet Respir. Med. 4, 59–72 (2016).
Kitsios, G. D. et al. Respiratory microbiome profiling for etiologic diagnosis of pneumonia in mechanically ventilated patients. Front. Microbiol. 9, 1413 (2018).
Mu, S. et al. Prospective evaluation of a rapid clinical metagenomics test for bacterial pneumonia. Front. Cell Infect. Microbiol. 11, 684965 (2021).
Natalini, J. G., Singh, S. & Segal, L. N. The dynamic lung microbiome in health and disease. Nat. Rev. Microbiol. https://doi.org/10.1038/s41579-022-00821-x (2022).
Dickson, R. P., Erb-Downward, J. R. & Huffnagle, G. B. Towards an ecology of the lung: New conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Respir. Med. 2, 238–246 (2014).
Bassis, C. M. et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. mBio 6, e00037 (2015).
Es, C. et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 184, 957–963 (2011).
Faner, R. et al. The microbiome in respiratory medicine: Current challenges and future perspectives. Eur. Respir. J. 49, 1602086 (2017).
Dickson, R. P., Erb-Downward, J. R. & Huffnagle, G. B. The role of the bacterial microbiome in lung disease. Expert Rev. Respir. Med. 7, 245–257 (2013).
Sole, M. L. et al. Pulmonary microbiome of patients receiving mechanical ventilation: Changes over time. Am. J. Crit. Care 30, 128–132 (2021).
Pettigrew, M. M., Tanner, W. & Harris, A. D. The lung microbiome and pneumonia. J. Infect. Dis. 223, S241–S245 (2021).
Metlay, J. P. et al. Diagnosis and treatment of adults with community-acquired pneumonia. An official clinical practice guideline of the American Thoracic Society and Infectious Diseases Society of America. Am. J. Respir. Crit. Care Med. 200, e45–e67 (2019).
Avalos-Fernandez, M. et al. The respiratory microbiota alpha-diversity in chronic lung diseases: First systematic review and meta-analysis. Respir. Res. 23, 214 (2022).
Sze, M. A. et al. The lung tissue microbiome in chronic obstructive pulmonary disease. Am. J. Respir Crit. Care Med. 185, 1073–1080 (2012).
Cuthbertson, L. et al. Lung function and microbiota diversity in cystic fibrosis. Microbiome 8, 45 (2020).
Heul, A. V., Planer, J. & Kau, A. L. The human microbiota and asthma. Clin. Rev. Allergy Immunol. 57, 350–363 (2019).
Richardson, H., Dicker, A. J., Barclay, H. & Chalmers, J. D. The microbiome in bronchiectasis. Eur. Respir. Rev. 28, 190048 (2019).
Smith, D. J. et al. Pyrosequencing reveals transient cystic fibrosis lung microbiome changes with intravenous antibiotics. Eur. Respir. J. 44, 922–930 (2014).
Drengenes, C. et al. Laboratory contamination in airway microbiome studies. BMC Microbiol. 19, 187 (2019).
Liu, H.-X. et al. Difference of lower airway microbiome in bilateral protected specimen brush between lung cancer patients with unilateral lobar masses and control subjects. Int. J. Cancer 142, 769–778 (2018).
Yu, G. et al. Characterizing human lung tissue microbiota and its relationship to epidemiological and clinical features. Genome Biol. 17, 163 (2016).
Hanada, S., Pirzadeh, M., Carver, K. Y. & Deng, J. C. Respiratory viral infection-induced microbiome alterations and secondary bacterial pneumonia. Front. Immunol. 9, 2640 (2018).
Yildiz, S., Mazel-Sanchez, B., Kandasamy, M., Manicassamy, B. & Schmolke, M. Influenza A virus infection impacts systemic microbiota dynamics and causes quantitative enteric dysbiosis. Microbiome 6, 9 (2018).
Han, Y., Jia, Z., Shi, J., Wang, W. & He, K. The active lung microbiota landscape of COVID-19 patients through the metatranscriptome data analysis. Bioimpacts 12, 139–146 (2022).
Lloréns-Rico, V. et al. Clinical practices underlie COVID-19 patient respiratory microbiome composition and its interactions with the host. Nat. Commun. 12, 6243 (2021).
Shen, Z. et al. Genomic diversity of severe acute respiratory syndrome–coronavirus 2 in patients with coronavirus disease 2019. Clin. Infect. Dis. 71, 713–720 (2020).
Vientós-Plotts, A. I., Ericsson, A. C., Rindt, H. & Reinero, C. R. Respiratory dysbiosis in canine bacterial pneumonia: Standard culture vs. microbiome sequencing. Front. Vet. Sci. 6, 354 (2019).
Dickson, R. P. et al. Lung microbiota predict clinical outcomes in critically ill patients. Am. J. Respir. Crit. Care Med. 201, 555–563 (2020).
de Roux, A. et al. Mixed community-acquired pneumonia in hospitalised patients. Eur. Respir. J. 27, 795–800 (2006).
Tikhomirova, A. & Kidd, S. P. Haemophilus influenzae and Streptococcus pneumoniae: Living together in a biofilm. Pathog. Dis. 69, 114–126 (2013).
Du, S. et al. Clinical factors associated with composition of lung microbiota and important taxa predicting clinical prognosis in patients with severe community-acquired pneumonia. Front. Med. 16, 389–402 (2022).
Bousbia, S. et al. Repertoire of intensive care unit pneumonia microbiota. PLoS One 7, e32486 (2012).
Rovery, C. et al. PCR detection of bacteria on cardiac valves of patients with treated bacterial endocarditis. J. Clin. Microbiol. 43, 163–167 (2005).
Funding
This study was supported by a grant of the Spanish Ministry of Health, Instituto de Salud Carlos III, FIS17/01463.
Author information
Authors and Affiliations
Contributions
J.M.M. and L.V. participated in the design of the study and writing the draft paper. M.E. and A.S. participated in performing metagenomic techniques and analysis of results. M.A. and N.A. participated in collecting epidemiological and microbiological data of patients with pneumonia. All authors contributed o the final writing and review of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of this Article contained an error in the spelling of the author Ane Sorarrain, which was incorrectly given as Ane Sorrarain.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marimón, J.M., Sorarrain, A., Ercibengoa, M. et al. Lung microbiome on admission in critically ill patients with acute bacterial and viral pneumonia. Sci Rep 13, 17724 (2023). https://doi.org/10.1038/s41598-023-45007-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-45007-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
