
Abstract
Multiplex PCR is an increasingly popular method for identifying species, investigating environmental diversity, and conducting phylogenetic analysis. The complexity and increasing availability of diverse templates necessitate a highly automated approach to design degenerate primer pairs for specific targets with multiple sequences. Existing tools for degenerate primer design suffer from poor maintenance, semi-automation, low adaptability, and low tolerance for gaps. We developed PMPrimer, a Python-based tool for automated design and evaluation of multiplex PCR primer pairs for specific targets using diverse templates. PMPrimer automatically designs optimal multiplex PCR primer pairs using a statistical-based template filter; performs multiple sequence alignment, conserved region identification, and primer design; and evaluates the primers based on template coverage, taxon specificity, and target specificity. PMPrimer identifies conserved regions using Shannon’s entropy method, tolerates gaps using a haplotype-based method, and evaluates multiplex PCR primer pairs based on template coverage and taxon specificity. We tested PMPrimer using datasets with diverse levels of conservation, sizes, and applications, including tuf genes of Staphylococci, hsp65 genes of Mycobacteriaceae, and 16S ribosomal RNA genes of Archaea. PMPrimer showed outstanding performance compared with existing tools and experimental validated primers. PMPrimer is available as a Python package at https://github.com/AGIScuipeng/PMPrimer.
Similar content being viewed by others

Introduction
The development of advanced sequencing technologies has made it easier to obtain diverse templates for specific targets, such as nucleotide sequences in NCBI1, 16S ribosomal RNA gene sequences in SILVA2, and SARS-COV-2 genome sequences in the GISAID database3. Based on these data, PCR-based methods are widely used for identifying species4, investigating environmental diversity5, and conducting phylogenetic analysis6. It is crucial to design multiplex PCR primer pairs in an unbiased manner to target specific regions of diverse templates. The complexity and rapid growth in the availability of diverse templates have made it necessary to automate the entire in silico design workflow, including data preprocessing, data analysis, primer design, and multiplex PCR primer pair evaluation7.
For accurate in silico design of multiplex PCR primer pairs, it is important to filter out low-quality (too short, too long, or abnormal) and redundant templates, align diverse templates using a multiple sequence alignment tool (for example, MUSCLE58), identify conserved regions based on sequence similarity (such as allele frequency or Shannon’s entropy9), and design primers for diverse templates (for example, Primer3 software10). It is also important to evaluate the primers for primer dimer formation, secondary structure (hairpin formation), melting temperature (Tm), and template coverage and to evaluate in silico multiplex PCR primer pairs or amplicons for template coverage, taxon specificity, and target specificity.
Existing tools for the design of multiplex PCR primer pairs have several shortcomings, such as poor maintenance, semi-automation, low adaptability, and low tolerance of gaps (Supplementary Table S1). DECIPHER is used to design primer pairs targeting a specific group of sequences from the whole sequence set, but its R-based package is time-consuming and its web-based tool cannot currently be accessed11. Many tools design primers using consensus sequences while ignoring minor alleles in consensus regions, such as PrimerDesign-M12, openPrimeR13, PhyloPrimer14, and rprimer15. PrimerDesign-M and rprimer require previous multiple sequence alignment coupled with information about the intended target regions12,15. The rprimer tool cannot identify conserved regions and produces many candidate multiplex PCR primer pairs, which makes it almost impossible for the user to directly obtain the optimal primer pairs15. PhyloPrimer is used to design primers for microbial sequences and preferentially produces non-degenerate primer pairs14. Furthermore, most primer design tools strip gaps for convenience, especially software based on consensus sequences. Finally, tools based on the R language, such as openPrimeR13 and rprimer15, are inefficient at processing massive amounts of data.
To overcome these shortcomings and to design multiplex PCR primer pairs automatically with improved performance, we developed PMPrimer, a Python-based tool for designing multiplex primers for specific targets using diverse templates. The only necessary input file is the target sequence file in FASTA format. The greatest strengths of PMPrimer are its abilities to identify conserved regions using Shannon’s entropy method, tolerate gaps using a haplotype-based method, and evaluate multiplex PCR primer pairs based on template coverage and taxon specificity. The target specificity of primer pairs can be assessed by using BLAST16.
Methods
PMPrimer
PMPrimer is a Python-based tool for designing multiplex PCR primer pairs for specific targets using diverse templates. This tool requires MUSCLE58, Primer310, and BLAST16 and includes four major modules: data preprocessing and alignment, conserved region identification, primer design and evaluation, and amplicon selection and evaluation (Fig. 1).

PMPrimer workflow. PMPrimer includes four main modules: data preprocessing and alignment, conserved region identification, primer design and evaluation, and amplicon selection and evaluation. The major steps are shown in the diagram.
Data preprocessing and alignment
PMPrimer starts with data quality assessment (identity duplication, record duplication, and sequence abnormality), filters out templates based on length distribution (too short or too long), and removes redundant templates with identical sequences in terminal taxa. MUSCLE58 is then utilized for multiple sequence alignment. The distance matrix can be calculated based on multiple sequence alignment to estimate differences within and between taxa.
Conserved region identification
PMPrimer assesses the degree of complexity of nucleotide types at each position of the alignment based on Shannon’s entropy. Shannon’s entropy is calculated based on the presence of four bases (A, T, C, G) and gap symbols (–). The smaller the Shannon’s entropy value, the higher the conservation. The presence of a region with Shannon’s entropy below the desired score (default Shannon’s entropy value = 0.12) triggers the process for conserved region identification, and the length is extended as the average Shannon’s entropy drops below the desired score. To help users set a threshold Shannon’s entropy score, the minimum frequency of a major allele for a conserved position is used as the initial parameter (default major allele frequency = 0.95), and Shannon’s entropy is calculated using this threshold. The adjacent conserved regions equal to or larger than the minimum length of the initial conserved region (default = 15 bp) are then combined as the proposed conserved region. The gap positions are then counted, and the effective length of the conserved region is obtained by subtracting the gap positions from the length of the conserved region. Finally, the conserved region that satisfies the minimum length (default = 15 bp) for primer design is used for downstream analysis.
Primer design and evaluation
Primer design comprises five steps: (1) extracting haplotype sequences in conserved regions, (2) generating optimal primers for each haplotype sequence, (3) selecting optimal primer regions and collecting all sequences in the primer regions, (4) recomputing physicochemical properties for each primer, and (5) evaluating the template coverage of multiplex primers.
Amplicon selection and evaluation
Using the primers in conserved regions, multiplex PCR primer pairs are selected based on amplicon length, haplotype number limit (default = 10), and maximum difference in Tm (default = 12.0 °C). The multiplex PCR primer pairs or amplicons are then evaluated for template coverage, taxon specificity, and target specificity.
Testing datasets
To systematically evaluate PMPrimer, we selected three datasets, 16S ribosomal RNA (rRNA) genes of Archaea, hsp65 genes of Mycobacteriaceae, and tuf genes of Staphylococci, for the following reasons. First, the level of conservation varies from 3.90% similarity in 16S rRNA genes at the domain level to 89.48% similarity in hsp65 genes at the family level to 91.73% similarity in tuf genes at the genus level. Second, the number of sequences in these datasets varies from thousands to tens of thousands (11,757 16S rRNA gene sequences, 6528 hsp65 gene sequences, and 2547 tuf gene sequences). Third, these three types of genes are frequently used in the assessment of environmental diversity (16S rRNA genes), species identification, and clinical diagnosis (hsp65 genes and tuf genes). The 16S rRNA gene is the most widely used region for bacterial taxonomy and identification in prokaryotes17. In mycobacteria, hsp65 genes have higher discrimination ability for species identification compared to 16S rRNA genes and rpoB genes18. tuf genes are the most widely used genes for species identification of Staphylococci compared to gap genes, hsp60 genes, and internal transcribed spacer regions based on searches in PubMed (https://pubmed.ncbi.nlm.nih.gov/) and Web of Science (https://www.webofscience.com) on September 7, 2023.
16S rRNA genes of Archaea
We retrieved 13,421 16S rRNA genes in Archaea from the non-redundant reference dataset (SSURef 108 NR) in SILVA (release 108, accessed November 2022)2. After removing sequences with degenerate bases, we used the 11,757 remaining sequences as a testing dataset for 16S rRNA genes. Taxonomic classification at the phylum, class, and order levels was used to evaluate template coverage and taxon specificity in this dataset.
hsp65 (groEL2) genes of Mycobacteriaceae
We retrieved 7188 RefSeq assemblies in the Mycobacteriaceae family from NCBI (accessed November 2022)1. Based on gene annotation, 12,705 possible hsp65 gene sequences were extracted from these genomes. After low-quality and redundant sequences were filtered out, 1161 possible hsp65 sequences remained. Gene clustering analysis was carried out based on sequence similarity, and 578 hsp65 (groEL2) and 583 groEL1 sequences were ultimately identified (Supplementary Fig. S1). Finally, 6528 hsp65 genes corresponding to 578 non-redundant hsp65 sequences in 221 species were used as a testing dataset.
tuf genes of Staphylococci
We retrieved 3624 tuf gene sequences in the Staphylococcus genus from the European Nucleotide Archive (ENA) of the European Bioinformatics Institute database (Coding release, accessed August 2022)19. The low-quality sequences (too short, too long, and redundant sequences) were filtered out, and multiple sequence alignment of the remaining sequences was performed using MUSCLE58. The distance matrix was obtained based on the alignment, and unusual sequences were further filtered out. Finally, 2547 tuf genes from 54 species in the Staphylococcus genus were obtained for downstream analysis.
Comparative analysis
To demonstrate the performance of PMPrimer, we compared this software tool with DECIPHER11, PrimerDesign-M12, PhyloPrimer14, rprimer15, and openPrimeR13 in terms of the analysis process, degree of automation, run time, and results. For the analysis process, modular design allows users to conveniently choose an appropriate module for subsequent analysis. Automation can be evaluated by comparing the entire analysis process and the usability of the results. Run time is an important indicator of software performance, especially in the context of big data. By comparing the template coverage and taxon specificity of the resulting multiplex PCR primer pairs, the efficiency and specificity of different software tools can be compared. The dataset for tuf genes from Staphylococci was used for software comparisons on Windows or WSL with Intel(R) Core(TM) i7-9700 CPU, 3.00 GHz, 16.0 GB RAM.
Results
PMPrimer software
PMPrimer is a Python-based tool for the automated design and evaluation of multiplex PCR primer pairs for specific targets using diverse templates (Fig. 1). To satisfy the need for automation, PMPrimer can directly extract taxonomic information at three levels of classification (genus, species, and subspecies) from input data in FASTA format downloaded from the NCBI or ENA databases. The length distribution of template sequences in the input data is calculated, and at least 90% of sequences with most common length are kept, whereas sequences that are too short or too long are removed. Redundant sequences are removed, but one representative redundant sequence is retained to preserve template diversity. For example, 578 sequences were chosen to represent 6528 hsp65 genes of Mycobacteriaceae. Multiple sequence alignment is then performed, and the conservation of each position is scored based on Shannon’s entropy. Unlike analysis of consensus bases, Shannon’s entropy analysis preserves all types of bases, including gaps, and effectively represents the complexity of each position. Conserved regions are first identified based on Shannon’s entropy, and adjacent conserved regions are then combined, if possible; this process can tolerate interruptions caused by the presence of positions with high diversity. Furthermore, the effective length of the conserved region is obtained by subtracting gap length from the length of the conserved region. Conserved regions satisfying the minimum length requirement are used for primer design. To ensure diversity of conserved regions, all haplotype sequences are used for primer design, and the optimal primer region is selected for multiplex primers. Finally, primer pairs are selected and evaluated by electronic PCR amplification. To balance primer number and template coverage, primers are sorted and selected based on their corresponding templates. In addition, taxon specificity is evaluated using our method, which represents discrimination efficiency among different taxa at different levels of classification.
Evaluation of PMPrimer using testing datasets
To test the performance of PMPrimer, three commonly used datasets were employed: 16S rRNA genes of Archaea, hsp65 genes of Mycobacteriaceae, and tuf genes of Staphylococci. These datasets have diverse template numbers (11,757, 6528, and 2547 sequences, respectively), redundancy rates of templates (0.20%, 91.14%, and 87.63%, respectively), average similarity scores (3.90%, 89.48%, and 91.73%, respectively), gap rates in alignments (92.87%, 0.95%, and 0.58%, respectively), and taxon levels (domain, family, and genus, respectively) (Table 1).
16S rRNA genes of Archaea
Based on 16S rRNA genes of Archaea, PMPrimer identified three conserved regions using the parameters “threshold for Shannon’s entropy = 0.26 (major allele frequency 0.85), gap threshold = 1.0, melting temperature = 45 °C, count of haplotype = 600” and other default parameters (Supplementary Table S2). In a previous study5, the best primer pair (A519F (S-D-Arch-0519-a-S-15) and 802R (S-D-Bact-0785-b-A-18)) was identified for Archaea. Here we identified one optimal degenerate primer in each of three conserved regions (Table 2). Primer 519–533 (7818–8175) is identical to previously described primer A519F, with 94.3% coverage, while primer 784–805 (10,365–11,009), with 90.4% coverage, is four bases longer than previously described primer 802R, with 90.7% coverage in our dataset. The difference between primers 784–805 and 802R is primarily due to the use of different templates (Archaea vs. Archaea and Bacteria). The 0.3% difference in coverage is due to the presence of degenerate bases in Archaea and Bacteria (in 802R, N at 789 stands for A, C, G, and T, and V at 788 stands for A, C, and G; in 784–805, H at 789 stands for A, C, and T, and S at 788 stands for C and G). Furthermore, a new primer 871–890 (12,677–13,180) was identified with 84.8% coverage in our dataset.
hsp65 (groEL2) genes of Mycobacteriaceae
Hsp65 is frequently used for species identification in Mycobacteriaceae using primer pair Tb11 and Tb124,18, which was designed 30 years ago20. To evaluate this primer pair and to design optimal primer pairs for mycobacteria, we built a dataset using hsp65 gene sequences from 6 genera, 221 species, and 234 subspecies. Based on in silico evaluation, Tb11 had only 83.0% (480/578) coverage and Tb12 had only 20.4% (118/578) coverage in our dataset. PMPrimer identified nine conserved regions with the parameters “count of haplotype = 70” and other default parameters and designed one optimal degenerate primer in each of these nine conserved regions (Supplementary Table S3). For convenience, we also provide numbering based on the hsp65 gene of Mycobacterium tuberculosis (NC_000962.3). In the alignment, Tb11 and Tb12 are located at positions 148–168 and 569–589, respectively. We identified two degenerate primers adjacent to Tb11 and Tb12 with higher coverage (163–181 (166–184) with 99.3% coverage and 514–531 (517–534) with 97.9% coverage). Using these degenerate primers, we identified primer pairs suitable for 300-bp fragments (short reads from Illumina and Ion Torrent sequencing), 600-bp fragments (long reads from Illumina and Ion Torrent sequencing), and > 600-bp fragments (from PacBio and NanoPore sequencing) (Table 3) for species identification and comparative analysis.
tuf genes of Staphylococci
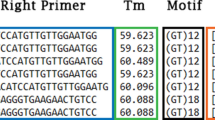
In an earlier study, the tuf gene was used for species identification of coagulase-negative Staphylococci using four primer pairs21. In the current study, we retrieved tuf gene sequences of Staphylococci from the ENA and ultimately obtained 2547 tuf gene sequences in this dataset, which included 54 species. Based on in silico evaluation, there was only 0.03% coverage in the original files for primer Tuf32. Therefore, we removed the amplicon produced by the Tuf32/900 primer pair from subsequent analysis. The coverage of the three other primer pairs/six primers was 100% (positions 57–77, 165–184, 339–360, 453–474, 698–717, and 835–852 in the alignment). PMPrimer identified 18 conserved regions with the parameters “threshold for Shannon’s entropy = 0.02 (major allele frequency 0.995), minimum length of initial conserved region = 5, merge = TRUE” and other default parameters (Supplementary Table S4). The six primers designed in the previous study were also identified by PMPrimer, with one base shift, one/two base differences, or divided into two primers due to highly variable positions. For convenience, we also provide numbering based on the tuf gene of Staphylococcus aureus (NC_007795.1). Based on the above degenerate primers, we also identified optimal primer pairs suitable for fragments of different lengths (Supplementary Table S5).
Software comparison
To date, several primer design tools based on multiple sequence alignment have been developed, but each of these tools was optimized for a specific application, making it difficult to compare them. Nevertheless, we selected DECIPHER11, PrimerDesign-M12, openPrimeR13, PhyloPrimer14, and rprimer15 for comparisons with PMPrimer as a whole or in part to demonstrate the strengths and weaknesses of PMPrimer. PhyloPrimer could not be run due to the lack of a user manual, so we did not include it in downstream analysis. Because all these selected tools cannot generate results from datasets for 16S rRNA genes of Archaea or hsp65 (groEL2) genes of Mycobacteriaceae within 12 h or exceeding the limit of the input file size, we only used the tuf dataset for comparison after removing redundant sequences.
PMPrimer can run with unaligned input sequences directly, rerun from any step based on modular design, and produce filtered results with evaluations for optimized primers or primer pairs. However, PrimerDesign-M and rprimer only support alignment sequences, PrimerDesign-M and openPrimeR require amplicon regions to be provided, and rprimer generates thousands of degenerate primers without selecting optimal primer pairs. Moreover, DECIPHER supports unaligned sequences and runs automatically and in a modular fashion, while all the other previously designed software tools do not. When we used the six primers with 100% coverage designed in a previous study as the true primer set21, PMPrimer identified all of these primers and additional novel primers, while rprimer and DECIPHER only identified 5 and 4 of these primers, respectively. To compare run times, we use multiple sequence alignment for all tools. rprimer ran the fastest, followed by PMPrimer and DECIPHER (Table 4).
Discussion
Designing primer pairs for specific targets using diverse templates is essential for various applications; although several tools have been developed for this purpose, none meets all the requirements of these applications. Here, we developed PMPrimer for designing primers aimed at specific targets using diverse templates in both an automatic and modular manner to fill the gap between requirements and existing tools.
Most current tools (except for PhyloPrimer14) require users to prepare templates without abnormal sequences manually, a complicated and time-consuming process. PMPrimer can process multiple sequences downloaded directly from public databases such as NCBI1 and ENA19 with preprocessing analysis models. Furthermore, some tools, such as PrimerDesign-M12, openPrimeR13, and rprimer15, cannot identify conserved regions automatically. PMPrimer can identify conserved regions based on Shannon’s entropy9 and combine adjacent conserved regions to form larger conserved regions. Most existing tools design primers using consensus sequences, which ignores low-frequency alleles and gaps. By contrast, PMPrimer designs primers using haplotype sequences, which can tolerate minor alleles and gaps simultaneously. Although existing tools can theoretically be used for any target sequences, none can support big data. We examined the power of PMPrimer to design primers for diverse targets using three datasets with different template numbers, rates of sequence redundancy and similarity, gap rates of alignments, and taxon levels. Degenerate primers have been used as multiplex PCR primer pairs. However, this approach increases the redundancy of primers by using all possible combinations of degenerate bases22. Therefore, PMPrimer also provides haplotype primer pairs in target sequences, providing the minimal set of primer pairs for target regions. For examples, in the tuf dataset, the forward degenerate primer 58–78 (58–78) (5'-CACGTTGACCAYGGTAAAACD-3') represents six primers, but two of these primers do not exist; the reverse degenerate primer 334–353 (340–359) (5′-TCACGMGTTTGWGGCATTGG-3′) represents four primers, but one of these primers does not exist. Consequently, the amplicon produced by the above degenerate primer pair is theoretically produced using 24 primer pair combinations, but 12 of these do not exist. In addition, PMPrimer provides several indicators to evaluate multiplex PCR primer pairs, such as template coverage, taxon specificity, and target specificity.
To handle the diverse characteristics of target sequences, PMPrimer has multiple built-in parameters to make the appropriate adjustments for data processing. For instance, different Shannon’s entropy scores are used for target sequences with different conservation levels, such as “threshold for Shannon’s entropy = 0.26 (major allele frequency 0.85)” for 16S rRNA genes of Archaea, “threshold for Shannon’s entropy = 0.12 (major allele frequency 0.95) (default)” for hsp65 genes of Mycobacteriaceae, and “threshold for Shannon’s entropy = 0.02 (major allele frequency 0.995)” for tuf genes of Staphylococci. When the target sequences are highly diverse, we recommend using “gaps parameter” to tolerate gaps and “merge parameter” to combine adjacent conserved regions.
PMPrimer has several limitations. First, it can only extract taxonomic information at the genus, species, and subspecies levels from input data automatically. We will add an analysis model to obtain taxonomic information for any continuous three level classification based on sequence description and taxonomy databases in the future. Second, some parameters must be set for specific target sequences manually. For convenience, we will automatically detect some important parameters by evaluating target sequences, such as Shannon’s entropy score and minimum allele frequency. Third, conserved region identification is the key to primer design. PMPrimer can currently identify conserved regions using specific parameters. However, we expect PMPrimer to be able to identify all possible conserved regions by iteration. Fourth, PMPrimer only provides in silico validation for newly designed primers or primer pairs. We plan to develop a tool to deal with amplicon sequencing data using information obtained during the primer design stage, such as haplotype primer pairs, distance matrix at different taxon levels, and specific positions with high variability. We will then evaluate newly designed primer pairs systematically using amplicon sequencing data.
Conclusions
PMPrimer can be used to design and evaluate multiplex PCR primers in an automatic, modular, efficient manner using diverse templates with different characteristics. In silico evaluation using three datasets demonstrated that PMPrimer can be used for various applications.
Data availability
Datasets analyzed in this study are available at https://github.com/AGISCuipeng/PMPrimer_datasets.
Code availability
PMPrimer software is freely available at https://github.com/AGIScuipeng/PMPrimer.
References
Wheeler, D. L. et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 35, D5–D12. https://doi.org/10.1093/nar/gkl1031 (2007).
Pruesse, E. et al. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. https://doi.org/10.1093/nar/gkm864 (2007).
Shu, Y. & McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 22, 30494. https://doi.org/10.2807/1560-7917.ES.2017.22.13.30494 (2017).
Dai, J., Chen, Y. & Lauzardo, M. Web-accessible database of hsp65 sequences from Mycobacterium reference strains. J. Clin. Microbiol. 49, 2296–2303. https://doi.org/10.1128/jcm.02602-10 (2011).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1. https://doi.org/10.1093/nar/gks808 (2013).
Sun, Q. et al. VarEPS: An evaluation and prewarning system of known and virtual variations of SARS-CoV-2 genomes. Nucleic Acids Res. 50, D888–D897. https://doi.org/10.1093/nar/gkab921 (2022).
Hendling, M. & Barišić, I. In-silico design of DNA oligonucleotides: Challenges and approaches. Comput. Struct. Biotechnol. J. 17, 1056–1065. https://doi.org/10.1016/j.csbj.2019.07.008 (2019).
Edgar, R. C. MUSCLE v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping. BioRxiv https://doi.org/10.1101/2021.06.20.449169 (2021).
Shannon, C. E. A mathematical theory of communication. Bell Syst. Tech. J. 27, 379–423. https://doi.org/10.1002/j.1538-7305.1948.tb01338.x (1948).
Untergasser, A. et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 40, e115. https://doi.org/10.1093/nar/gks596 (2012).
Wright, E. S. Using DECIPHER v2.0 to analyze big biological sequence data in R. R. J. 8, 352. https://doi.org/10.32614/RJ-2016-025 (2016).
Yoon, H. & Leitner, T. PrimerDesign-M: A multiple-alignment based multiple-primer design tool for walking across variable genomes. Bioinformatics 31, 1472–1474. https://doi.org/10.1093/bioinformatics/btu832 (2015).
Kreer, C. et al. openPrimeR for multiplex amplification of highly diverse templates. J. Immunol. Methods 480, 112752. https://doi.org/10.1016/j.jim.2020.112752 (2020).
Varliero, G., Wray, J., Malandain, C. & Barker, G. PhyloPrimer: A taxon-specific oligonucleotide design platform. PeerJ 9, e11120. https://doi.org/10.7717/peerj.11120 (2021).
Persson, S., Larsson, C., Simonsson, M. & Ellström, P. rprimer: An R/bioconductor package for design of degenerate oligos for sequence variable viruses. BMC Bioinform. 23, 1–18. https://doi.org/10.1186/s12859-022-04781-0 (2022).
Camacho, C. et al. BLAST+: Architecture and applications. BMC Bioinform. 10, 1–9. https://doi.org/10.1186/1471-2105-10-421 (2009).
Petti, C. A. Interpretive Criteria for Identification of Bacteria and Fungi by DNA Target Sequencing; Approved Guideline. 2nd ed. (2018).
Jouet, A. et al. Deep amplicon sequencing for culture-free prediction of susceptibility or resistance to 13 anti-tuberculous drugs. Eur. Respir. J. https://doi.org/10.1183/13993003.02338-2020 (2021).
Amid, C. et al. The European nucleotide archive in 2019. Nucleic Acids Res. 48, D70–D76. https://doi.org/10.1093/nar/gkz1063 (2020).
Telenti, A. et al. Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J. Clin. Microbiol. 31, 175–178. https://doi.org/10.1128/jcm.31.2.175-178.1993 (1993).
Van Reckem, E., De Vuyst, L., Leroy, F. & Weckx, S. Amplicon-based high-throughput sequencing method capable of species-level identification of coagulase-negative staphylococci in diverse communities. Microorganisms 8, 897. https://doi.org/10.3390/microorganisms8060897 (2020).
Sambo, F. et al. Optimizing PCR primers targeting the bacterial 16S ribosomal RNA gene. BMC Bioinform. 19, 1–10. https://doi.org/10.1186/s12859-018-2360-6 (2018).
Funding
Funding was provided by Agricultural Science and Technology Innovation Project (Grant number: CAAS-ZDRW202105), and STI 2030-Major Projects.
Author information
Authors and Affiliations
Contributions
L.Y.: methodology, software, data curation, formal analysis, writing—original draft. F.D.: methodology, data curation, formal analysis. Q.L.: methodology, data curation, formal analysis. J.H.X.: resources, formal analysis. W.F.: formal analysis. F.Y.D.: conceptualization, supervision. P.C.: conceptualization, supervision, funding acquisition. W.F.L.: conceptualization, supervision, writing—original draft, writing—reviewing and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yang, L., Ding, F., Lin, Q. et al. A tool to automatically design multiplex PCR primer pairs for specific targets using diverse templates. Sci Rep 13, 16451 (2023). https://doi.org/10.1038/s41598-023-43825-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-43825-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
