
Abstract
Thiopental sodium (Tho) is an intravenous anesthetic. The current study aimed to find a rapid RP-HPLC method of Tho analysis with high linearity, repeatability, sensitivity, selectivity, and inexpensive. In our developed method, there is no need to use special chemical reagents, a high percentage of organic solvent, a high flow rate, or a further guard column. The chromatographic system consists of an ODS column (150 mm × 4.6 mm × 5 μm). The mobile phase was prepared by mixing KH2PO4 solution: methanol (40:60) with a flow rate of 1.2 mL/min at a detection wavelength of 230 nm, at room temperature using an injection volume of 10 μL. The method manifested a satisfied linearity regression R2 (0.9997) with a good repeatability precision range (0.16–0.47%) with LOD and LOQ; 14.4 μg/mL and 43.6 μg/mL respectively. Additionally, the method proved its efficiency via system suitability achievement in robustness and ruggedness, according to the validation guidelines. The shorter analysis time makes the method very valuable in quality control to quantify the commercial Tho in pharmaceutical preparations. This improved HPLC method has been successfully applied for Tho analysis for Thiopental UP Pharma 500 mg vials and Thiopental Eipico 1.0 g vials in our routine finished and stability studies testing laboratories. Additionally, the detection limit of Tho has been tested in our quality control lab to detect the smallest amount of traces that may be present after the cleaning process of the production machines for cephalosporins preparations. The method has shown positive results for Tho in low-level raw materials and pharmaceutical formulations.
Similar content being viewed by others

Introduction
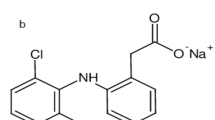
Thiopental sodium (Tho) has the IUPAC name 4,6(1H,5H)-Pyrimidinedione, 5-ethyldihydro-5-(1methylbutyl)-2-thioxo-, monosodium salt, (±)-; Sodium (±)-5-ethyl-5-(1-methylbutyl)-2-thiobarbiturate1. Tho has the chemical molecular formula C11H17N2O2SNa in molar mass “264.32 g/mole” as manifested in (Fig. 1).

Structure of Thiopental sodium “C11H17N2O2SNa in molar mass “264.32 g/mole”.
Tho could be taken by injection as intravenous anesthesia. It could be used for the induction of general anesthesia, as well as relaxation during balanced anesthesia with other anesthetic drugs such as analgesics and muscle relaxants2. As a reinforcement for the management of refractory convulsive disorders that are genetically diverse, such as those produced by local anesthetics. It is also used to minimize intracranial pressure in individuals who have high intracranial pressure if managed respiration is administered. Tho also has great importance in forensic medicine and criminology as it is used in investigations related to drug crimes3.
Tho is sold under more than one brand name, such as Farcopental, Thiopental, and Anapental, with different strengths of 1.0 g and 0.5 g vials. Tho also be found in a sterile mixture of thiopental sodium with or without anhydrous sodium carbonate as a buffer. Its finished product vial container must contain 93.0–107.0% of the labeled amount of thiopental sodium of the mixture according to the United States-Pharmacopoeia (USP 44–NF 39 2021). But the British-Pharmacopoeia (BP–2022–VOL3) issued another limit of the Tho assay as pure thiopental at 77.0–92.0% of the stated amount of the mixture of thiopental sodium and anhydrous sodium carbonate in the finished product vial container1.
The wide spectrum of this drug makes it important in the field of pharmaceutical trade, which necessitates the need to find effective, simple, easy, and rapid methods for assay determination. In addition, a sensitive method should be conducted at low concentrations of this drug preparation when this method is used to estimate Tho after washing, cleaning machines, and production lines. The sensitive method should be conducted to ensure the effectiveness of the cleaning method to remove the drug residual effects of this drug that may be entered into the next product in the production process, causing a completely unacceptable cross-contamination process4,5,6,7,8,9,10,11,12,13,14. This type of contamination is according to the quality standards mentioned in the rules of good manufacturing practice (GMP)5,6,14.
Label-free quantitation using an HPLC conjugated with their detectors is one of the simplest and most economical and cost-effective methods to differentiate the expression of proteins and their byproducts as metabolites. In label-free technology, each sample is measured separately. The necessity for high recovery and accuracy performance label-free quantitation methods is still high in the bioactive constituent, especially in the drug research field15.
There are many different methods with more than one technique in the analysis tools being conducted for the assay determination of Tho, some of which include the electrochemical method of the glassy carbon electrode by cyclic voltammetry16, cathodic stripping voltammetry17, HPTLC2, GC18,19,20, GC–EI–MS method21, LC–MS/MS22, HPLC3,23,24,25,26,27,28, UV spectrophotometry as USP 44–NF 39 2021, gravimetric analysis as constant weight using chloroform for the extraction as BP–2022–VOL3 issued1.
Most of the disadvantages of the previous analysis techniques and methods were detailed as more expensive2,18,19,21,25, time-consuming1,20, hazardous as chloroform1,6,26, sodium fluoride22, iodine-azide reaction27, needing special conditions or reagents as tetrabutyl ammonium bromide26 or using pre-column20,28, EDTA, CN column, sodium hydroxide as a solvent, and post-column27, sometimes derivatization was required. Also, they lack sufficient selectivity and specificity, especially with UV methods, and adjusted pH buffer solutions22,25,27. These factors are used to get the optimum peak shape with ideal tailing29,30. Supplementary Table 1 shows a brief comparison of different assay methods including the important parameters.
The field of scientific research has recently tended to purify industrial wastewater, pharmaceutical factories, and hospitals. So, finding easy, fast, sensitive, accurate, and economical methods has become an urgent necessity4,5,6,7,8,9,10,14,27,31,32,33.
The current paper aims to introduce an inexpensive, efficient, simple, and rapid method for the assay determination of Tho. It is because of the scarcity of quick and simple analytical methods to determine the thiopental assay in recent years that most of the system suitability and method validation parameters have been implemented. Furthermore, inexpensive chemicals with integrational analysis method parameters were used to obtain the detection and identification of Tho with accurate selectivity and high specificity via reasonable resolution.
Methodology and experimental analysis
Thiopental sodium and sodium carbonate working standards were supplied as a gift sample from UP Pharma (Assuit, Egypt). Methanol HPLC-grade, potassium dihydrogen phosphate, hydrochloric acid (37%), sodium hydroxide, and hydrogen peroxide (30%) (Scharlau, Spain). Water for injection (WFI) was used in the analysis and passed through a 0.45 μm nylon membrane filter before use. A phosphate solution was prepared by weighing about 5.82 g of potassium dihydrogen phosphate in 1000 mL of WFI.
Chromatographic system configuration
Compared with the previously conducted HPLC methods and the current analysis method, we did not use a dedicated pH solution adjustment or special chemical reagent to realize the optimum separation for the ideal system suitability achievement.
Tho assay determination was conducted using the HPLC model HP 1100 series with a variable wavelength. The current method was conducted with the RP C18 ODS column (150 mm × 4.6 mm × 5 μm) (Thermo Scientific). The mobile phase was prepared as KH2PO4 solution “(5.82 g in 1000 mL of WFI)”: methanol in a ratio of 40:60 v/v, at a flow rate of 1.2 mL/min with a detection wavelength of 230 nm at room temperature and an injection volume of 10 μL.
Parameters of method validation
The HPLC validation method was performed according to the International Conference on Harmonization (ICH) guidelines concerning parameters including system suitability, range of linearity, the limit of detection (LOD), the limit of quantification (LOQ), repeatability (precision), recovery and accuracy, robustness, ruggedness, the stability of the solution, specificity, and selectivity11,12,13,34,35,36.
System suitability check
System suitability was performed by injecting six replicate injections of the same working standard solution, which was prepared by dissolving a quantity of thiopental sodium with a sodium carbonate mixture of about 1.080 g, equivalent to 1.0 g of Tho [stock solution], in 200 mL of WFI, then diluting 10 mL into a 100 mL volumetric flask using WFI to get a solution with a concentration about 500 µg/mL.
Range & linearity
The analytical approach is deemed to be linear if there is a substantial portion between the response and the claimed working concentration, starting at the lowest point in the tested range and increasing to the highest point with R2 ≥ 0.9995,6,11,12,13,14,34,35,37.
Regression linearity equation:
where Y represents the response of the average peak area, X represents the claimed working concentration in (%), a represents the slope, and b is the intercept of the calibration curve where the target concentration (100%) represents 500 µg/mL.
The linearity parameter was submitted using different concentrations in the range (20–150%) of the Tho working standard. The claimed working concentrations were prepared using WFI as a solvent from the stock solution at a concentration of 5000 µg/mL. Every solution was injected into duplicates.
Limit of detection (LOD)
It was defined as the lowest specified analyte concentration in the matrix that could be identified using the detection of the instrument. Furthermore, it should not be included in the accuracy, precision, and linearity ranges11,12,13,34,35.
Limit of quantitation (LOQ)
It was defined as the lowest specified analyte concentration in the matrix that could be identified using the detection of the instrument. Furthermore, it must be included in the accuracy, precision, and linearity ranges11,12,13,34,35.
LOD and LOQ could be calculated according to the slope and standard error data from the linearity of the calibration as follows:
where σ: is the standard error of X & Y arrays and S: represents the slope of the linearity calibration curve.
Accuracy and recovery
Both recovery and accuracy are used alternately. The measurement's accuracy is defined as the proximity of the actual concentration (measured value) to the theoretical concentration (true value)11,12,13,34,35.
Accuracy was implemented by weighing three individual Tho working standards to give theoretical concentrations at (350 µg/mL), (500 µg/mL), and (600 µg/mL). Accuracy% could be estimated using the linearity equation:
Where 85.75% is the working standard assay as is%
Repeatability and precision
Repeatability was conducted using six different preparations individually of the target concentration of the intended method 100% (500 µg/mL) of Tho using the same equipment on the same day via the same analyst or compared with another analyst as inter–precision11,12,13,34,35,38.
Robustness
Robustness was submitted using designed small changes including slight changes, in the temperature, composition of the mobile phase, etc.
The designed small changes were conducted in a different organic solvent ratio (Methanol) at (600 ± 60 mL) and a flow rate of 1.2 mL/min ± 0.1 mL/min).
Ruggedness
Ruggedness was submitted using designed and major observable changes, including analyst-to-analyst, column-to-column, and day-to-day while maintaining all of the analysis method parameters and conditions as they are without changes.
Stability of sample solution (at the HPLC rack)
This test was performed to judge the stability of the solution after preparation over time. The test was conducted at the target concentration of 100% (500 µg/mL) over 48 h. The test is valid for use at room temperature over 48 h if the RSD% of the peak area is ≤ 2.0% from the start to the end of the conducted experiments.
Specificity and selectivity
Forced degradation studies were performed to indicate the stability-indicating properties. Accelerated degradation was implemented using acid hydrolysis at 0.1 M HCl for 30 min, base hydrolysis at 0.1 M NaOH for 30 min, stress oxidation degradation at 3.0% w/v of H2O2 for 30 min, and light degradation over 3 h.
Test of the validated method
Tho analysis of the different commercial dosage forms in the Egyptian local market
Thiopental UP Pharma 500 mg vials and Thiopental Eipico 1.0 g vials were tested using the validated method of Tho.
Thiopental UP pharma 500 mg vials batch number (220741) after the constitution stability studies
The after-constitution stability study was conducted using the WFI, saline 0.9% wt/v, and glucose 5% wt/v at zero time, 6 h at room temperature 30 ± 2 °C, and in the refrigerator at temperature 5 ± 3 °C for 24 h.
The constituted vial was performed using 10 mL of solvent, and the constituted vial was reserved for the desired time under the described storage conditions. Then, all of the content of the vial was transferred into a 1000 mL volumetric flask, completed to the mark using WFI to get a solution concentration (0.5 mg/mL), and introduced to the HPLC assay.
Results and discussions
System suitability check
The thiopental peak appeared about 4.6 min at the optimum parameters of the analysis method (Fig. 2a) and in the range of 4.3–6.7 min over all the parameter changes (Fig. 2b–i). Table 1 showed high performance for the intended analysis method, where the RSD % < 2.0%, USP tailing < 2.0, and theoretical plates ≥ 20005,6,11,12,13,14,34,35. So, according to the output data of the system suitability parameters, the method manifested superior validity through a wide range of retention times.



(a) Tho chromatogram at a flow rate 1.2 mL/min, Column—1, Aqueous phase 40% and Methanol 60%. (b) Tho chromatogram at a flow rate 1.1 mL/min, Column—1, Aqueous phase 40% and Methanol 60%. (c) Tho chromatogram at a flow rate 1.3 mL/min, Column—1, Aqueous phase 40% and Methanol 60%. (d) Tho chromatogram at a flow rate 1.2 mL/min, Column—1, Aqueous phase 46% and Methanol 54%. (e) Tho chromatogram at a flow rate 1.2 mL/min, Column—1, Aqueous phase 34% and Methanol 66%. (f) Tho chromatogram at a flow rate 1.2 mL/min, Column—1, Aqueous phase 40% and Methanol 60%, Day—2. (g) Tho chromatogram at a flow rate 1.2 mL/min, Column—2, Aqueous phase 40% and Methanol 60%, Day—2. (h) Tho chromatogram at a flow rate 1.2 mL/min, Column—3, Aqueous phase 40% and Methanol 60%, Day—2. (i) Tho chromatogram at a flow rate 1.2 mL/min, Column—1, Aqueous phase 40% and Methanol 60%, Day—3.
Russo et al.28 reported their method for Tho determination earlier, where they used a high flow rate of 2.0 mL/min with 2 columns showing results without any data about the system suitability and a very short introduction to method validation parameters showing a long retention time of 8 min compared to our method. Despite their use of a low concentration in the calibration curve of 1–100 µg/mL. Also, they used two different injection volumes of 20 μL, and 50 μL to achieve high linearity.
Also, many authors introduced their papers to separate and determine the Tho assay in the past two decades, from 1979 to 1998, but the conducted methods were deficient and lacked the implementation of several method validation factors, especially system suitability. With all of these scientific contributions, we can’t deny their efforts. We show a rapid survey so the reader doesn’t get bored. These approaches manifested long at the retention time, absence of tailing factor, column efficiency, theoretical plates, resolution, specificity, recovery, theoretical plates, resolution, specificity, recovery, precision, or use of hazards, chemical reagents, and special conditions. Also, high flow rates, two columns, high or more than one organic modifier, or mobile phase pH adjustment may be used in the separation process, which is not present in our current method39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57.
The HPLC method by Beril et al.3 was introduced using acetonitrile (A), methanol (B), 50 mM pH = 2.7 phosphate buffer (C) as mobile phase in the ratio (40:5:55) of A: B: C (4.00 mL/min). The retention time was introduced as 10.57 (± 0.010) min, LOD and LOQ 0.56, 1.69 ppm with RSD repeatability of 4.98%. The method used a high flow rate and adjusted the buffer solution to more acidic conditions with two different organic modifiers, acetonitrile and methanol. Also, the method did not show any details about the system suitability parameters except for linearity, LOD, LOQ, and repeatability, with very bad repeatability results.
Coppa and his co-workers reported their HPLC method for Tho determination with a very shortfall in analytical method validation parameters, especially in selectivity, specificity, and system suitability. Additionally, they used a tertiary system of mobile phase consisting of two organic modifiers, methanol and acetonitrile, with a flow rate of 1.5 mL/min to get a retention time of about 9.5 min which is time-consuming compared with our method23.
Recently, Elmansi et al.25 reported a new method for Atracurium and Thiopental by HPLC/ UV method using the factorial model in one chromatogram where Tho LOD was 0.02 μg/mL. However, from our point of view, this method requires special conditions, such as using a high ratio of methanol to 80% of the mobile phase and needing to adjust the phosphate buffer to pH 6.5. Also, the method uses a high content of methanol as a solvent and further dilution to prepare the calibration solution. In addition, this method lacked the application of many parameters of the method validation, such as system suitability parameters such as tailing or symmetry factor, theoretical plates, RSD%, accuracy and recovery ruggedness, and specificity. Colatutto et al.20 also didn’t introduce satisfactory parameters for the method validation and didn’t mention a full analysis of the method parameters of the HPLC system in their paper.
Range and linearity
The main aim of our study is the Tho assay determination in the lab in the pharmaceutical dosage form. The results manifested high linearity with R2 = 0.9997 at the working concentrations in the range (20–150%) as we can see in Fig. 3 and Table 2.

Linearity calibration curve of Tho.
LOD and LOQ
LOD and LOQ limits could be determined simply using the linearity calibration data of Tho. LOD was found to be 14.4 µg/mL whereas LOQ was 43.6 µg/mL.
Accuracy and recovery
Table 3 showed that the accuracy results of the tested range (70–120% of the target concentration of 100% (500 µg/mL) were found to be within the acceptance criteria (98–102%)5,6,11,12,13,14,34,35. Compared with Russo et al.28, our method manifested high accuracy and recovery percentages of the intended method, whereas their method showed 87.9 ± 6.2% recovery with a poor percentage of 93.2% at 50 µg/mL.
Repeatability and precision
The RSD% of peak areas was used for judgment on the repeatability of the analyte using six different preparations at the same target at 500 µg/mL concentration as in Tables 4 and 5. It was found to be 0.39% and 0.34% within the intra–precision and 0.92% of inter–precision as it demanded in the repeatability requirements as RSD% < 2.0%5,6,11,12,13,14,34,35.
Robustness
The results of conscious small changes included a flow rate ± 0.1 mL/min and methanol (± 2.5%) were determined using RSD%. The RSD% was found to be < 2% in all cases, as shown in Tables 6 and 7. It is clear for man there is a reverse proportion between the retention time and the ratio of the organic modifier in the methanol. Where the retention time increases by decreasing the organic ratio, and vice versa. This assures the principle chromatographic rule “likes dissolve likes or likes attract likes”11,13,34,35.
Ruggedness
The results of conscious, major, and observable changes include day-to-day and column-to-column. The data was presented as shown in Tables 8 and 9. RSD% was found to be < 2% in all cases11,12,13.
Stability of sample solution (at the HPLC rack)
The experimental results revealed that the tested solution of standard Tho could be given repeatable and precise peak response data over 48 h at room temperature, as in Table 10. Our investigation relegated the conclusion that the Tho is stable only at the special condition of − 20 °C as Anilanmert et al., conducted22 where they reported that Tho is not stable at room temperature or in a refrigerator. Also, Russo et al.28 showed very little solution stability at 27 °C, of Tho where they reported that the recovery percentage range from 5 to 75 μg/mL over 2 h was 42.2 ± 4.7% to 94.5 ± 2.7%.
Specificity and selectivity
The current method supplied us with highly specific data about the resolution and separation performance of the adjacent co-eluted peaks for the Tho principle peak with a resolution parameter of at least 2.77 as in Table 11 and Fig. 4a–c. It was observed that the acid hydrolysis on the Tho solution could not be applied, and a white precipitate was formed at the time of the addition without any signal response of the Tho after injection of the filtrate after centrifugation.

(a) Forced degradation using base hydrolysis 0.1 M NaOH for 30 min. (b) Forced degradation using H2O2 3% w/v hydrolysis for 30 min. (c) Light forced degradation after 3 h.
Test of the validated method
Tho analysis of the different commercial dosage forms in the Egyptian local market
The Tho assay results of Thiopental UP Pharma 500 mg vials, and Thiopental Eipico 1.0 g vials revealed good results, manifested in the following Table 12. Where the method was found to be selective, specific, and resoluted for the Tho peak.
Thiopental UP pharma 500 mg vials batch number (220741) after the constitution stability studies
The tabulated results in Table 13 confirmed the stability and validity of the use of the Tho solutions after constitution using WFI, saline 0.9% wt/v, and glucose 5% wt/v at 30 ± 2 °C and 5 ± 3 °C for 6 h and 24 h, respectively. Where the assay percentage was found to be within the acceptance criteria and did not exceed 3.0% from the starting assay at zero time. Also, the results manifested that the method did not affect the composition of the different initiators of the solvent on the retention time over the study.
Conclusion
The method was found to be accurate from concentration levels 350–600 µg/mL with high accuracy (100.4–101.4%), precise and repeatable for the same analyst over three days with intra–precision of 0.31%, inter–precision in the range 0.36–0.47% and inter-precision between two analysts of 0.16%. The linearity of the method was conducted in the range of 100–750 µg/mL with an excellent regression coefficient R2 = 0.9997. It proves its high capability to achieve the requirements of the chromatographic system suitability as follows: theoretical plates and column efficiency as 3412–5962, USP tailing at 0.96–1.07, and the RSD% of the peak areas at 0.05–0.74%. Finally, the selectivity and specificity of the current method were confirmed by realizing the minimum resolution between the thiopental principal peak and the most adjacent related impurity peak at 2.77. The validated method proved its performance capability in the separation of the thiopental principle peak from any other appearance-forced degradation peaks. The method revealed a wide accepted retention time range of 4.3–6.7 min, with an optimum retention time of 4.6 ± 0.3 min.
Data availability
All data generated or analyzed during this study are included in this article.
References
McLeish, E. Thiopental sodium for injection. Br. Pharmacopoeia 3, 1412–1413 (2022).
Sanganalmath, P. U., Nagaraju, P. M. & Mohan, B. M. HPTLC method for the assay of thiopental in post-mortem blood in a fatal case of suicide. J. Pharm. Biomed. Anal. 80, 89–93 (2013).
Beril, A., Ibrahim, N., Ibrahim, A. & Munevver, A. Fast chromatographic screening method for 7 drugs of potential threat in drug facilitated crimes. Pak. J. Pharm. Sci. 31, 231–235 (2018).
Saddik, M. S. et al. A novel C@Fe@Cu nanocomposite loaded with doxorubicin tailored for the treatment of hepatocellular carcinoma. Pharmaceutics 14(9), 1845 (2022).
Al-Hakkani, M. F., Gouda, G. A., Hassan, S. H. A., Mohamed, M. M. A. & Nagiub, A. M. Environmentally azithromycin pharmaceutical wastewater management and synergetic biocompatible approaches of loaded azithromycin@hematite nanoparticles. Sci. Rep. 12, 10970 (2022).
Al-Hakkani, M. F. et al. Cefotaxime removal enhancement via bio- nanophotocatalyst α-Fe2O3 using photocatalytic degradation technique and its echo-biomedical applications. Sci. Rep. 12, 11881 (2022).
Saddik, M. S. et al. Tailoring of novel azithromycin-loaded zinc oxide nanoparticles for wound healing. Pharmaceutics 14(1), 111 (2022).
Al-Hakkani, M. F., Hassan, S. H. A., Saddik, M. S., El-Mokhtar, M. A. & Al-Shelkamy, S. A. Bioengineering, characterization, and biological activities of C@Cu2O@Cu nanocomposite based-mediated the Vicia faba seeds aqueous extract. J. Mater. Res. Technol. 14(5), 1998–2016 (2021).
Al-Hakkani, M. F., Gouda, G. A. & Hassan, S. H. A. A review of green methods for phytofabrication of hematite (α-Fe2O3) nanoparticles and their characterization, properties, and applications. Heliyon 7(1), e05806 (2021).
Al-Hakkani, M. F. Biogenic copper nanoparticles and their applications: A review. SN Appl. Sci. 2(3), 505 (2020).
Al-Hakkani, M. F. A rapid, developed and validated RP-HPLC method for determination of azithromycin. SN Appl. Sci. 1(3), 222 (2019).
Al-Hakkani, M. F. Guideline of inductively coupled plasma mass spectrometry “ICP–MS”: Fundamentals, practices, determination of the limits, quality control, and method validation parameters. SN Appl. Sci. 1(7), 791 (2019).
Al-Hakkani, M. F. Forced degradation study with a developed and validated RP-HPLC method for determination of cefpodoxime proxetil in the bulk and finished pharmaceutical products. J. Iran. Chem. Soc. 16(7), 1571–1578 (2019).
Al-Hakkani, M. F., Gouda, G. A., Hassan, S. H. A., Mohamed, M. M. A. & Nagiub, A. M. Cefixime wastewater management via bioengineered Hematite nanoparticles and the in-vitro synergetic potential multifunction activities of Cefixime@Hematite nanosystem. Surf. Interfaces 30, 101877 (2022).
Kartanas, T. et al. Label-free protein analysis using liquid chromatography with gravimetric detection. Anal. Chem. 93(5), 2848–2853 (2021).
Najafi, M. Determination of thiopental in aqueous media using catechol electrocatalytic properties. J. Adv. Def. Sci. 5(3), 179–185 (2014).
Ali, A. M., Farghaly, O. A. & Ghandour, M. Determination of thiopentone sodium in aqueous and biological media by cathodic stripping voltammetry. Anal. Chim. Acta 412(1–2), 99–110 (2000).
Van Hamme, M. & Ghoneim, M. A sensitive gas chromatograph assay for thiopentone in plasma. Br. J. Anaesth. 50(2), 143–145 (1978).
Külpmann, W. Gas-chromatographic method for the determination of thiopental in serum. Z. Anal. Chem. 311(4), 409–409 (1982).
Colatutto, A. et al. Comparison between a high-perfomance liquid chromatographic method and a gas chromatographic method for the quantification of serum thiopental for the diagnosis of brain death in critically ill patients. Riv. Ital. Med. Lab. J. 8(3), 162–167 (2012).
Adamowicz, P. & Kała, M. Simultaneous screening for and determination of 128 date-rape drugs in urine by gas chromatography–electron ionization-mass spectrometry. Forens. Sci. Int. 198(1), 39–45 (2010).
Anilanmert, B. et al. Simultaneous analysis method for GHB, ketamine, norketamine, phenobarbital, thiopental, zolpidem, zopiclone and phenytoin in urine, using C18 poroshell column. J. Chromatogr. B 1022, 230–241 (2016).
Coppa, G. et al. Fast, simple and cost-effective determination of thiopental in human plasma by a new HPLC technique. Clin. Chim. Acta 305(1), 41–45 (2001).
Hosotsubo, H. Determination of thiopental in human serum and plasma by reversed-phase high-performance liquid chromatography. Chromatographia 25(10), 887–890 (1988).
Elmansi, H., Elsayed, N. & Belal, F. Novel approach for chromatographic separation of atracurium and thiopental using factorial model. Microchem. J. 170, 106751 (2021).
Najafi, M. & Pashabadi, A. Ultrasound-assisted surfactant-enhanced emulsification microextraction-HPLC determination of low-levels of thiopental in serum and urine samples. J. Rep. Pharm. Sci. 4(1), 30 (2015).
Zakrzewski, R. & Ciesielski, W. Determination of thiopental in urine sample with high-performance liquid chromatography using iodine–azide reaction as a postcolumn detection system. J. Chromatogr. B 824(1), 327–332 (2005).
Russo, H., Allaz, J. L. & Bressolle, F. High-performance liquid chromatographic assay for thiopental in human plasma: Application to pharmacokinetic studies. J. Chromatogr. B: Biomed. Sci. Appl. 694(1), 239–245 (1997).
Abdelaleem, E. A., Naguib, I. A., Zaazaa, H. E. & Hussein, E. A. Development and validation of HPLC and HPTLC methods for determination of cefoperazone and its related impurities. J. Chromatogr. Sci. 54(2), 179–186 (2015).
Abd-El-Aziz-Shama, S., Abd-El-Azim, S., Elham, A. & Shaimaa, H. N. A Simultaneous, validated RP-HPLC method for determination of eight cephalosporins in pharmaceutical formulations. Syst. Rev. Pharm. J. 12(2), 1–12 (2021).
Al-Hakkani, M. F., Gouda, G. A., Hassan, S. H. A. & Nagiub, A. M. Echinacea purpurea mediated hematite nanoparticles (α-HNPs) biofabrication, characterization, physicochemical properties, and its in-vitro biocompatibility evaluation. Surf. Interfaces 24, 101113 (2021).
Saddik, M. S. et al. Biosynthesis, characterization, and wound-healing activity of phenytoin-loaded copper nanoparticles. AAPS Pharm. Sci. Tech. 21(5), 1–12 (2020).
Saddik, M. S. et al. Novel green biosynthesis of 5-fluorouracil chromium nanoparticles using harpullia pendula extract for treatment of colorectal cancer. Pharmaceutics 13(2), 226 (2021).
Al-Hakkani, M. F., Gouda, G. A., Hassan, S. H. A., Farghaly, O. A. & Mohamed, M. M. A. Fully investigation of RP- HPLC analytical method validation parameters for determination of Cefixime traces in the different pharmaceutical dosage forms and urine analysis. Acta Pharm. Sci. 59(1), 97–111 (2021).
Al-Hakkani, M. F. HPLC analytical method validation for determination of Cefotaxime in the bulk and finished pharmaceutical dosage form. Sustain. Chem. Eng. 1, 33–42 (2020).
Davoudi, S. & Marahel, F. Determination of sulfacetamide in blood and urine using PBS quantum dots sensor and artificial neural networks. J. Anal. Chem. 77(11), 1448–1457 (2022).
Karsakova, I. V., Frolova, A. I., Tikhomirova, T. I. & Tsizin, G. I. Determination of phenols by liquid chromatography-mass spectrometry with sorption preconcentration on magnetic hydrophobized silica. J. Anal. Chem. 77(12), 1540–1546 (2022).
Leninskii, M. A., Savelieva, E. I., Karakashev, G. V., Vasilieva, I. A. & Samchenko, N. A. Determination of the conversion products of toxic organophosphorus substances in construction materials using high-performance liquid chromatography with tandem mass spectrometry. J. Anal. Chem. 77(13), 1694–1704 (2022).
King, D., Stewart, J. & Venkateshwaran, T. HPLC determination of propofol-thiopental sodium and propofol-ondansetron mixtures. J. Liq. Chromatogr. Relat. Technol. 19(14), 2285–2294 (1996).
Kuroda, N., Inoue, K., Mayahara, K., Nakashima, K. & Akiyama, S. Application of 3-(1, 8-naphthalimido) propyl-modified silyl silica gel as a stationary phase in high performanc liquid chromatography of barbiturates and diastereomeric compounds. J. Liq. Chromatogr. Relat. Technol. 19(17–18), 2867–2881 (1996).
Kakinohana, O. & Okuda, Y. The plasma concentration measurement of local anesthetics, thiobarbiturates and mexiletine by HPLC with automatic pre-treatment system. Jpn. J. Anesthesiol. 44(8), 1165–1170 (1995).
Meier, P. & Thormann, W. Determination of thiopental in human serum and plasma by high-performance capillary electrophoresis—micellar electrokinetic chromatography. J. Chromatogr. A 559(1–2), 505–513 (1991).
Schmid, R. W. & Wolf, C. Simultaneous determination of thiopental and its metabolite, pentobarbital, in blood by high-performance liquid chromatography and post-column photochemical reaction. J. Pharm. Biomed. Anal. 7(12), 1749–1755 (1989).
Mangin, P., Lugnier, A. & Chaumont, A. A polyvalent method using HPLC for screening and quantification of 12 common barbiturates in various biological materials. J. Anal. Toxicol. 11(1), 27–30 (1987).
Gruhl, H. & Mayer, H. Simple and rapid determination of thiopental in serum by HPLC. J. Clin. Chem. Clin. Biochem. 22(5), 385–388 (1984).
Nicot, G., Lachatre, G., Valette, J., Merle, L. & Nouaille, Y. Dosage plasmatique du phénobarbital, pentobarbital et penthiobarbital par chromatographie liquide à haute performance. Therapie 39(4), 361–367 (1984).
Sharman, J. R. & Ahern, K. M. A simple method for determining thiopental in plasma. J. Anal. Toxicol. 7(1), 37–39 (1983).
Sauerbrey, R. & Reiber, H. Analysis of thiopental in serum and cerebrospinal fluid by HPLC. Fresenius’ J. Anal. Chem. 311(4), 412–412 (1982).
Christensen, J. H. & Andreasen, F. Determination of thiopental by high pressure liquid chromatography. Acta Pharmacol. Toxicol. 44(4), 260–263 (1979).
Hannak, D., Scharbert, F. & Kattermann, R. Stepwise binary gradient high-performance liquid chromatographic system for routine drug monitoring. J. Chromatogr. A 728(1–2), 307–310 (1996).
Celardo, A. & Bonati, M. Determination of thiopental measured in human blood by reversed-phase high-performance liquid chromatography. J. Chromatogr. B: Biomed. Sci. Appl. 527, 220–225 (1990).
Marty, P., Arnoux, P. & Cano, J. Micromethod for determination of thiopental in biological fluids by high performance liquid chromatography. Application to therapeutic follow-up and pharmacokinetic study. Anal. Lett. 21(12), 2241–2252 (1988).
Avram, M. J. & Krejcie, T. C. Determination of sodium pentobarbital and either sodium methohexital or sodium thiopental in plasma by high-performance liquid chromatography with ultraviolet detection. J. Chromatogr. B: Biomed. Sci. Appl. 414, 484–491 (1987).
Houdret, N., Lhermitte, M., Lalau, G., Izydorczak, J. & Roussel, P. Determination of thiopental and pentobarbital in plasma using high-performance liquid chromatography. J. Chromatogr. B: Biomed. Sci. Appl. 343, 437–442 (1985).
Björkman, S. & Idvall, J. A high-performance liquid chromatographic method for methohexital and thiopental in plasma or whole blood. J. Chromatogr. B: Biomed. Sci. Appl. 307, 481–487 (1984).
Premel-Cabic, A., Turcant, A., Cailleux, A. & Allain, P. Micromethod for determination of thiopental in human plasma by high-performance liquid chromatography. J. Chromatogr. B: Biomed. Sci. Appl. 276, 451–455 (1983).
Shiu, G. K. & Nemoto, E. M. Simple, rapid and sensitive reversed-phase high-performance liquid chromatographic method for thiopental and pentobarbital determination in plasma and brain tissue. J. Chromatogr. B: Biomed. Sci. Appl. 227(1), 207–212 (1982).
Acknowledgements
The corresponding author gratefully acknowledges UP Pharma Industrial for its valuable support.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). UP Pharma Industrial is the resource of raw materials, chemicals, working standards, and instrumental analysis.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.F.A. and M.H.A.H. Methodology, software: M.F.A. and N.A. Data curation: M.F.A. and M.H.A.H. Writing—original draft preparation: M.F.A. and M.H.A.H. Visualization, investigation: M.F.A., N.A. Supervision: M.F.A. and M.H.A.H. Software, validation: M.F.A. and M.H.A.H. Writing—reviewing and editing: M.F.A. and M.H.A.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Al-Hakkani, M.F., Ahmed, N. & Hassan, M.H.A. Rapidly, sensitive quantitative assessment of thiopental via forced stability indicating validated RP-HPLC method and its in-use stability activities. Sci Rep 13, 10294 (2023). https://doi.org/10.1038/s41598-023-37329-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-37329-0
This article is cited by
-
Biosynthesis approach of copper nanoparticles, physicochemical characterization, cefixime wastewater treatment, and antibacterial activities
BMC Chemistry (2023)
-
Cefoperazone rapidly and sensitive quantitative assessment via a validated RP-HPLC method for different dosage forms, in-use stability, and antimicrobial activities
BMC Chemistry (2023)
-
Biosynthesis approach of zinc oxide nanoparticles for aqueous phosphorous removal: physicochemical properties and antibacterial activities
BMC Chemistry (2023)
-
A new validated facile HPLC analysis method to determine methylprednisolone including its derivatives and practical application
Scientific Reports (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
