
Abstract
Crystal chemical design principles were applied to synthesise novel U4+ dominant and titanium excess betafite phases Ca1.15(5)U0.56(4)Zr0.17(2)Ti2.19(2)O7 and Ca1.10(4)U0.68(4)Zr0.15(3)Ti2.12(2)O7, in high yield (85–95 wt%), and ceramic density reaching 99% of theoretical. Substitution of Ti on the A-site of the pyrochlore structure, in excess of full B-site occupancy, enabled the radius ratio (rA/rB = 1.69) to be tuned into the pyrochlore stability field, approximately 1.48 ≲ rA/rB ≲ 1.78, in contrast to the archetype composition CaUTi2O7 (rA/rB = 1.75). U L3-edge XANES and U 4f7/2 and U 4f5/2 XPS data evidenced U4+ as the dominant speciation, consistent with the determined chemical compositions. The new betafite phases, and further analysis reported herein, point to a wider family of actinide betafite pyrochlores that could be stabilised by application of the underlying crystal chemical principle applied here.
Similar content being viewed by others

Introduction
Ceramic materials are considered as leading candidate wasteforms for the immobilisation and geological disposal of long lived actinides arising from nuclear fuel cycles and medical radioisotope production1,2,3,4,5,6,7. Titanate ceramics with the pyrochlore structure are of particular interest for such applications, given the long term stability of natural mineral analogues with substantial uranium or thorium inventory1,2,4,8,9. Pyrochlore-group minerals with the prototypical formula of A2-mB2O6(O,OH,F)1−n, comprise three subgroups (pyrochlore, microlite and betafite) classified according to B-site composition; the betafite subgroup is defined as having 2 TiB ≥ (Nb + Ta)B10. Naturally occurring minerals of the pyrochlore group have been shown to be stable under environmental conditions retaining actinides effectively over geological time periods, in excess of 1 billion years, much longer than the performance period of a geological disposal facility11,12,13.
Taking the simplified formula A2B2O6O’, the pyrochlore structure may be described as interpenetrating B2O6 and (anti-cristobalite) A2O’ networks, with corner sharing BO6 octahedra and distorted AO8 scalenohedra. The pyrochlore structure is related to the fluorite structure (comparable formula A2B2O8), by ordering of both cations and oxygen vacancies, leading to a 2 × 2 × 2 superstructure, relative to the fluorite unit cell (ap = 2 af). The pyrochlore structure is stabilised, under ambient conditions, within the approximate radius ratio range: 1.46 ≲ rA/rB ≲ 1.78: below this threshold, a defect fluorite phase is stabilised, with cation and oxygen vacancy disorder; whereas, above the threshold, a monoclinic structure is stabilised, typified by La2Ti2O78,14.
The archetype betafite CaUTi2O7 is of specific interest as ceramic wasteform for actinide disposition, it is a component of the multiphase Synroc F wasteform and a ceramic phase assemblage designed to immobilise U-rich waste from 99Tc production4,15,16,17,18,19. There is a consensus that solid state synthesis of near single phase CaUTi2O7, with ≳ 95 wt% yield, is problematic16,17,18,19. Dickson et al. noted that CaUTi2O7 “invariably coexisted with substantial portions of perovskite (CaTiO3) and uraninite (UO2)”16; and Vance et al. reported “several days failed to assure complete reaction… and the pyrochlore yields did not exceed ~ 75 wt%”18. These results are perhaps not altogether surprising given, that the radius ratio of CaUTi2O7, rA/rB = 1.75, is on the cusp of the pyrochlore stability field (herein, Shannon’s effective ionic radii are employed20). Interestingly, the radius ratio may be tuned into the stability field of the pyrochlore structure by oxidation of U4+ to U5+/U6+, with coupled charge substitution, for example: Ca1.4U0.7Ti2O7, with U4.5+ and rA/rB = 1.7121. However, U4+ is the preferred speciation for wasteform applications, given the lower solubility and compatibility with reducing groundwaters, expected at depth, in a geological disposal facility. Vanderah et al. established that the pyrochlore structure may be stabilised for relatively large A-site cations, by substitution on the A-site of typical B-site cations, in excess of full B-site occupancy; remarkably, up to 25% substitution on the A-site may be tolerated22. We therefore applied this crystal chemical design principle to hypothesise novel U4+ dominant, and titanium excess, betafite compositions with radius ratio, rA/rB = 1.69, within the pyrochlore stability field, nominally Ca1.00U0.50Zr0.20Ti2.30O7 and Ca0.96U0.72Zr0.17Ti2.15O7. Herein, we report the successful synthesis and characterisation of such betafite compounds. Our hypothesis was guided by the observation of a nominally B-site stoichiometric pyrochlore phase by Vance et al. in the zirconolite solid solution of CaZr1−xUxTi2O7 with x = 0.7, apparently co-existing with a zirconolite 4M phase18. The novel betafite phases designed and reported herein, point to a wider family of actinide pyrochlores that could be stabilised by application of the same crystal chemical principle, which we hope will be more extensively investigated.
Results and discussion
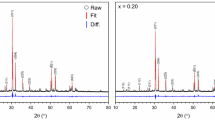
Powder X-ray diffraction (PXRD) analysis of the synthesised products demonstrated the formation of pyrochlore structured compounds (space group Fd \(\overline{3 }\) m) with the presence of only minor or trace secondary phases (Fig. 1). The clear presence of (111) and (311) reflections, indexed in Fd \(\overline{3 }\) m, at 2θ ≈ 15° and 2θ ≈ 30°, respectively, was diagnostic of cation and oxygen vacancy ordering, characteristic of a pyrochlore structure. The calculated lattice parameters of nominal Ca1.00U0.50Zr0.20Ti2.30O7 and Ca0.96U0.72Zr0.17Ti2.15O7, a = 10.1215(2) Å and a = 10.1374(1) Å, respectively, were slightly smaller than those of the previously reported betafite phase, Ca0.92U1.08Ti1.99O7, a = 10.1579 Å16. This is in accordance with the comparably greater radius ratio of Ca0.92U1.08Ti1.99O7, rA/rB = 1.74.

PXRD patterns of the product after sintering of (a) nominal Ca1.00U0.50Zr0.20Ti2.30O7, (b) nominal Ca0.96U0.72Zr0.17Ti2.15O7, and (c) 10wt% Fe addition to nominal Ca0.96U0.72Zr0.17Ti2.15O7 (d) 10wt% Ni addition to nominal Ca0.96U0.72Zr0.17Ti2.15O7; compounds were synthesised at 1320 °C in flowing N2. Vertical marks are Bragg reflections determined from the reported structure of Ca0.92U1.08Ti1.99O716.
For the nominal Ca1.00U0.50Zr0.20Ti2.30O7 composition, TiO2 (rutile) and CaTiO3 (perovskite) were detected as the minor phases, as shown in Fig. 1a. Whereas, for the nominal Ca0.96U0.72Zr0.17Ti2.15O7 content, only trace impurities of TiO2 and UTi2O6 (brannerite) were observed, as shown Fig. 1b. These data imply that an intermediate composition between these end members should yield a truly single phase material. Addition of 10 wt%. Fe and Ni was made to the target composition Ca0.96U0.72Zr0.17Ti2.15O7, for the purpose of scavenging potential trace oxygen from the nitrogen gas atmosphere used in synthesis (see "Methods" section). Addition of 10 wt.% Fe to nominal composition Ca0.96U0.72Zr0.17Ti2.15O7, led to the formation of Fe2TiO4 (ulvospinel) in addition to CaTiO3, as shown in Fig. 1c. Addition of 10 wt.% Ni to nominal composition Ca0.96U0.72Zr0.17Ti2.15O7, was found to not influence the phase assemblage, and unreacted Ni metal was retained, Fig. 1d. No free uranium oxides were detected in the XRD of any product, which, together with the well sintered microstructures (see below), suggested that the solid state reactions were not kinetically hindered.
The microstructures of sintered betafite ceramics are shown in Fig. 2, and were fully consistent with the phase assemblage determined from XRD data. The Energy Dispersive X-ray (EDX) determined compositions of constituent phases are presented in Table 1 and Table S1(supplementary material); EDX spectra are presented in Figs. S1–S4.

Scanning Electron Microscopy (SEM) observation in backscattered electron mode of the polished surface (a) nominal Ca1.00U0.50Zr0.20Ti2.30O7, (b) nominal Ca0.96U0.72Zr0.17Ti2.15O7, and (c) 10wt% Fe addition to nominal Ca0.96U0.72Zr0.17Ti2.15O7 (d) 10wt% Ni addition to nominal Ca0.96U0.72Zr0.17Ti2.15O7. The inset of (d) shows the presence of Ni phase in low magnification. Labels highlight: B the betafite major phase, R rutile, P perovskite, Br brannerite, U ulvospinel, and Ni nickel.
Nominal composition Ca1.00U0.50Zr0.20Ti2.30O7 exhibited a dense microstructure, with little porosity observed (Fig. 2a). From greyscale contrast, it was evident that the microstructure comprised three distinct phases. The major phase (labelled B) was identified as betafite, as determined by the coincidence of U, Ca and Ti signals in EDX spectra (Fig. S1). The presence of the Zr Lα emission line at ca. 2 keV was indicative of the solid solution of Zr in the betafite phase of all products. Minor phases were determined to be TiO2 and CaTiO3 (labelled R and P, respectively; EDX spectra presented in Fig. S1). Nominal composition Ca0.96U0.72Zr0.17Ti2.15O7 also presented a dense microstructure, Fig. 2b, that comprised a majority betafite phase with minor TiO2 and UTi2O6 (labelled Br; EDX spectra presented in Fig. S2). The addition of 10wt% Fe was found to have a significant impact on the phase assemblage and microstructure of nominal composition Ca0.96U0.72Zr0.17Ti2.15O7. As shown in Fig. 2c, in addition to a major betafite phase, minor CaTiO3 and Fe2TiO4 (ulvospinel, labelled U) were observed, together with considerable porosity (EDX spectra presented in Fig. S3). 10wt% Ni addition to nominal composition Ca0.96U0.72Zr0.17Ti2.15O7 also produced a dense microstructure, Fig. 2d, that comprised a majority betafite phase with minor TiO2 and UTi2O6, and residual Ni metal (EDX spectra presented in Fig. S4). The observed size of the Ni phase (see inset to Fig. 2d) was consistent with that of the starting Ni metal reagent. This, and the absence of any Ni Kα emission line in the EDX spectra of Fig. S4a–c, demonstrated no detectable reaction of the Ni metal had occurred.
The EDX chemical compositions of the major betafite phases were close to those targeted and evidenced an excess of B-site cations within precision: Ca1.15(5)U0.56(4)Zr0.17(2)Ti2.19(2)O7 and Ca1.10(4)U0.68(4)Zr0.15(3)Ti2.12(2)O7, for nominal Ca1.00U0.50Zr0.20Ti2.30O7 and Ca0.96U0.72Zr0.17Ti2.15O7, respectively (see Table 1). The EDX determined compositions implied average uranium oxidation states of 4.04 + and 4.00 +, respectively, assuming Ti4+ speciation (note: synthesis conditions were not considered sufficiently reducing to afford significant reduction to Ti3+). The composition of the betafite phase in nominal Ca0.96U0.72Zr0.17Ti2.15O7 with 10 wt.% Fe addition was Ca0.90(5)U0.71(5)Zr0.15(2)Ti1.97(3)Fe0.28(6)O7. Evidently, Fe was incorporated into the crystal structure; assuming speciation as Fe3+, the composition implied an average uranium oxidation state of 4.05 +. The chemical composition of the betafite phase in nominal Ca0.96U0.72Zr0.17Ti2.15O7 with 10wt.% Ni addition, was determined to be Ca1.03(2)U0.64(3)Zr0.14(2)Ti2.17(3)O7, with an implied uranium oxidation state of 4.21 + . The EDX determined composition was close to that of the Ni free counterpart composition.
Rietveld analysis of PXRD data was performed to allow quantitative phase analysis (QPA) of the products and the results are summarised in Table 2; target compositions were used as the structural model. The largest fraction of betafite was found for the nominal Ca1.00U0.50Zr0.20Ti2.30O7 composition: betafite—94.58 wt.%, rutile—2.14 wt.%, brannerite—3.29 wt.%; for other compositions, the betafite phase comprised approximately 85 wt.% of the phase assemblage. Combining QPA, EDX analyses, and measured bulk densities, it was possible to estimate the relative density of the ceramic materials, which are presented in Table S2.
The nominal Ca1.00U0.50Zr0.20Ti2.30O7 and Ca0.96U0.72Zr0.17Ti2.15O7 ceramics were estimated to have a relative density of approximately 99% of theoretical, whereas the estimated relative densities of the ceramics with 10wt.% Fe and 10wt.% Ni were somewhat lower. This was consistent with the observation or absence of porosity in the corresponding microstructures shown in Fig. 2 and discussed above. The average bulk uranium oxidation state in the betafite ceramics was investigated by analysis of X-ray absorption near edge structure (XANES) at the U L3-edge; data are presented in Fig. 3. The oxidation state was determined by the linear regression method as has been previously proposed23,24, using the edge position, E0, of reference compounds of known oxidation state to establish a calibration line, as shown in Fig. 4. The bulk average uranium oxidation states determined by this method evidenced the presence of only U4+ in the betafite ceramics, within experimental error, as shown in Table 3). The difference in features of the white line maximum and near-edge structure of the U L3-XANES, for compounds with the same nominal oxidation state shown in Fig. 3, reflect sensitivity to the specific local environment of the U absorber in the reference compounds.

U L3-edge XANES data of (a) nominal Ca1.00U0.50Zr0.20Ti2.30O7, (b) nominal Ca0.96U0.72Zr0.17Ti2.15O7, and (c) 10wt.% Fe addition to nominal Ca0.96U0.72Zr0.17Ti2.15O7 composition (d) 10wt.% Ni addition to nominal Ca0.96U0.72Zr0.17Ti2.15O7 composition. Also shown are spectra of reference compounds: U4+ in UO2, UTi2O6; U5+ in U0.5Y0.5Ti2O6, USbO5, LaUO4, UMo2O8; and U6+ in CaUO4.

The oxidation state as a function of edge position (E0) for uranium compounds together with a model linear fit (the dashed line; R2 = 0.913). Data points correspond to (a) nominal Ca1.00U0.50Zr0.20Ti2.30O7, (b) nominal Ca0.96U0.72Zr0.17Ti2.15O7. Reference compounds represent: U4+ in UO2, UTi2O6; U5+ in U0.5Y0.5Ti2O6, USbO5, LaUO4, UMo2O8; and U6+ in CaUO4.
XANES data were also analysed by combinatorial linear combination fitting (LCF)25,26, using the library of reference compounds to estimate the fraction of contributing oxidation states. Significance tests of the goodness of fit R-factor were undertaken using the Hamilton R-factor ratio test, with a significance level of α = 0.0527. From these fits the weighted mean oxidation state and associated root mean square error approximation were calculated. The plots of best fit are shown in Fig. S5. The results of the combinatorial LCF, summarised in Table 3, also evidenced a dominant average bulk uranium oxidation state of U4+, but with a minor U5+ contribution; no significant U6+ contribution was determined.
The bulk average oxidation state of uranium in nominal Ca1.00U0.50Zr0.20Ti2.30O7 and Ca0.96U0.72Zr0.17Ti2.15O7 was further investigated using X-ray Photoelectron Spectroscopy (XPS). As shown in Fig. 5A, the spectra of both compositions presented two main peaks, U 4f7/2 and U 4f5/2 (separated by ca. 11.0 eV, due to spin–orbit splitting) and two satellite peaks, Sat.7/2 and Sat.5/2. Fitting of Sat.5/2 was used to assess the contributing uranium oxidation states; for both compositions, the Sat.5/2 peak could be fitted by a majority U4+ contribution with a minor U5+ contribution, as shown in Fig. 5B; no contribution from U6+ was apparent. Similarly, deconvolution of the U 4f7/2 peak for nominal Ca1.00U0.50Zr0.20Ti2.30O7 and Ca0.96U0.72Zr0.17Ti2.15O7 evidenced a majority contribution from U4+ and a minor contribution from U5+, as shown in Fig. 6; no U6+ contribution was evidenced. The positions of two major components, with separations of 0.9 eV (U4+—U5+ in the deconvolution of U 4f7/2), are in agreement with those observed in the literature for mixed valence in uraninite28. The fractions of U4+ and U5+, as determined by peak deconvolution of U 4f7/2, are reported in Table 3.

(A) The U 4f7/2 and U 4f5/2 regions of the XPS spectra for (a) nominal Ca1.00U0.50Zr0.20Ti2.30O7 and (b) nominal Ca0.96U0.72Zr0.17Ti2.15O7. (B) Satellite peaks in the range of 393–405 eV with the fitting of the components of U4+ and U5+ for (a) nominal Ca1.00U0.50Zr0.20Ti2.30O7 and (b) nominal Ca0.96U0.72Zr0.17Ti2.15O7.

Deconvolution of the U 4f7/2 photoelectron peaks for (a) nominal Ca1.00U0.50Zr0.20Ti2.30O7 and (b) nominal Ca0.96U0.72Zr0.17Ti2.15O7. The dashed lines are the fitting curves of U4+ and U5+ components. The dotted line and the solid line are the measured data and fitted contributions, respectively. The solid line at the bottom is baseline for the curve fitting.
As can be seen, a greater proportion of U4+ speciation was found in nominal Ca1.00U0.50Zr0.20Ti2.30O7 (91% U4+, 9% U5+) whereas uranium in nominal Ca0.96U0.72Zr0.17Ti2.15O7 was marginally more oxidised (U4+ 88%, U5+ 12%). Overall, the average bulk oxidation states determined from U L3-edge XANES and U 4f7/2 XPS are in good agreement, and evidence dominant U4+ speciation with a minor U5+ contribution of around 10%. These bulk analyses are consistent with the dominant U4+ oxidation state inferred from EDX analyses of the betafite phase, within which the uranium is overwhelmingly partitioned. Our estimation of the U5+ content determined from XPS is based on curve fitting, assuming intrinsically symmetrical U 4f7/2 lines. In reality these lines are slightly asymmetrical (multiplets) and the Shirley background is also only an approximation. These assumptions are expected to result in a small overestimation of the U5+ content. We can therefore conclude that the uranium speciation in the betafite phases is primarily U4+, with a U5+ contribution of no more than 10%.
Subsequent to this study, Blackburn et al. investigated the zirconolite solid solution CaZr1-xThxTi2O7, and discovered the formation of a new betafite phase, for x > 0.4; a single phase was produced for x = 0.6, with a determined composition of Ca1.00(2)Zr0.33(2)Th0.54(1)Ti2.13(2)O729. The radius ratio of this phase is rA / rB = 1.70, within the pyrochlore stability field, and identical to that of the betafite phases designed and reported here. In contrast, McCauley and Hummel, reported synthesis of the end member composition CaThTi2O7, to be unsuccessful30. Indeed, this is consistent with a radius ratio, rA/rB = 1.79, outside of the pyrochlore stability field. Therefore, Ca1.00(2)Zr0.33(2)Th0.54(1)Ti2.13(2)O7 may also be considered an example of a pyrochlore structure stabilised by Ti excess on the B-site and partial occupancy of the A-site. This example, and those reported herein, point to a wider family of actinide pyrochlores that could be stabilised by application of the underlying crystal chemical principle applied here.
Conclusion
Novel U4+ dominant and titanium excess betafite phases, Ca1.15(5)U0.56(4)Zr0.17(2)Ti2.19(2)O7 and Ca1.10(4)U0.68(4)Zr0.15(3)Ti2.12(2)O7, were successfully synthesised in high yield (85 – 95 wt%), by application of the crystal chemical design principle of targeting excess B-site cations to the A-site in the pyrochlore structure. This design strategy enabled the radius ratio to be effectively tuned into the pyrochlore stability field, and the synthesis of U4+ betafite ceramics in high yield, and with high relative density (> 99% theoretical), for the first time. U L3-edge XANES and U 4f7/2 and U 4f5/2 XPS data evidenced U4+ as the dominant speciation, consistent with EDX determined compositions. Reconsideration of the recently reported thorium betafite phase, Ca1.00(2)Zr0.33(2)Th0.54(1)Ti2.13(2)O7 established that this compound is also effectively stabilised by the same crystal chemical design principle applied here. More broadly, this example, and the novel betafite phases designed and reported herein, point to a wider family of actinide pyrochlores that could be stabilised by application of same crystal chemical principle. The observed incorporation of Fe within Ca0.90(5)U0.71(5)Zr0.15(2)Ti1.97(3)Fe0.28(6)O7 demonstrates a further degree of chemical flexibility which could be exploited in terms of this crystal chemical design principle, with partial Ti4+ occupancy of the pyrochlore A-site facilitated by co-substitution on the B-site of a suitable cation.
Methods
Caution
Uranium is an alpha emitter. Manipulations, synthesis and characterisation were performed in a materials radiochemistry laboratory in a controlled area, using HEPA filtered fume hoods and a dedicated glove box, following risk assessments and monitoring procedures31.
Betafite ceramics were produced by solid-state reaction – sintering between stoichiometric quantities of CaTiO3 (Sigma-Aldrich, purity ≥ 99% trace metals basis), ZrO2 (Sigma-Aldrich, purity ≥ 99%) and TiO2 (Sigma-Aldrich, purity ≥ 99%) and UO2 (purity > 99%). UO2 with a small particle size of 1 μm was selected as a reagent, given previous suggestion that pyrochlore synthesis may be kinetically hindered by the use of UO216,18. The target betafite compositions were Ca1.00U0.50Zr0.20Ti2.30O7 and Ca0.96U0.72Zr0.17Ti2.15O7, as discussed in the Introduction section. The mixture of reagents was ball milled for 16 h in high-density-polyethylene pots containing calcium stabilised zirconia media and isopropanol as a carrier fluid. The media were separated from the milled slurry and dried overnight at 90 °C. The master batch of Ca0.96U0.72Zr0.17Ti2.15O7 precursor was divided into three parts; 10 wt% of metallic Fe (Acros Organics, purity ≥ 99%) or Ni (Acros Organics, purity ≥ 99.9%) was added to one part of the precursor, by mixing in a mortar and pestle. These compositions were fabricated to investigate the potential for metallic Fe and Ni to maintain U4+ by scavenging trace oxygen. Batched material was uniaxially pressed in a 10 mm steel die under uniaxial pressure of 180 MPa and sintered at 1320 °C for 2 h, with a ramp rate of 5 °C·min−1, in flowing high purity nitrogen (250 mL·min−1).
The density of the sintered ceramics was measured based on Archimedes displacement method. For phase analysis, sintered ceramics were sectioned using a diamond saw and a small segment was ground to a fine powder in a mortar and pestle. Examination of the phase assemblage was performed by powder X-ray diffraction (PXRD; D2 Phaser, Bruker, Karlsruhe, Germany) with a Cu Kα source, Ni Kβ filter operating voltage of 30 kV and current of 10 mA. Quantitative phase analysis (QPA) was performed by Rietveld refinement using the GSAS software package and the ExpGUI interface32. The microstructure of sintered pellets was examined by Scanning Electron Microscopy (SEM) in backscattered electron mode using a Hitachi TM3030 microscope coupled with a Bruker Quantax 70 EDX system. Samples were prepared for analysis by polishing sections of ceramic to a 0.25 µm finish using SiC paper and progressively finer diamond pastes. Semi-quantitative compositions were acquired by Energy Dispersive X-ray spectroscopy (EDX) based on 10 EDX data points; a stoichiometry of 7 O atoms per formula unit was assumed, given the low accuracy of EDX to light element determination.
Average uranium oxidation states were determined by analysis of U L3-edge X-ray absorption near edge structure (XANES). The ceramic products and reference compounds for XANES measurement were prepared by homogenously mixing powder specimens with polyethylene glycol and uniaxially pressing to form 13 mm diameter pellets of approximately one absorption length. XANES data were acquired on Beamline B18 at Diamond Light Source (DLS; Oxford, UK). The beamline configuration comprised a water cooled vertically collimated Si mirror, a double crystal Si(111) monochromator, a double toroidal focusing mirror, and harmonic rejection mirrors. Uranium L3-edge XANES spectra were recorded in transmission mode between 17,000 and 17,410 eV. To improve data quality, the beam spot size was defocused to ca. 1.0 mm and multiple scans were acquired and averaged. Data reduction and linear combination fitting were performed using the Athena software package25.
X-ray photoelectron spectroscopy (XPS) data of uranium were recorded at room temperature using a SPECS Phoibos 150 hemispherical analyser, using monochromated X-rays (SPECS, microfocus source, 15 kV, 50 W, spot size: 0.3 mm). Samples were glued with 2-component epoxy glue (Dynaloy 325) and stored under vacuum at room temperature for 7 days to allow cure finishing and avoid surface oxidation. Samples were scraped by a diamond file under vacuum (1 × 10−7 mbar) to produce bulk representative surfaces. The energy scale for XPS was calibrated with the Au 4f7/2 (84.0 eV) and Cu 2p3/2 (932.7 eV) emissions. The vacuum in the photoemission chamber were 1.2 × 10−10 mbar. Charge compensation was performed by a flood gun (1 eV, 15 mA). The obtained spectra were deconvoluted using Gaussian function and the baseline subtracted with a Shirley function. The location and full-width at half-maximum (FWHM) of the components were allowed to vary freely, but the width of the components was set to be equal in each fit.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Ewing, R. C. Nuclear waste forms for actinides. Proc. Natl. Acad. Sci. USA 96, 3432–3439 (1999).
Ewing, R. C. Ceramic matrices for plutonium disposition. Prog. Nucl. Energy 49, 635–643 (2007).
Lee, W. E., Ojovan, M. I., Stennett, M. C. & Hyatt, N. C. Immobilisation of radioactive waste in glasses, glass composite materials and ceramics. Adv. Appl. Ceram. 105, 3–12 (2006).
Carter, M. L., Li, H., Zhang, Y., Vance, E. R. & Mitchell, D. R. G. Titanate ceramics for immobilisation of uranium-rich radioactive wastes arising from 99Mo production. J. Nucl. Mater. 384, 322–326 (2009).
Hyatt, N. C. Safe management of the UK separated plutonium inventory: A challenge of materials degradation. npj Mater. Degrad. 4, 28 (2020).
Blackburn, L. R. et al. Review of zirconolite crystal chemistry and aqueous durability. Adv. Appl. Ceram. 120, 69–83 (2021).
Sun, S. K., Bailey, D. J., Gardner, L. J. & Hyatt, N. C. Ceramic-based stabilization/solidification of radioactive waste. In Low Carbon Stabilization and Solidification of Hazardous Wastes (eds Tsang, D. C. W. & Wang, L.) 449–468 (Elsevier, 2022).
Ewing, R. C., Weber, W. J. & Lian, J. Nuclear waste disposal—Pyrochlore (A2B2O7): Nuclear waste form for the immobilization of plutonium and “minor” actinides. J. Appl. Phys. 95, 5949–5971 (2004).
McMaster, S. A., Ram, R., Faris, N. & Pownceby, M. I. Radionuclide disposal using the pyrochlore supergroup of minerals as a host matrix—A review. J. Hazard. Mater. 360, 257–269 (2018).
Hogarth, D. D. Classification and nomenclature of the pyrochlore group. Am. Mineral. 62, 403–410 (1977).
Lumpkin, G. R. & Ewing, R. C. Geochemical alteration of pyrochlore group minerals: Microlite subgroup. Am. Mineral. 77, 179–188 (1992).
Lumpkin, G. R. & Ewing, R. C. Geochemical alteration of pyrochlore group minerals: Pyrochlore subgroup. Am. Mineral. 80, 732–743 (1995).
Lumpkin, G. R. & Ewing, R. C. Geochemical alteration of pyrochlore group minerals; betafite subgroup. Am. Mineral. 81, 1237–1248 (1996).
Reid, D. P., Stennett, M. C. & Hyatt, N. C. The fluorite related modulated structures of the Gd2(Zr2−xCex)O7 solid solution: An analogue for Pu disposition. J. Solid State Chem. 191, 2–9 (2012).
Kesson, S. E. & Ringwood, A. E. Safe disposal of spent nuclear fuel. Radioact. Waste Manag. Nucl. Fuel Cycle 4, 159–174 (1983).
Dickson, F. J., Hawkins, K. D. & White, T. J. Calcium uranium titanate—A new pyrochlore. J. Solid State Chem. 82, 146–150 (1989).
Matzke, H.-J. et al. Incorporation of transuranic elements in titanate nuclear waste ceramics. J. Am. Ceram. Soc. 73, 370–378 (1990).
Vance, E. R. et al. Incorporation of uranium in zirconolite (CaZrTi2O7). J. Am. Ceram. Soc. 85, 1853–1859 (2002).
Vance, E. R., Carter, M. L., Moricca, S. & Eddowes, T. L. Titanate ceramics for immobilization of U-rich waste. In Environmental Issues and Waste Management Technologies in the Ceramic and Nuclear Industries X Vol. 168 (eds Vienna, J. et al.) 225–232 (Wiley, 2005).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A32, 751–767 (1976).
James, M. et al. Crystal chemistry and structures of (Ca, U) titanate pyrochlores. J. Am. Ceram. Soc. 93, 3464–3473 (2010).
Vanderah, T. A., Levin, I. & Lufaso, M. W. An unexpected crystal-chemical principle for the pyrochlore structure. Eur. J. Inorg. Chem. 2005, 2895–2901 (2005).
Colella, M., Lumpkin, G. R., Zhang, Z., Buck, E. C. & Smith, K. L. Determination of the uranium valence state in the brannerite structure using EELS, XPS, and EDX. Phys. Chem. Miner. 32, 52–64 (2005).
Kosog, B., La Pierre, H. S., Denecke, M. A., Heinemann, F. W. & Meyer, K. Oxidation state delineation via U LIII-edge XANES in a series of isostructural uranium coordination complexes. Inorg. Chem. 51, 7940–7944 (2012).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Bès, R. et al. Use of HERFD–XANES at the U L3- and M4-edges to determine the uranium valence state on [Ni(H2O)4]3[U(OH,H2O)(UO2)8O12(OH)3]. Inorg. Chem. 55, 4260–4270 (2016).
Hamilton, W. Significance tests on the crystallographic R factor. Acta Crystallogr. 18, 502–510 (1965).
Schindler, M., Hawthorne, F. C., Freund, M. S. & Burns, P. C. XPS spectra of uranyl minerals and synthetic uranyl compounds. I: The U 4f spectrum. Geochim. Cosmochim. Acta 73, 2471–2487 (2009).
Blackburn, L. R. et al. Synthesis, structure, and characterization of the thorium zirconolite CaZr1-xThxTi2O7 system. J. Am. Ceram. Soc. 104, 2937–2951 (2021).
McCauley, R. A. & Hummel, F. A. New pyrochlores of the charge-coupled type. J. Solid State Chem. 33, 99–105 (1980).
Hyatt, N. C. et al. The HADES facility for high activity decommissioning engineering and science: Part of the UK national nuclear user facility. IOP Conf. Ser. Mater. Sci. Eng. 818, 012022 (2020).
Toby, B. EXPGUI, a graphical user interface for GSAS. J. Appl. Crystallogr. 34, 210–213 (2001).
Acknowledgements
NCH, SKS, CLC and LMM are grateful for financial support from the Nuclear Decommissioning Authority and EPSRC under grant numbers EP/M026566/1, EP/S01019X/1, EP/N017870/1, EP/R511754/1 and EP/N017374/1. SKS acknowledge the fund from NSFC (52172064). LMM acknowledge the studentships from EPSRC (EP/R513313/1 and EP/N509735/1). This research utilised the MIDAS/HADES facility at The University of Sheffield established with financial support from EPSRC and BEIS, under grant EP/T011424/1. We acknowledge Diamond Light Source for allocation of beam-time at Beamline B18 (Proposal SP17782 and SP24074-1). XPS measurement was performed in European Commission, Joint Research Centre (JRC), Institute for Transuranium Elements (ITU).
Author information
Authors and Affiliations
Contributions
S.K.S.: Investigation, formal analysis, writing—original draft, review and editing. L.M.M.: Software, data curation, writing—original draft. T.G.: investigation, writing—original draft, review and editing. M.C.A.: Methodology, data curation, writing—review and editing. N.C.H.: Supervision, project administration, resources, funding acquisition, methodology, investigation, writing—original draft, review and editing. C.L.C.: Writing—review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sun, SK., Mottram, L.M., Gouder, T. et al. Crystal chemical design, synthesis and characterisation of U(IV)-dominant betafite phases for actinide immobilisation. Sci Rep 13, 10328 (2023). https://doi.org/10.1038/s41598-023-36571-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-36571-w
This article is cited by
-
Long-term, sustainable solutions to radioactive waste management
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
