
Abstract
Indium (In) is a neutron absorbing additive that could feasibly be used to mitigate criticality in ceramic wasteforms containing Pu in the immobilised form, for which zirconolite (nominally CaZrTi2O7) is a candidate host phase. Herein, the solid solutions Ca1-xZr1-xIn2xTi2O7 (0.10 ≤ x ≤ 1.00; air synthesis) and Ca1-xUxZrTi2-2xIn2xO7 (x = 0.05, 0.10; air and argon synthesis) were investigated by conventional solid state sintering at a temperature of 1350 °C maintained for 20 h, with a view to characterise In3+ substitution behaviour in the zirconolite phase across the Ca2+, Zr4+ and Ti4+ sites. When targeting Ca1-xZr1-xIn2xTi2O7, single phase zirconolite-2M was formed at In concentrations of 0.10 ≤ x ≤ 0.20; beyond x ≥ 0.20, a number of secondary In-containing phases were stabilised. Zirconolite-2M remained a constituent of the phase assemblage up to a concentration of x = 0.80, albeit at relatively low concentration beyond x ≥ 0.40. It was not possible to synthesise the In2Ti2O7 end member compound using a solid state route. Analysis of the In K-edge XANES spectra in the single phase zirconolite-2M compounds confirmed that the In inventory was speciated as trivalent In3+, consistent with targeted oxidation state. However, fitting of the EXAFS region using the zirconolite-2M structural model was consistent with In3+ cations accommodated within the Ti4+ site, contrary to the targeted substitution scheme. When deploying U as a surrogate for immobilised Pu in the Ca1-xUxZrTi2-2xIn2xO7 solid solution, it was demonstrated that, for both x = 0.05 and 0.10, In3+ was successfully able to stabilise zirconolite-2M when U was distributed predominantly as both U4+ and average U5+, when synthesised under argon and air, respectively, determined by U L3-edge XANES analysis.
Similar content being viewed by others


Introduction
Zirconolite (ideally monoclinic CaZrTi2O7; space group C2/c; Z = 8) is a candidate wasteform material for the immobilisation of actinides arising from reprocessing of spent nuclear fuel (SNF), such as U and Pu1,2,3,4. The United Kingdom inventory of separated Pu is forecast to reach approximately 140 teHM (tonnes equivalent heavy metal) upon completion of the ongoing reprocessing campaign, which terminated in July 2022. At present, the UK policy favours reuse; material not suitable for this purpose should be immobilised to place beyond reach5. Should this policy not prove implementable, the technology to immobilize and dispose of the inventory would be required6. At present, the most technically feasible immobilisation pathway would see the conversion of the bulk inventory into solid ceramic monoliths, produced either by a campaign of conventional cold-press sintering (CPS) or hot isostatic pressing (HIP), prior to disposal in a geological disposal facility (GDF)7,8.
It may be desirable for wasteforms containing a high fissile content to incorporate a suitable quantity of neutron absorbing additives to mitigate the potential for a criticality event to occur9,10. The role of the neutron absorber within the wasteform is to ensure that the probability of a criticality event is mitigated by reducing the internal neutron flux via absorption. Accordingly, during wasteform development and composition scoping trails, during which potential solid solution regimes between the actinide portion and the host material are devised and characterised, it is desirable to ensure that sufficient concentrations of neutron absorbers can be co-accommodated. At present, a range of such additives and their respective incorporation mechanisms for zirconolite have been devised, mainly deploying Gd3+ and/or Hf4+2,11,12,13,14,15. Whilst Gd3+ is typically accommodated over the Ca2+ and/or Zr4+ sites, requiring charge balance via a charge balancing species typically substituted for Ti4+ e.g. Ca1-xZr1-xGd2xTi2O7 or Ca1-xGdxZrTi2-xAlxO7, Hf4+ can be directly substituted for Zr4+ on the basis of near identical ionic radii (0.78 and 0.76 Å in sevenfold coordination, respectively), resulting in a very minor variation in unit cell volume16. However, there are a range of relatively uninvestigated additives that have been proposed as neutron poisons, such as Sm, Dy, Cd, B and In. At present, there is little to no reported data of In, Cd or B substitution within zirconolite and related titanate phases.
The incorporation of In3+ within zirconolite was reported by Begg et al., in which In3+ was deployed as a Ti3+ simulant17. A solid solution targeting CaZrTi2-xInxO7-2/x was fabricated by a cold press and sinter route, with a 1400 °C sintering temperature maintained for 20 h in air, with In3+ introduced as In2O3, targeting x = 0.25, 0.50 and 1.00. It was observed that zirconolite became unstable relative to perovskite and fluorite with increased substitution of In, with a zirconolite yield of 90%, 65% and 0% observed for x = 0.25, 0.50 and 1.00, respectively. It should be noted that In3+ substitution in the above solid solution was not charge balanced, with In3+ included to simulate the effects of Ti3+ ingrowth under reducing conditions rather than as a neutron poison. Another reported example of In-substitution in a zirconolite-like structure was KIn0.33Ti0.67Te2O7 fabricated by Lee et al., however this structure was shown to crystallise with orthorhombic unit cell symmetry in the space group Cmcm18. This material was reported to form a 3D framework in which each In3+ cation was bonded to 6 O atoms in octahedral coordination, again indicating that In3+ cations may preferentially occupy the Ti4+ sites in the CaZrTi2O7 structure. Herein, two novel zirconolite systems were investigated, the first of which was Ca1-xZr1-xIn2xTi2O7, whereby In3+ was targeted across both Ca2+ and Zr4+ cation sites in equimolar quantity, resulting in a self-charge balancing solid solution. As will later be discussed, the first part of this study demonstrated that In3+ preferentially occupied the Ti4+ site in zirconolite; hence, the Ca1-xUxZrTi2-2xIn2xO7 solid solution was also investigated, processed under both Ar and air, with the view to determine whether In3+ could successfully charge balance U4+ and U5+, respectively, at moderately low concentrations of U (x = 0.05 and 0.10).
Experimental methodology
Materials synthesis
Caution
Uranium is an alpha emitter. Manipulations, synthesis and characterisation was performed in a materials radiochemistry laboratory in a controlled area, using HEPA filtered fume hoods and a dedicated glovebox, following risk assessments and monitoring procedures.
Two solutions targeting Ca1-xZr1-xIn2xTi2O7 (0.10 ≤ x ≤ 1.00, Δx = 0.10) and Ca1-xUxZrTi2-2xIn2xO7 (x = 0.05, 0.10) were fabricated by a conventional mixed oxide synthesis route, whereby the precursors CaTiO3 (Sigma Aldrich, 99.9% trace metals basis), ZrO2 (Sigma Aldrich, 99.9% trace metals basis), TiO2 (anatase, Sigma Aldrich, 99.9% trace metals basis), In2O3 (Sigma Aldrich, 99.9% trace metals basis) and UO2 (ABSCO Ltd., depleted) were intimately mixed by planetary milling. Each reagent was dried at 800 °C prior to weighing, and added to a 45 mL ZrO2-lined milling jar with isopropanol and ZrO2 milling media. Each sample was homogenised at 400 rpm for 20 min, with the direction of milling reversed after 10 min intervals. The powder slurries were decanted and allowed to dry overnight at 80 °C to evaporate excess solvent; dried powders were further mixed by hand to break up agglomerates and pressed into the walls of a hardened steel die (ø = 10 mm) under 3 tonnes of uniaxial force to form green bodies. These pellets were then placed onto a ZrO2 crucible and sintered at 1350 °C for 20 h (air for Ca1-xZr1-xIn2xTi2O7; flowing Ar and air for Ca1-xUxZrTi2-2xIn2xO7).
Materials characterisation
A portion of each sintered pellet was retained and finely ground for powder X-ray diffraction (XRD) using a Bruker D2 Phaser (Cu Kα source: λ = 1.5418 Å, Ni Filter) fitted with a Lynxeye Position Sensitive Detector, operating at 30 kV and 10 mA. Data were collected in the range 10° ≤ 2θ ≤ 70° with a stepsize of 0.02° s−1. Phase identification and peak indexing was achieved using the PDF4+ database and Rietveld analysis was performed using the Bruker TOPAS package. Scanning electron microscopy (SEM) analysis was performed using a Hitachi TM3030 operating with a 15 kV accelerating voltage at a working distance of 8 mm, fitted with a Bruker Quantax 70 spectrometer for Energy Dispersive X-ray Spectrometry (EDS). Samples were prepared for SEM–EDS analysis by mounting in cold setting epoxy resin and curing for 24 h, prior to grinding using incremental grades of SiC paper, before polishing to a 1 μm optical finish using diamond suspension. In K-edge X-ray absorption spectroscopy (XAS) data were acquired at BL01B1, SPring-8 (Hyogo, Japan). The storage ring energy was operated at 8 GeV with a typical current of 100 mA. Measurements were obtained using a Si (311) double-crystal monochromator in transmission mode at room temperature. Data were collected alongside In2O3 and In foil reference compounds. U L3-edge fluorescence and Zr K-edge transmission XAS data were collected at Diamond Light Source Beamline B18 (Oxford, UK), alongside a selection of reference compounds, containing U and Zr in a variety of coordination environments and U-oxidation states. A Si(111) double crystal monochromator was used to fine-tune incident synchrotron radiation, with the intensity of the incident and transmitted beam measured using ionization chambers, filled with a mixture of N2 and He gas, operated in a stable region of the I/V curve. Data reduction and analysis was achieved using the Demeter software package19. Bond valence sums were calculated using the in-built function in the Artemis software package. Here, the bond valence sum parameters are derived from the work of Altermatt and Brown20,21 and use the following equation:
where sij is the bond valence, Rij is the bond distance between atoms i and j and R′ij and b are the empirically determined parameters by Altermatt and Brown.
Results and discussion
Phase evolution in the Ca1-xZr1-xIn2xTi2O7 system
Powder XRD data for the formulations x = 0.10 and 0.20 were consistent with the formation of single phase zirconolite-2M. Unit cell dimensions were obtained by Rietveld analysis of powder diffraction data (Fig. 1, Table 1) corresponding to a decrease in the unit cell volume with increased In content. The unit cell parameters of pure CaZrTi2O7 have been previously reported as: a = 12.4462 Å, b = 7.2717 Å, c = 11.3813 Å, β = 100.5587° and V = 1012.60 Å322. There was therefore a consistent reduction in the lattice constants of the zirconolite-2M with respect to In substitution. No reflections representative of common secondary phases (ZrO2, CaTiO3) were evidenced in the diffraction patterns. Moreover, combined SEM–EDS analyses indicate the formation of high density microstructures comprised of a matrix of zirconolite-2M, with no phase separation visible by variation in backscattered electron contrast (Fig. 2). EDS analysis of the zirconolite-2M matrix formed for x = 0.10 and 0.20 confirmed the presence of In, indicated by the prominence of the In Lα and In Lβ emission lines present in the EDS spectrum (Fig. S1). The average composition of the zirconolite phase(s) was determined by semi-quantitative EDS analysis, normalised to seven oxygen atoms; the observed compositions are in good agreement with the targeted formulation (Table S1).

Rietveld refinement data for x = 0.10 (left) and x = 0.20 (right) compositions in the Ca1-xZr1-xIn2xTi2O7 system fit to the zirconolite-2M structural model.

BSE images for x = 0.10 (left) and x = 0.20 (right) in the Ca1-xZr1-xIn2xTi2O7 solid solution.
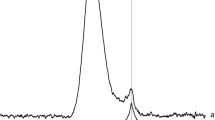
The In K-edge XANES spectra for single phase zirconolite-2M (x = 0.10 and 0.20) compositions are presented in Fig. 3. Data were collected alongside In2O3 and In foil reference compounds, representing In3+ and In0 oxidation states, respectively. For both In-containing zirconolite-2M compositions, the XANES spectra when overlaid were practically indistinguishable, indicative of identical speciation and coordination of In cations when targeting both x = 0.10 and 0.20. These spectra were comprised of a single intense absorption feature (E0 = 27,945.5 eV) followed by a weak post-edge oscillation, with maxima at 28,000 eV. It was clear that position of the absorption edge of x = 0.10 and 0.20 compounds was identical to that of the In2O3 reference compound (space group Ia\(\overline{3 }\)) which contains trivalent In3+ distributed across two sites, both of which are sixfold coordinated to O2−, inferring uniform In3+ speciation within the zirconolite-2M phase. Moreover, linear combination fitting of the XANES spectra for x = 0.10 and 0.20 confirmed 100% In3+ speciation relative to In2O3 and In-foil reference compounds, with R-factors of 0.00469 and 0.00531, respectively.

In K-edge XANES spectra for In-doped zirconolite-2M at concentrations x = 0.10 and 0.20, alongside In2O3 and In foil reference compounds.
Fitting of the EXAFS spectra (Fig. 4, Table 2) for both the x = 0.10 and 0.20 compositions produced good fits and utilised very similar models that corresponded well to the expected zirconolite-2M structure. For x = 0.10, the best fit model (R-factor = 0.0078) included 6 O backscatterers at 2.16(1) Å, 2 Ti backscatterers at 3.33(1) Å, and 2 Ti backscatterers at 3.57(1) Å. For x = 0.20, the best fit model (R-factor = 0.0106) included a split first O shell, with 3 O backscatterers at 2.11(6) Å and 3 O backscatterers at 2.21(6) Å, 2 Ti backscatterers at 3.34(2) Å, and 2 Ti backscatterers at 3.58(2) Å. Both fits match well with the expected structure of zirconolite-2M according to Whittle et al.23, with the exception of the second Ti shell at ~ 3.58 Å exhibiting an increase in the expected degeneracy (2 instead of the expected 1) since the Ca backscatterer at the same interatomic distance (Reff = 3.57 Å) was not modelled to avoid over-parameterising the model. Given both the second Ti shell and the Ca backscatterers manifest at very similar areas of the EXAFS spectrum, it is likely that significant interference makes fitting the expected shells and their corresponding degeneracies (i.e. 1 Ti and 1 Ca as opposed to the best fit model 2 Ti) challenging. Similar challenges in fitting have been observed in other doped zirconolite systems24 whereby the delineation of the exact type and number of backscatterers in the approximate range 3.5–3.6 Å was found to be not possible.

In K-edge XAS spectra for x = 0.10 and 0.20 compositions in the Ca1-xZr1-xIn2xTi2O7 system. Left—k3-weighted EXAFS. Right—Fourier transform of the k3-weighted EXAFS, using a Hanning window function. Black lines are data and red lines are the best fit models for the data.
The total degeneracy for the first O shell was found to be six in both samples (6 O at 2.16(1) Å for x = 0.10 and a split O shell of 3 O at 2.11(6) Å and 3 O at 2.21(6) Å for x = 0.20). The sixfold coordination of In3+, determined by these EXAFS analyses, implies that some compositional rearrangement is required to maintain charge balance across the single phase zirconolite-2M samples, for example (Ca1-xTix)(Zr1-xTix)Ti2-2xIn2xO7, consistent with the known defect mechanisms in non-stoichiometric zirconolite25. Indeed, the only previous study that explored In doping of zirconolite found that In readily expresses a preference for the fivefold Ti site17. The total degeneracy, and by extension the inferred mixture of In3+ coordination environments, correspond well to the bond valence sums (BVS) which sum to approximately 3 (2.973 for x = 0.10 and 3.014 for x = 0.20) for both samples. When trialing other models, with In3+ solely on the alternative Ti site (total O degeneracy of 5), the Ca site (total O degeneracy of 8) or Zr site (total O degeneracy of 7) the calculated BVS diverged markedly from the expected value of 3, and the overall model produced a qualitatively and quantitatively worse fit to the data.
As the targeted In3+ concentration was progressed beyond x ≥ 0.20, zirconolite-2M was no longer isolated as a single phase, with a number of ancillary In-bearing phases clearly distinguished by both XRD and combined SEM–EDS analyses. Nevertheless, zirconolite was detected by XRD up to x = 0.80, albeit at very low concentration (approximately 3 wt% at x = 0.80; see quantitative phase analysis in Table 3). Figure 5 shows the powder XRD data for compositions in the range 0.30 ≤ x ≤ 1.00. When targeting x = 0.30, a number of additional reflections corresponding to TiO2 (P42/mnm) and InTi0.75Ca0.25O3.25 (C2/m) were clearly observed, accounting for 2.3 ± 0.3 and 12.9 ± 0.3 wt% of the overall phase assemblage, respectively. SEM analysis (Fig. 6) confirmed the presence of all phases identified by XRD in the microstructure. In the compositional interval 0.40 ≤ x ≤ 0.60, an additional In-bearing cubic ZrO2 phase was also observed (highlighted by arrows in Fig. 5), plateauing with a maximum fraction of ~ 24 wt% of the overall phase assemblage at x = 0.50. The relative concentrations of InTi0.75Ca0.25O3.25 and TiO2 steadily increased, with a corresponding decrease in the zirconolite-2M phase. It was interesting to note that zirconolite did not appear to undergo any structural transformations to the 4M or 3T polytypes, as is typically observed with excessive doping in the Ca2+ and/or Zr4+ sites1. When targeting the end-member In2Ti2O7 composition (i.e. x = 1.00), a two phase mixture of In2TiO5 (79.4 ± 0.34 wt%) and TiO2 (20.6 ± 0.34 wt%) was identified by XRD and confirmed by SEM–EDS analyses, consistent with previous data26. The crystal structure of the cubic pyrochlore phase is characterized as an anion-deficient fluorite superstructure, adopting the generic A2B2O7 stoichiometry, resulting in two distinct cation sites; A cations are typically larger trivalent atoms (e.g. REE3+ = Dy, Y, Sm, Gd) whereas the B site is comprised of smaller, higher valence cations such as Ti4+ and Hf4+, although a number of variations do exist27. The stability of the cubic pyrochlore phase (space group Fd \(\overline{3 }\) m) is dictated by the size ratio of rA/rB whereby the A and B sites are eight and sixfold coordinated to O2−, respectively. Varying A and B site cations such that rA/rB > 1.78 results in the formation of a monoclinic structure, whereas rA/rB < 1.46 promotes the defect-fluorite structure type, in which oxygen vacancies are disordered across the sub-lattice. Considering the respective ionic radii of In3+ and Ti4+ in eight and sixfold coordination (0.92 Å and 0.605 Å) and the corresponding ratio (rA/rB = 1.52) the In2Ti2O7 phase should form the cubic pyrochlore structure on the basis of ionic radius ratio, nevertheless, no yield of In2Ti2O7 was observed at 1350 °C. Despite further attempts to form this phase with an increased sintering temperature of 1700 °C and extended dwell time of 24 h, we noted that these conditions were sufficient to completely volatilise the pellet during sintering and no yield was obtained.

Powder XRD data for 0.30 ≤ x ≤ 1.00 compositions in the Ca1-xZr1-xIn2xTi2O7 system. TiO2 reflections are labelled with closed circles. In2TiO5 reflections are labelled with stars. The prominent reflections contributing to In-doped c-ZrO2 are indicated with arrows.

SEM analysis of x = 0.30, 0.50, 0.70 and 0.90 compositions in the Ca1-xZr1-xIn2xTi2O7 solid solution.
U L3 and Zr K-edge X-ray absorption near edge structure (XANES) analysis of the Ca1-xUxZrTi2-2xIn2xO7 system
As In adopted the In3+ oxidation state when substituted in the Ca1-xZr1-xIn2xTi2O7 solid solution, preferentially occupying the Ti site, it may be feasible for In3+ to act as a neutron poison and charge balancing species within the Ti4+ site. Moreover, to our knowledge, the only other reported instance of In3+ substitution in zirconolite was consistent with occupation in the Ti4+ site as a simulant for the reduced Ti3+ species17. Hence, two compositions in the Ca1-xUxZrTi2-2xIn2xO7 system targeting x = 0.05 and 0.10 were also fabricated, with U deployed as a surrogate for Pu. Samples were first synthesised under argon, with a view to maintain the preferred U4+ valence configuration, as this is generally the target oxidation state for immobilised Pu in zirconolite. Powder XRD data for both compositions sintered under flowing Ar gas are shown in Fig. 7, and are consistent with the formation of near single phase zirconolite-2M (a small fraction of ZrO2 was also observed) at both targeted concentrations of U, indicating the In3+ was successfully accommodated in solid solution. The unit cell parameters of the zirconolite-2M phase(s) were calculated by Rietveld analysis of powder XRD data and are listed in Table 3. A yield of near single phase zirconolite-2M (< 2 wt% free ZrO2) was also obtained when compositions were synthesised in air, indicating that In3+ may capable of charge balancing U valence states greater than U4+.

Powder XRD data for Ca0.95U0.05ZrTi1.90In0.10O7 (x = 0.05) and Ca0.90U0.10ZrTi1.80In0.20O7 (x = 0.10) synthesised under both air and argon.
In order to determine the formal oxidation state of U, X-ray absorption near edge structure (XANES) spectra were collected at the U L3-edge (~ 17,166 eV) in transmission mode (Fig. 8) alongside a series of reference compounds containing U with known oxidation state and coordination environment. Scanning across the U L3 absorption edge probes the dipole transition from a core 2p3/2 shell to the partially occupied 6d valence shell. The observed spectra for all measured compounds were comprised of a single intense absorption feature with additional resonance features present above the main absorption edge; the energy position of the absorption edge is correlated to the formal oxidation state of the absorbing atom in many actinide species.

Normalised U L3-edge XANES for x = 0.05 (left) and x = 0.10 (right) compositions in the Ca1-xUxZrTi2-2xIn2xO7 solid solution sintered in argon and air. Data are displayed alongside UO2 (dashed) and CaUO4 (dotted) reference compounds, representing U4+ and U6+, respectively.
The E0 position of compositions sintered under flowing Ar was measured as the energy for the normalised absorbance μx = 0.5 (i.e. the energy at half the absorption edge), and was found to be 17,166.0(5) eV for both x = 0.05 and 0.10 compounds, indicative of uniform U speciation at both concentrations. These spectra displayed a clear resemblance to the UO2 reference compound with respect to the normalised edge intensity, position and post-edge oscillation feature, consistent with U4+ speciation in the zirconolite-2M compounds. The E0 position of the U L3-edge in the air-synthesised compounds was shifted to higher energy relative to the UO2 reference compound, measured to be 17,167.7(5) eV and 17,167.8(5) eV for x = 0.05 and 0.10, respectively, indicative of oxidised U. To establish the average oxidation states of U in both sets of compounds, a linear regression plot of the E0 position (defined as μx = 0.5) relative to a large suite of reference compounds was constructed (Fig. 9). The oxidation states of U in the x = 0.05 and 0.10 were calculated to be 4.1 and 4.1 in argon; 5.2 and 5.3 in air. These data are strong evidence to support the use of zirconolite as a potential wasteform for U/Pu, as the zirconolite-2M structure is clearly capable of accommodating changes in oxidation state without phase separation. Moreover, it is clear that In3+ is capable of charge balancing both U4+ and U5+ within the zirconolite structure, in the case of the latter, implying a charge balancing mechanism consistent with Ca1-xU5+xZrTi2-2xIn2xO7+x (Table 4).

Oxidation state of U in Ca1-xUxZrTi2-2xIn2xO7 (x = 0.05, 0.10; air and argon) calculated by linear regression of E0 position relative to reference compounds (U4+O2, U4+Ti2O6, U4+SiO4, U5+SbO5, U5+MoO4, LaU5+O5, CaU6+O4, Ca3U6+O6 and MgU6+O4).
XAS spectra were acquired at the Zr K-edge for x = 0.05 and 0.10 in the Ca1-xUxZrTi2-2xIn2xO7 solid solution (for both air and argon) alongside a variety of reference compounds containing Zr in distinctive coordination environments (Figs. 10, 11). There were notable differences in the XANES spectra of Zr-containing compounds depending on the coordination of Zr atoms. However, the absorption features for each measured zirconolite compound were observed to be near-identical regardless of targeted U concentration or processing environment, and, in excellent agreement with those previously reported in the literature, including synthetic CaZrTi2O722,28 and annealed metamict (Ca,Th)ZrTi2O7 zirconolite29,30. The XANES spectra are presented alongside a series of reference compounds, with variation in pre-edge intensity and main white-line absorption features consistent with Zr in a variety of coordination environments31. It was observed that the zirconolite and m-ZrO2 compounds exhibited rounded features at the crest of the white line, which is typical of sevenfold coordinate Zr, whereas the ZrSiO4, t-ZrO2 and Li2ZrO3 compounds exhibited notable peak splitting which is typical of octahedral coordination, or, as in the case of ZrSiO4, highly distorted eightfold coordination22. Qualitative analysis of the XANES region was consistent with the Zr atoms in zirconolite occupying the expected sevenfold coordination environment, as there are clear parallels with the m-VIIZrO2 reference compound. The Zr-K edge k3-weighted EXAFS modelling of the first oxygen coordination shell for each zirconolite specimen is presented in Fig. 10 (Table S2). The same model could be fit to all samples for both atmospheres (x = 0.05 and 0.10 for both argon and air) and was consistent with 7 O backscatterers present around the Zr central atom. In all fits, the first oxygen coordination shell was split into 4 O backscatterers at 2.10–2.11 Å and 3 O at 2.24–2.25 Å. These fits are in good agreement with the expected Zr coordination environment for zirconolite, in which a spread of 7 O backscatterers are present from 2.045 to 2.339 Å23. A distinct yet low intensity pre-edge feature was observed for the zirconolite materials and Zr compounds, attributed to the formally forbidden 1s→4d transition; the relative prominence of this feature is increased for compounds for which Zr is located in a distorted, non-centrosymmetric coordination environment. The inset of Fig. 10 highlights the pre-edge region of zirconolite (represented by the x = 0.10 (argon) composition), consistent with sevenfold coordination, alongside ZrSiO4, m-ZrO2 and Li2ZrO3, representing eight, seven and sixfold coordinate Zr, respectively. There is a clear trend in the intensity of the pre-edge feature with reduced coordination, exhibited most clearly by the difference between Li2VIZrO3 and VIIIZrSiO4. The increased intensity of this feature is attributed to a lack of centrosymmetry around the absorbing Zr atom. Furthermore, the pre-edge shape and intensity of the zirconolite and m-ZrO2 appears similar, further supporting the existence of sevenfold Zr in the U/In-doped zirconolite materials.

Zr K-edge XAS spectra for Ca1-xUxZrTi2-2xIn2xO7 (x = 0.05, 0.10) processed under argon and air. Left - Zr K-edge XANES data. Right - k3-weighted EXAFS of the Zr K-edge using a Hanning function window. The fit shown is for the first coordination shell only; black lines are data and red lines are the best fit models for the data.

Zr K-edge XANES data for Ca1-xUxZrTi2-2xIn2xO7 (x = 0.05, 0.10) processed under argon and air. Data are presented alongside a series of Zr reference compounds.
Conclusions
Herein, we have presented a novel examination of the solid solution behaviour of indium within the zirconolite phase, as a potential neutron absorbing additive. Two distinct substitution regimes were devised, targeting Ca1-xZr1-xIn2xTi2O7 (targeting In3+ distributed equimolar across Ca2+ and Zr4+ sites) and Ca1-xUxZrTi2-2xIn2xO7 (targeting In3+ across the Ti4+ site). In K-edge X-ray spectroscopy data confirmed In was uniformly present in the In3+ oxidation state, yet the prevailing In coordination environment was consistent with accommodation in the Ti4+ site in sixfold coordination, contrary to the targeted formulation. Nevertheless, single phase zirconolite-2M was able to form in the Ca1-xZr1-xIn2xTi2O7 system for 0.10 ≤ x ≤ 0.20. Progressive substitution of In3+ beyond x ≥ 0.20 promoted the formation of several In-titanate phases, namely In2TiO5 and InTi0.75Ca0.25O3.25, alongside TiO2 and c-ZrO2. In3+ was also utilised to successfully charge compensate U4+ and U5+ resulting in the formation of near single phase zirconolite-2M (accompanied by ~ 1–2 wt% ZrO2) in the Ca1-xUxZrTi2-2xIn2xO7 system at x = 0.05 and 0.10. The dominant oxidation states of U4+ (argon synthesis) and U5+ (air synthesis) were determined by U L3-edge XANES analysis. Analysis of the XANES and EXAFS region of the Zr K-edge in these materials was consistent with Zr4+ occupying sevenfold coordinated sites in the zirconolite structure, as would be expected. These data form a useful contribution towards ongoing efforts to design, characterise and performance test titanate wasteform materials substituted with appropriate quantities of neutron poisoning additives.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Blackburn, L. R. et al. Review of zirconolite crystal chemistry and aqueous durability. Adv. Appl. Ceram. 120(2), 1–15 (2021).
Vance, E. R. et al. Excess Pu disposition in zirconolite-rich synroc. Mat. Res. Soc. Symp. Proc. 412, 41–47 (1996).
Ewing, R. C. Nuclear waste forms for actinides. Proc. Natl. Acad. Sci. USA 96, 3432–3439 (1999).
Lumpkin, G. R. et al. Alpha-recoil damage in zirconolite (CaZrTi2O7). J. Mater. Res 1(4), 564–576 (1986).
Hyatt, N. C. Plutonium management policy in the United Kingdom: The need for a dual track strategy. Energy Policy 101, 303–309 (2017).
Hyatt, N. C. Safe management of the UK separated plutonium inventory: A challenge of materials degradation. npj Mater. Degrad. 4, 28 (2020).
Ewing, R. C., Whittleston, R. A. & Yardley, B. W. D. Geological disposal of nuclear waste: A primer. Elements 12(4), 233–237 (2016).
Nuclear Decommissioning Authority (NDA). Progress on Plutonium Consolidation, Storage and Disposition (2019).
Hicks, T. W. Criticality Safety Assessment for Waste Packages Containing High-enriched Uranium (2007).
Solano, J. M., Page, T., Hicks, T. W. & Thorne, P. The use of Neutron-absorbing Materials in ILW and Spent Fuel Packages for Criticality Control (2012).
Zhang, Y. B. et al. Phase evolution, microstructure and chemical stability of Ca1-xZr1-xGd2xTi2O7 (0.0 ≤ x ≤ 1.0) system for immobilizing nuclear waste. Ceram. Int. 44(12), 13572–13579 (2018).
Li, H. et al. Characterisation of stainless steel-synroc interactions under hot isostatic pressing (HIPing) conditions. J. Nucl. Mater. 355(1–3), 136–141 (2006).
Ma, S. et al. Effects of ionic radius on phase evolution in Ln-Al co-doped Ca1-xLnxZrTi2-xAlxO7 (Ln = La, Nd, Gd, Ho, Yb) solid solutions. Ceram. Int. 44(13), 15124–15132 (2018).
Putnam, R. L., Navrotsky, A., Woodfield, B. F., Shapiro, J. L., Stevens, R. & Boerio-Goates, J. Thermochemistry of Hf-zirconolite, CaHfTi2O7. In Mat. Res. Soc. Symp. Proc. no. 556 (2011).
Caurant, D., Loiseau, P. & Bardez, I. Structural characterization of Nd-doped Hf-zirconolite Ca1-xNdxHfTi2-xAlxO7 ceramics. J. Nucl. Mater. 407(2), 88–99 (2010).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. 32, 751–767 (1976).
Begg, B. D., Vance, E. R., Hunter, B. A. & Hanna, J. V. Zirconolite transformation under reducing conditions. J. Mater. Res. 13(11), 3181–3190 (1998).
Lee, D. W. et al. KIn0.33IIITe0.67VITe2IVO7: Zirconolite-like mixed-valent metal oxide with a 3D framework. Inorg. Chem. 60(20), 15091–15095 (2021).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Altermatt, D. & Brown, I. D. The automatic searching for chemical bonds in inorganic crystal structures. Acta Crystallogr. Sect. B 41(4), 240–244 (1985).
Brown, I. D. & Altermatt, D. Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystallogr. Sect. B 41(4), 244–247 (1985).
McCaugherty, S. & Grosvenor, A. P. Low-temperature synthesis of CaZrTi2O7 zirconolite-type materials using ceramic, coprecipitation, and sol-gel methods. J. Mater. Chem. C. 7, 177 (2019).
Whittle, K. R. et al. Combined neutron and X-ray diffraction determination of disorder in doped zirconolite-2M. Am. Mineral. 97, 291–298 (2012).
Blackburn, L. R. et al. Phase evolution in the CaZrTi2O7–Dy2Ti2O7 system: A potential host phase for minor actinide immobilization. Inorg. Chem. 61(15), 5744–5756 (2022).
Gatehouse, B. M., Grey, I. E., Hill, R. J. & Rossell, H. J. Zirconolite, CaZrxTi3-xO7; structure refinements for near-end-member compositions with x = 0.85 and 1.30. Acta Cryst. B37(1974), 306–312 (1981).
Brown, F., Kimizuka, N. & Michiue, Y. Orthorhombic In(Fe1-xTix)O3+x/2 (0.50 ≤ × ≤ 0.69) and monoclinic In(Fe1-xTix)O3+x/2 (0.73 ≤ × ≤ 0.75) in the system InFeO3-In2Ti2O7 at 1300°C in air. II. Synthesis and crystal structures. J. Solid State Chem. 157(1), 13–22 (2001).
Mccauley, R. A. Structural characteristics of pyrochlore formation. J. Appl. Phys. 51(1), 290–294 (1980).
Mountjoy, G. et al. Changes in the Zr environment in zirconia-silica xerogels with composition and heat treatment as revealed by Zr K-edge XANES and EXAFS. Phys. Chem. Chem. Phys. 2(10), 2455–2460 (2000).
Farges, F., Ewing, R. C. & Brown, G. E. The structure of aperiodic, metamict (Ca, Th)ZrTi2O7 (zirconolite): An EXAFS study of the Zr, Th, and U sites. J. Mater. Res. 8(8), 1983–1995 (1983).
Farges, F., Ponader, C. W. & Brown, G. E. Structural environments of incompatible elements in silicate glass/melt systems: I. Zirconium at trace levels. Geochim. Cosmochim. Acta 55(6), 1563–1574 (1991).
Connelly, A. J. et al. The structural role of Zr within alkali borosilicate glasses for nuclear waste immobilisation. J. Non. Cryst. Solids 357(7), 1647–1656 (2011).
Hyatt, N. C., Corkhill, C. L., Stennett, M. C., Hand, R. J., Gardner, L. J. & Thorpe, C. L. The HADES facility for high activity decommissioning engineering & science: Part of the UK national nuclear user facility. In IOP Conf. Series: Materials Science and Engineering, vol. 818, 1–8 (2020).
Acknowledgements
This research utilised the HADES/MIDAS facility at the University of Sheffield established with financial support from EPSRC and BEIS, under grant EP/T011424/132. We acknowledge Diamond Light Source for access to Beamline B18 under Proposal No. SP17243. L. R. Blackburn is grateful to EPSRC for funding through the Doctoral Prize Fellowship scheme, under grant EP/T517835/1. C. L. Corkhill is grateful to EPSRC for funding through the award of an Early Career Research Fellowship (EP/N017374/1).
Author information
Authors and Affiliations
Contributions
The authors contributed to the manuscript in the following manner: L.B.: designed the experiment, fabricated the samples, collected data, performed analysis and drafted the manuscript. L.T.: formal analysis and manuscript preparation. M.D.W., T.I., M.K., S-K.S., A.M., L.G.: formal analysis. M.S.: formal analysis and funding acquisition. C.C., N.H.: supervision and funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Blackburn, L.R., Townsend, L.T., Dixon Wilkins, M.C. et al. Underpinning the use of indium as a neutron absorbing additive in zirconolite by X-ray absorption spectroscopy. Sci Rep 13, 9329 (2023). https://doi.org/10.1038/s41598-023-34619-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-34619-5
This article is cited by
-
Long-term, sustainable solutions to radioactive waste management
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
