
Abstract
Synaptotagmin-1 is a vesicular protein and Ca2+ sensor for Ca2+-dependent exocytosis. Ca2+ induces synaptotagmin-1 binding to its own vesicle membrane, called the cis-interaction, thus preventing the trans-interaction of synaptotagmin-1 to the plasma membrane. However, the electrostatic regulation of the cis- and trans-membrane interaction of synaptotagmin-1 was poorly understood in different Ca2+-buffering conditions. Here we provide an assay to monitor the cis- and trans-membrane interactions of synaptotagmin-1 by using native purified vesicles and the plasma membrane-mimicking liposomes (PM-liposomes). Both ATP and EGTA similarly reverse the cis-membrane interaction of synaptotagmin-1 in free [Ca2+] of 10–100 μM. High PIP2 concentrations in the PM-liposomes reduce the Hill coefficient of vesicle fusion and synaptotagmin-1 membrane binding; this observation suggests that local PIP2 concentrations control the Ca2+-cooperativity of synaptotagmin-1. Our data provide evidence that Ca2+ chelators, including EGTA and polyphosphate anions such as ATP, ADP, and AMP, electrostatically reverse the cis-interaction of synaptotagmin-1.
Similar content being viewed by others


Introduction
Exocytosis is the process of vesicle fusion and neurotransmitter release regulated by soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins, which are currently considered to be the catalysts of the fusion reaction1,2. Neuronal SNARE proteins are selectively expressed in neurons and neuroendocrine cells, and regulate release of neurotransmitters and hormones3. Neuronal SNARE proteins consist of syntaxin-1 and SNAP-25 in the plasma membrane, and vesicle-associated membrane protein-2 (VAMP-2) (also called synaptobrevin-2) in the vesicle membrane1. Synaptotagmin-1 is a Ca2+ sensor for fast Ca2+-dependent exocytosis as an electrostatic switch4. The C2AB domain of synaptotagmin-1 coordinates Ca2+ binding, and the Ca2+-bound C2AB domain penetrates negatively-charged anionic phospholipids by electrostatic interaction2. Several different models of synaptotagmin-1 to describe the process of Ca2+-dependent vesicle fusion have been proposed, but the molecular mechanisms of synaptotagmin-1 remain controvertial5.
Synaptotagmin-1 is a vesicular protein and interacts with anionic phospholipids electrostatically5. Native vesicles contain ~ 15% anionic phospholipids including phosphatidylserine (PS) and phosphatidylinositol (PI)6, so Ca2+ induces synaptotagmin-1 binding to its own vesicle membrane, i.e., the cis-interaction7,8. Ca2+ fails and even slightly reduces vesicle fusion in the in-vitro reconstitution system, because synaptotagmin-1 preferentially interacts with vesicle membranes due to the physical proximity and this cis-membrane interaction prevents the trans-interaction of synaptotagmin-1 with the target membranes7,8,9. We have reported that ATP reverses this inactivating cis-interaction of synaptotagmin-1 by the electrostatic effect, and the trans-membrane interaction of synaptotagmin-1 only occurs to trigger vesicle fusion in-vivo10. This ATP effect on the cis-membrane interaction of synaptotagmin-1 has been confirmed independently: in a vesicle sedimentation assay a few hundred μM ATP electrostatically prevents a cis-configuration of synaptotagmin-111, and in a fusion assay using a colloidal probe microscopy and pore-spanning membranes ATP accelerates full fusion by preventing the cis-interaction without affecting the trans-interaction of synaptotagmin-112. However, the electrostatic regulation of the cis- and trans-membrane interaction of synaptotagmin-1 to trigger Ca2+-dependent vesicle fusion has not been described in detail.
Although synaptotagmin-1 is a conserved Ca2+ sensor for synchronous release of diverse vesicles including synaptic vesicles, large dense-core vesicles (LDCVs), and other secretory granules, the mechanism by which Ca2+-cooperativity is regulated is not clear. The Hill coefficient (n) in the Ca2+ dose–response curves for exocytosis represents Ca2+-cooperativity and the Hill coefficient varies depending on cell types from 2 to 5; e.g. calyx-of-Held synapses (n, 4.2)13,14,15, neuromuscular junctions (n, 3.8)16, bipolar cells (n, 4)17, pituitary melanotrophs (n, 2.5)18, and chromaffin cells (n, 1.8)19. The Hill coefficient is the intrinsic property of each cell type and factors that regulate Ca2+-cooperativity are poorly understood.
Synaptotagmin-1 binds to anionic phospholipids by electrostatic interaction and the Ca2+-binding loops of the C2 domains penetrate anionic phospholipids by reducing repulsion between anionic phospholipids and acidic residues in the C2AB domain4. The polybasic patch in the C2B domain electrostatically interacts with PIP2 in a Ca2+-independent manner20, and thereby increases the Ca2+-sensitivity of synaptotagmin-1 membrane binding10,21. Given that the C2AB domain has five possible Ca2+-binding sites22,23 and therefore may have the Hill coefficient up to 4–5, but whether local PIP2 concentrations regulate Ca2+-cooperativity is not known.
Here we provide an assay to monitor the cis- and trans-membrane interaction of synaptotagmin-1 by using native LDCVs and the plasma membrane-mimicking liposomes (PM-liposomes). Ca2+ chelators, including EGTA and polyphosphate anions such as ATP, ADP, and AMP, electrostatically reverse the cis-interaction of synaptotagmin-1. Both ATP and EGTA, as Ca2+ chelators, have a similar effect to prevent the cis-membrane interaction of synaptotagmin-1 in free [Ca2+] of 10–100 μM, but ATP, which has a good buffering capacity in the range of 10–500 μM free [Ca2+], is an excellent Ca2+ buffer to study vesicle fusion and synaptotagmin-1 membrane binding. When the trans-membrane interaction of synaptotagmin-1 only occurs, high PIP2 concentrations in the PM-liposomes decrease the Hill coefficient of vesicle fusion and synaptotagmin-1 membrane binding to ~ 2, suggesting that local PIP2 concentrations might control Ca2+-cooperativity of synaptotagmin-1.
Material and methods
Purification of large dense-core vesicles (LDCVs)
LDCVs, also known as chromaffin granules, were purified from bovine adrenal medullae by using continuous sucrose gradient, then resuspended in a solution of 120 mM K-glutamate, 20 mM K-acetate, and 20 mM HEPES.KOH, pH 7.4, as described elsewhere24.
Protein purification
All SNARE and the C2AB domain of synaptotagmin-1 constructs based on rat sequences were expressed in E. coli strain BL21 (DE3) and purified by Ni2+-NTA affinity chromatography followed by ion-exchange chromatography as described elsewhere10,20. The stabilized Q-SNARE complex consists of syntaxin-1A (aa 183–288) and SNAP-25A (no cysteine, cysteines replaced by alanines) in a 1:1 ratio by the C-terminal VAMP-2 fragment (aa 49–96), and was purified as described earlier25. The C2AB domain of synaptotagmin-1 (aa 97–421) and soluble form of VAMP-2 lacking the transmembrane domain (VAMP-21–96) were purified using a Mono S column (GE Healthcare, Piscataway, NJ) as described previously26. The stabilized Q-SNARE complex was purified by Ni2+-NTA affinity chromatography followed by ion-exchange chromatography on a Mono Q column (GE Healthcare, Piscataway, NJ) in the presence of 50 mM n-octyl-β-d-glucoside (OG)10. The point mutated C2AB domain (S342C) was labelled with Alexa Fluor 488 C5 maleimide (C2ABA488)26.
Lipid composition of liposomes
All lipids were obtained from Avanti Polar lipids (Alabaster, AL). Lipid composition (mol, %) of the PM-liposomes that contain the Q-SNARE complex was 45% PC (l-α-phosphatidylcholine, Cat. 840055), 15% PE (l-α-phosphatidylethanolamine, Cat. 840026), 10% PS (l-α-phosphatidylserine, Cat. 840032), 25% Chol (cholesterol, Cat. 700000), 4% PI (l-α-phosphatidylinositol, Cat. 840042), and 1% PI(4,5)P2 (PIP2, Cat. 840046). When PIP2 concentrations were changed, PI contents were adjusted accordingly. For FRET-based lipid-mixing assays, 1.5% 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl (NBD-DOPE) as a donor dye and 1.5% 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-lissamine rhodamine B sulfonyl ammonium salt (Rhodamine-DOPE) as an acceptor dye were incorporated in the PM-liposomes (accordingly 12% unlabelled PE).
Preparation of proteoliposomes
Incorporation of the Q-SNARE complex into large unilamellar vesicles (LUVs) was achieved by OG-mediated reconstitution, called the direct method, i.e. incorporation of proteins into preformed liposomes10,20. Briefly, lipids dissolved in a 2:1 chloroform–methanol solvent were mixed according to lipid composition. The solvent was removed using a rotary evaporator to generate lipid film on a glass flask, then lipids were resuspended in 1.5 mL diethyl ether and 0.5 mL buffer containing 150 mM KCl and 20 mM HEPES/KOH pH 7.4. The suspension was sonicated on ice (3 × 45 s), then multilamellar vesicles were prepared by reverse-phase evaporation using a rotary evaporator as diethyl ether was removed. Multilamellar vesicles (0.5 mL) were extruded using polycarbonate membranes of pore size 100 nm (Avanti Polar lipids) to give uniformly-sized LUVs. After the preformed LUVs had been prepared, SNARE proteins were incorporated into them using OG, a mild non-ionic detergent, then the OG was removed by dialysis overnight in 1 L of buffer containing 150 mM KCl and 20 mM HEPES/KOH pH 7.4 together with 2 g SM-2 adsorbent beads. Proteoliposomes had protein-to-lipid molar ratio of 1:500.
Vesicle fusion assay
A FRET-based lipid-mixing assay was applied to monitor vesicle fusion in-vitro10,20. LDCV fusion reactions were performed at 37 °C in 1 mL fusion buffer containing 120 mM K-glutamate, 20 mM K-acetate, 20 mM HEPES–KOH (pH 7.4), 1 mM MgCl2, and 3 mM ATP (Fig. 4b). Fusion buffer in Fig. 3a,b contains no ATP, but EGTA; 120 mM K-glutamate, 20 mM K-acetate, 20 mM HEPES–KOH (pH 7.4), 5 mM MgCl2, and 10 μM EGTA. ATP should be made freshly before all experiments, because it is easily destroyed by freezing and thawing. Free Ca2+ concentration in the presence of Mg2+ and ATP or EGTA was calibrated using the MaxChelator simulation program.
The PM-liposomes that contain NBD-DOPE and Rhodamine-DOPE as a donor and an acceptor dye, respectively, were incubated with LDCVs, thus leading to dequenching of donor fluorescence (NBD) as a result of lipid dilution with unlabelled vesicle membrane10,20. The fluorescence dequenching signal of vesicle fusion was measured using wavelength of 460 nm for excitation and 538 nm for emission. Fluorescence values were normalized as a percentage of maximum donor fluorescence (i.e., total fluorescence) after addition of 0.1% Triton X-100 at the end of experiments.
Fluorescence anisotropy measurements
The C2AB fragments (20 nM, S342C) were labelled with Alexa Fluor 48826. Anisotropy was measured at 37 °C in 1 mL of buffer containing 120 mM K-glutamate, 20 mM K-acetate, and 20 mM HEPES–KOH (pH 7.4), 5 mM MgCl2, 10 μM EGTA. First, 1 mM Ca2+ was applied, then ATP or EGTA was accordingly added to chelate Ca2+ and reverse the membrane binding of the C2AB domain; each time ATP or EGTA was uniformly mixed by pipetting and a magnetic stirring setup with dilution factor of 1:500 in 1 mL buffer. (Fig. 2). Excitation wavelength was 495 nm and emission was measured at 520 nm. Anisotropy (r) was calculated using the formula r = (IVV − G × IVH)/(IVV + 2 × G × IVH), where IVV indicates the fluorescence intensity with vertically polarized excitation and vertical polarization on the detected emission and IVH denotes the fluorescence intensity when using a vertical polarizer on the excitation and horizontal polarizer on the emission. G is a grating factor used as a correction for the instrument’s differential transmission of the two orthogonal vector orientations. Lipid composition of the PM-liposomes (protein-free) was identical to those used in a fusion assay except labelled PE (45% PC, 15% PE, 10% PS, 25% Chol, 4% PI, and 1% PIP2).
Ca2+ calibration
ATP contains negatively charged oxygen atoms which bind to Mg2+, Ca2+, or Sr2+, thereby chelating divalent cations27. Ca2+ concentrations were calibrated with Fluo-5N, pentapotassium salt, cell impermeant, a low-affinity Ca2+ indicator with a Kd of 90 μM. Fluo-5N (500 nM) was included in buffer containing 120 mM K-glutamate, 20 mM K-acetate, 20 mM HEPES–KOH (pH 7.4), 5 mM MgCl2, and 10 μM EGTA. 5 mM ATP, ADP, or AMP (sodium salt, Sigma-Aldrich) was added to chelate free Ca2+. The fluorescence signal was measured at 37 °C with wavelength of 494 nm for excitation and 516 nm for emission. The following equation was used to measure free Ca2+ concentrations:
where Fmin is the fluorescence intensity in the absence of calcium with 10 mM EGTA, Fmax is the maxium fluorescence with 5 mM CaCl2, and F is the fluorescence of intermediate Fluo-5N. Fluo-5N experimental data with 5 mM ATP were correlated with the MaxChelator simulation program that calculates the free [Ca2+].
Statistical analysis
All quantitative data are mean ± SD from ≥ 3 independent experiments. Dose–response curves were fitted using four-parameter logistic equations (4PL) (GraphPad Prism) to calculate Hill slope and EC50.
Results
Calibration of free [Ca2+] using Fluo-5N and simulation program in the presence of ATP
Ca2+ is a triggering factor of vesicle fusion and intracellular Ca2+ concentration ([Ca2+]i) is typically ~ 100 nM, but local [Ca2+]i and Ca2+ microdomains at the vesicle-release sites close to voltage-gated calcium channels increase to ~ 300 μM15,28,29. We used ATP, which is a low affinity Ca2+ buffer, to maintain ~ 10 ≤ free [Ca2+] ≤ ~ 300 μM for in-vitro assays10,20,24,30,31. ATP has a dissociation constant (Kd) ~ 230 μM [Ca2+]27, so ATP is an excellent Ca2+ buffer in the range of 10–500 μM free [Ca2+]32,33. We used Fluo-5N to measure free [Ca2+] in the presence of ATP to confirm the predictions of [Ca2+] and to determine how much total [Ca2+] is required to achieve a desired free [Ca2+](Fig. 1a–c). Fluo-5N is a low-affinity Ca2+ indicator (Kd of 90 μM)34, which is good for measuring around 100 μM free [Ca2+], because Kd of Ca2+ chelators should be close to the desired free [Ca2+]35. EGTA (10 μM) was included to remove contaminating Ca2+ for the calibration of free [Ca2+]. An initial total 113 μM free [Ca2+] was reduced to 26 μM in the presence of 5 mM ATP by its chelation of Ca2+ (Fig. 1a,b).

Calibration of free [Ca2+] using Fluo-5N and simulation in the presence of ATP. (a) Free [Ca2+] calibration in the presence of ATP, ADP, or AMP using Fluo-5N, a Ca2+ indicator with Kd = 90 μM. 5 mM of ATP, ADP, or AMP was applied (arrow). Representative trace of free [Ca2+] from four independent experiments. (b) Comparison of free [Ca2+] in the presence of 5 mM ATP between Fluo-5N and the MaxChelator simulation program, which calculates free [Ca2+] in the presence of ATP and Mg2+. (c) ADP and AMP chelate free [Ca2+], but the Ca2+-chelating efficiency is less than that of ATP. Data in (b,c) are mean ± SD from three to four independent experiments (n = 3–4).
Then we compared this experimental data of free [Ca2+] with the MaxChelator, which is a computer simulation programs32,35 that enables calculation of appropriate stoichiometric concentrations of Ca2+ and Mg2+ in the presence of different Ca2+ chelators such as EGTA and ATP, and thereby provides detailed infomation to obtain the desired free [Ca2+]35. The MaxChelator program included 5 mM Mg2+ and 10 μM EGTA, and assumed 37 °C as in the Ca2+ calibration experiments (Fig. 1a). Indeed the MaxChelator calculated free [Ca2+] = 29 μM in the presence of 5 mM ATP with 113 μM total [Ca2+] at pH 7.4. This agreement with the measured free [Ca2+] = 26 μM confirms that the MaxChelator can predict free [Ca2+] obtained in experiments that use a Fluo-5N fluorescent Ca2+ indicator (Fig. 1b).
Negatively-charged oxygen atoms of ATP chelate divalent cations such as Mg2+, Ca2+, or Sr2+27. In the experiments, 5 mM ADP or 5 mM AMP chelated Ca2+, thereby reducing free [Ca2+] from 122 to 57 μM and from 126 to 99 μM, respectively (Fig. 1c). Increasing the number of phosphate groups in Adenosine increases Ca2+ affinity and lowers Kd by increasing the number of Ca2+ ions that are bound27,33. ATP, ADP, and AMP have distinct ranges of Ca2+-buffering capacity and distinct Kd values33, so Ca2+-chelating effect is ATP > ADP > AMP (Fig. 1a–c). Altogether, the predictions of free [Ca2+] in the complex buffer solutions including Mg2+, ATP and EGTA were confirmed using a fluorescent Ca2+ indicator.
Monitoring the cis- and trans-membrane interaction of synaptotagmin-1
Synaptotagmin-1 interacts with anionic phospholipids by electrostatic interaction. Native vesicles contain ~ 15% anionic phospholipids, including phosphatidylserine (PS) and phosphatidylinositol (PI)6. Therefore, Ca2+ induces synaptotagmin-1 to bind to its own vesicle membrane, i.e., cis-interaction, which prevents trans-interaction to the plasma membranes and thereby inactivates the ability of synaptotagmin-1 to trigger fusion7,8,9.
Ca2+-bound synaptotagmin-1 is inserted to native vesicle membranes such as synaptic vesicles and large dense-core vesicles (LDCVs) that contain anionic phospholipids10. However, ATP electrostatically prevents the cis-interaction of synaptotagmin-1, whereas the trans-interaction of synaptotagmin-1 to the plasma membrane remains active to mediate Ca2+-dependent vesicle fusion, because PIP2 overcomes the inhibitory effect of ATP by increasing the membrane-binding affinity of the C2AB domain10,11,12.
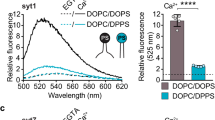
We tested an assay that uses fluorescence anisotropy measurement to monitor the cis- and trans-membrane interaction of synaptotagmin-1 (Fig. 2). Direct measurement of the cis- and trans-membrane interaction of endogenous synaptotagmin-1 in native vesicle membranes is impossible, so we monitored the binding of an exogenously-added C2AB domain of synaptotagmin-1 (Syt97-421), which was labelled with Alexa Fluor 488 at S342C (Fig. 2a). We took advantage of a single fluorescent labelling for anisotropy measurement to monitor the interaction of the C2AB domain with native vesicles or liposomes; the membrane-bound C2AB domain leads to increase of fluorescence anisotropy due to a reduction in the rotational mobility10 (Fig. 2a,b). It is noted that our experiments using the cytoplasmic C2AB domain are intended to shed light on the cis- and trans-interactions, but the geometry is not truly being imitated.

Monitoring cis- and trans-membrane interaction of synaptotagmin-1. (a) Binding of the C2AB domain of synaptotagmin-1 to the membrane of native vesicles (i.e., LDCVs) was monitored using fluorescence anisotropy in which the C2AB domain (Syt-197–421) was labelled with Alexa Fluor 488 at S342C (green dots). (Left) The C2AB domain has Ca2+-binding sites (magenta) and the Ca2+-bound C2AB domain is inserted to membrane, thus decreasing the rotational mobility. LDCVs contain anionic phospholipids, equivalent to around 15% PS10. (Right) A dose of 1 mM Ca2+ was applied to induce binding of the C2AB domain to LDCVs, then 1 mM ATP was added five times (arrows) to reverse this binding. The final total of 5 mM ATP disrupted membrane binding of the C2AB domain (red). (b) C2AB domain binding to the PM-liposomes. 1 mM ATP was added thirteen times (arrows) and the final total of 13 mM ATP reversed membrane binding of the C2AB domain; the C2AB binding remained in 5 mM ATP (red). (c) In-vitro reconstitution of LDCV fusion using a lipid-mixing assay. Purified LDCVs were incubated with the PM-liposomes that incorporate the stabilized Q-SNARE complex. 1 mM Ca2+ in the presence of 5 mM ATP accelerated LDCV fusion. (d,e) Binding of the C2AB domain to LDCVs (d) and the PM-liposomes (e) was monitored using fluorescence anisotropy as in a,b. First, 1 mM Ca2+ was applied to induce binding of the C2AB domain, then 100 μM EGTA was added ten times (arrows) to reverse this binding. A total dose of 800 μM EGTA disrupted C2AB binding to LDCV (red, d) and a final total dose of 1 mM EGTA reversed C2AB binding to liposomes (red, e). (f,g) LDCV fusion was increased by 1 mM Ca2+ in the presence of 800 μM EGTA (f), but was not affected in the presence of 1 mM EGTA (g).
We first monitored the cis-membrane interaction between the C2AB domain and the LDCV membranes (Fig. 2a). The presence of 1 mM Ca2+ increased fluorescence anisotropy; this change indicates that the C2AB domains bind to LDCV membranes in a Ca2+-dependent manner. Five sequential applications of 1 mM ATP gradually decreased the anisotropy signal by chelating Ca2+; this result suggests dissociation of the C2AB domain from LDCVs (Fig. 2a). 5 mM ATP in the presence of 1 mM Ca2+ almost completely disrupted the cis-membrane interaction of the C2AB domain with the LDCV membranes (Fig. 2a); free [Ca2+] in the presence of Mg2+, ATP and EGTA was calibrated using the MaxChelator simulation program and free [Ca2+] was 351 µM in case of 5 mM ATP and 1 mM Ca2+ (Table 1).
Next, we tested the trans-membrane interactions between the C2AB domain and the PM-liposomes; 10% PS, 4% PI, and 1% PIP2 were included in the PM-liposomes (Fig. 2b). The C2AB domain of synaptotagmin-1 bound to liposomes in response to 1 mM Ca2+, and this trans-membrane interaction was reduced by ATP, 1 mM applied thirteen times sequentially (Fig. 2b). Free [Ca2+] in different ATP concentrations was summarized in Table 1. Ca2+-dependent vesicle fusion is accelerated by the increase of the trans-interactions and the decrease of the cis-membrane interaction of synaptotagmin-110,20, so we hypothesized that 5 mM ATP in the presence of 1 mM Ca2+ is appropriate to observe Ca2+-dependent fusion (red in Fig. 1a,b).
To test this hypothesis and examine the effect of the cis- and trans-membrane interaction of synaptotagmin-1 on vesicle fusion, we applied a reconstitution system of vesicle fusion by using native LDCVs10,20,24. The PM-liposomes contain the stabilized Q-SNARE complex (syntaxin-1A and SNAP-25A in a 1:1 molar ratio25). Indeed, 5 mM ATP in the presence of 1 mM Ca2+ (i.e., 351 µM free [Ca2+] according to the MaxChelator program (Table 1)) dramatically accelerated LDCV fusion, which was completely blocked by the soluble VAMP-2 (VAMP-21–96); this results indicates SNARE-dependent vesicle fusion (Fig. 2c). We have previously shown that 300–400 μM free [Ca2+] in the absence of ATP fails to enhance vesicle fusion, but rather slightly inhibits fusion, because the cis-membrane interaction of the C2AB domain to native vesicle membranes becomes robust from 100 μM up to 3 mM10. ATP prevents this cis-membrane interaction by charge screening and competing with the vesicle membrane, thus allowing synaptotagmin-1 to interact in trans with the plasma membrane10.
Polyphosphates such as ATP reverse an inactivating cis-interaction of synaptotagmin-1 by an electrostatic effect (Fig. 2a–c). Next, we tested whether other Ca2+ chelators, e.g., EGTA, can have a similar inhibitory effect on the cis-membrane interaction. Anisotropy measurement was performed to monitor the cis- and trans-membrane interaction of the C2AB domain (Fig. 2a,b). EGTA was applied 10 times (100 μM each in the presence of 1 mM Ca2+) to reverse the cis-interaction of the C2AB domain to LDCVs (Fig. 2d). Application of 800 μM EGTA dramatically disrupted the cis-interaction in the presence of total 1 mM Ca2+ (red in Fig. 2d); free [Ca2+] was 200 μM (Table 1). However, the trans-membrane interactions of the C2AB domain to the PM-liposomes remained robust in the presence of 800 μM EGTA with 1 mM Ca2+ (200 μM free [Ca2+], Fig. 2e), whereas 1 mM EGTA significantly disrupted both the cis- and trans-membrane interactions of the C2AB domain (Fig. 2d,e); free [Ca2+] was 12 μM (Table 1).
Anisotropy measurement is useful to find a Ca2+-buffering condition to observe Ca2+-dependent vesicle fusion, where the cis-membrane interaction is prevented and the trans-interaction remains active. The presence of 800 μM EGTA with 1 mM Ca2+ (200 μM free [Ca2+], Table 1) significantly reversed the cis-interaction (Fig. 2d), but had a minor effect on the trans-interaction (Fig. 2e). Indeed, 800 μM EGTA with 1 mM Ca2+ reproduced Ca2+-dependent LDCV fusion (Fig. 2f). 1 mM EGTA with 1 mM Ca2+ (12 μM free [Ca2+], Table 1) failed to accelerate fusion, because the trans-interaction of the C2AB domain was dramatically disrupted by 1 mM EGTA (red in Fig. 2e); it is mainly because of low free [Ca2+]. Taken together, we established an anisotropy assay to monitor the cis- and trans-membrane interaction of synaptotagmin-1 by using native LDCVs and the PM-liposomes. Our data suggest that Ca2+ chelators such as EGTA, in addition to polyphosphates such as ATP, can prevent the cis-membrane interaction of synaptotagmin-1 by the electrostatic effect in a certain range of free [Ca2+].
EGTA reproduces the biphasic regulation of Ca2+ on LDCV fusion
We have previously reported the biphasic regulation of Ca2+ on LDCV fusion; 10–100 μM free Ca2+ exponentially accelerates native vesicle fusion, but > 300 μM free [Ca2+] progressively reduces Ca2+-dependent fusion, showing biphasic regulation of Ca2+ on LDCV fusion in a bell-shaped dose-dependence20. ATP was used for Ca2+-buffering to maintain free [Ca2+] in the range of 10–500 μM20. We examined whether EGTA reproduces the biphasic regulation of Ca2+ on LDCV fusion (Fig. 3a,b). Instead of ATP, 10 μM EGTA was included in fusion buffer and free [Ca2+] was calculated using the MaxChelator program. As expected, biphasic regulation of Ca2+ on LDCV fusion was observed, where Ca2+-dependent fusion progressively increased until [Ca2+] = ~ 100 μM, and gradually decreased at [Ca2+] from 300 μM to 1 mM (Fig. 3a,b).

EGTA reproduces ATP effect on Ca2+-dependent LDCV fusion and the C2AB binding to LDCVs. (a,b) LDCV fusion using a lipid-mixing assay as described in Fig. 2c at different concentrations of Ca2+ in the presence of 10 μM EGTA, instead of ATP. (a) Representative trace of dequenching of donor fluorescence (NBD). (b) Dose–response curve of LDCV fusion at various free [Ca2+]. Fusion is normalized as a percentage of control (No Ca2+). (c) Ca2+ dose–response curve for C2AB binding to LDCVs in the presence of 10 μM EGTA using anisotropy as described in Fig. 2a. Data in (b,c) are mean ± SD from three independent experiments (n = 3). Free [Ca2+] were calibrated using the MaxChelator simulation program. (a–c) ATP was not included in buffer: 120 mM K-glutamate, 20 mM K-acetate, 20 mM HEPES–KOH (pH 7.4), 5 mM MgCl2, and 10 μM EGTA.
Biphasic regulation of Ca2+ on LDCV fusion is mediated by two different mechanisms: (1) millimolar range of [Ca2+] decreases the trans-interaction of synaptotagmin-1 by shielding PIP2 and (2) sub-millimolar range of [Ca2+] above 300 μM increases the cis-interaction of synaptotagmin-1 to its own vesicle membrane20. To further confirm the cis-interaction at higher [Ca2+], we performed anisotropy measurement (Fig. 2a,d) to study the Ca2+ dose–response of the cis-interaction of synaptotagmin-1 in the presence of EGTA instead of ATP (Fig. 3c). Indeed, the cis-membrane interaction of the C2AB domain gradually increased from 300 μM [Ca2+] and remained robust at millimolar [Ca2+](Fig. 3c). Note that ATP and EGTA give rise to different kinetics of the Ca2+ dose–response curves of vesicle fusion and the cis-interaction of synaptotagmin-120, because ATP effectively buffers free [Ca2+] in the range of 10–500 μM, but EGTA cannot efficiently buffer free [Ca2+] in this range.
PIP2 concentration regulates Ca2+ cooperativity of synaptotagmin-1
Synaptotagmin-1 binds to anionic phospholipids by electrostatic interaction and the Ca2+-binding loops of the C2 domains are inserted to anionic phospholipids in a Ca2+-dependent manner; aspartate residues of the Ca2+-binding loops in the C2-domains together with anionic membrane lipids coordinate Ca2+-ions21,23,36. PIP2 enhances Ca2+-sensitivity of synaptotagmin-1 by interacting with the polybasic patch in the C2B domain10,21. Ca2+-cooperativity of synaptotagmin-1 varies among cell types, with the Hill coefficients ranging from ~ 2 to ~ 5. We tested that PIP2 also regulates Ca2+-cooperativity of synaptotagmin-1 for membrane binding (Fig. 4a, Table 2) and vesicle fusion (Fig. 4b, Table 2). Increases of PIP2 concentration from 1 to 5% in the PM-liposomes shifted Ca2+ titration curves for membrane binding to the left side; this change indicates increased Ca2+ sensitivity, but reduced Ca2+ cooperativity (Fig. 4a, Table 2).

PIP2 concentration regulates Ca2+ sensitivity and cooperativity of synaptotagmin-1. (a) Membrane binding of the C2AB domain of synaptotagmin-1 was monitored using anisotropy as in Fig. 2b. Ca2+ dose–response curve for C2AB binding to the PM-liposomes that include PS and PIP2. C2AB binding is presented as a percentage of maximum C2AB binding. (b) Ca2+ dose–response curve for LDCV fusion with the PM-liposomes containing different PIP2 concentrations. Fusion is normalized as a percentage of maximum fusion. Data in (a,b) are mean ± SD from three independent experiments (n = 3). 3 mM MgCl2 and 1 mM ATP were included in buffer, and free [Ca2+] was calibrated using the MaxChelator simulation program.
Next, we observed that Ca2+-cooperativity of synaptotagmin-1 for vesicle fusion was also reduced by increasing PIP2 concentration, correlating with the Ca2+-cooperativity of synaptotagmin-1 for membrane binding. The Ca2+ dose–response curve for LDCV fusion was shifted leftward as PIP2 concentration was increased in the PM-liposomes (Fig. 4b, Table 2). Taken together, high PIP2 concentration increases the sensitivity of synaptotagmin-1 to Ca2+, but lowers Ca2+ cooperativity. These changes imply that increasing the negative electrostatic potential in the plasma membranes attracts Ca2+-bound synaptotagmin-1 with low Ca2+ cooperativity, in which the total numbers of Ca2+ ions coordinated to one synaptotagmin-1 might be reduced to 2–3 (see section “Discussion”).
Discussion
The cis-binding of synaptotagmin-1 occurs in native vesicles such as LDCVs and synaptic vesicles, and inactivates Ca2+-dependent vesicle fusion by preventing the trans-interaction of synaptotagmin-1. Independent groups have confirmed that ATP at physiological concentrations disrupts such cis-interaction of synaptotagmin-111,12,37. Here we show that Ca2+ chelators, including EGTA and polyphosphate anions such as ATP, ADP, and AMP, electrostatically reverse the cis-interaction of synaptotagmin-1. We propose that Ca2+ chelators compete with vesicle membranes that contain anionic phospholipids in binding to Ca2+ and disrupt the cis-interaction of synaptotagmin-1 by charge screening10. However, PIP2 overcomes this inhibitory effect of ATP, because PIP2 dramatically enhances the Ca2+-binding affinity of synaptotagmin-121,38; this high Ca2+ affinity of the C2AB domain to PIP2-containing membranes is not affected by ATP10.
EGTA and 1,2-bis(o-aminophenoxy)ethane-N,N,N0,N0-tetraacetic acid (BAPTA) are well-known and reliable Ca2+ buffers in the range of 10 nM–1 μM [Ca2+] at the typical intracellular pH of 7.233,35. Given that EGTA and BAPTA have a Kd of 67 nM and 192 nM [Ca2+] at pH 7, respectively, and have a higher affinity for Ca2+ than for Mg2+35, both EGTA and BAPTA effectively buffer free [Ca2+] only at concentrations < 1 μM33,39, which is close to intracellular free [Ca2+]. However, EGTA is sensitively dependent on pH35, and BAPTA family has a strong dependence on ionic strength40; importantly, because EGTA and BAPTA have nanomolar-level Kd, they poorly buffer free [Ca2+] in the range of 10–500 μM. In contrast, ATP has Kd 230 µM27 and is an excellent buffer for free [Ca2+] in the range of 10–500 μM33.
Synaptotagmin-1 is a low-affinity Ca2+ sonsor; 10–100 μM [Ca2+] exponentially induce synaptotagmin-1 binding to membrane that contain PS and PIP2 with Kd ~ 50 μM21,26. Therefore, ATP is an appropriate and better Ca2+ buffer than EGTA or BAPTA to study the synaptotagmin-1 activity to bind membrane and trigger vesicle fusion. Indeed, we oberserved that ATP and EGTA result in different kinetics of the Ca2+ dose–response curves of vesicle fusion and of the cis-interaction of synaptotagmin-110,20 (Fig. 3b,c), because ATP has a different Ca2+-buffering capacity than EGTA.
The Kd of low-affinity Ca2+ indicator dyes can vary depending on ionic strength and is changed by anions such as ATP41; e.g., the Kd of low-affinity Ca2+ indicator dyes is increased by ATP and slightly decreased by excess Mg2+. The Kd of Fluo-5N can be altered by the presence of ATP/Mg2+, which makes it difficult to accurately measure free [Ca2+]. ATP binds both Ca2+ and Mg2+ with a different affinity27,33, so computer simulation programs32,35 like the MaxChelator are useful to calibrate free [Ca2+] in the presence of Mg2+, ATP or EGTA by calculating free [Mg2+], [Ca-ATP], and [Mg-ATP]35. We confirmed the MaxChelator-based predictions using a Fluo-5N fluorescent Ca2+ indicator (Fig. 1b).
Both the C2A and C2B domains of synaptotagmin-1 have highly cooperative Ca2+-dependent binding to membranes that contain anionic phospholipids26,42,43,44,45. Furthermore, synaptotagmin-1 contains a polybasic region within the C2B domain that binds to PIP2 in an Ca2+-independent manner46,47 and enhances Ca2+ sensitivity of synaptotagmin-1 membrane binding21 and exocytosis48. The C2AB domain has five possible Ca2+-binding sites22,23; negatively charged oxygen atom from acidic aspartate residues in the C2AB domain and negatively charged oxygen atom from anionic phospholipids provide complete coordination sites for Ca2+23,36. Ca2+ cooperativity of the C2AB domain seems reasonable when the Hill coefficient is ~ 4 to 5, but what regulates Ca2+ cooperativity remains poorly understood, e.g., low Hill coefficient (n, 2–3) in neuroendocrine cells such as pituitary melanotrophs (n, 2.5)18 and chromaffin cells (n, 1.8)19, but high Hill coefficient in synapses including calyx-of-Held synapses (n, 4.2)13,14,15, neuromuscular junctions (n, 3.8)16, and bipolar cells (n, 4)17. We overserved that increasing PIP2 concentration reduces the Hill coefficient, which represents Ca2+ cooperativity (Fig. 4). Our data support that local PIP2 concentration might control Ca2+ cooperativity by allosterically-stabilized dual binding of synaptotagmin-1 to Ca2+ and PIP238.
In this study, we investigate the electrostatic regulation of C2AB binding to vesicle membrane and the PM-liposomes. We have previously observed that Ca2+-independent interactions of the C2AB domain with the PM-liposomes containing anionic phospholipids (10% PS/1% PIP2) is significantly disrupted in the presence of physiological concentration of ATP/Mg2+, but this Ca2+-independent interaction remains strong when the PM-liposomes contain high PIP2 (10% PS/5% PIP2), suggesting that high PIP2 concentrations are required for Ca2+-independent binding of the C2AB domain in physiological ionic strength20. Here, we have used 10% PS/1% PIP2 in the PM-liposomes to selectively examine the Ca2+-dependent membrane interaction and binding of the C2AB domain. However, in the pre-fusion state for vesicle docking and priming, the C2AB domain of synaptotagmin-1 is most likely bound to the plasma membrane through the PIP2-interacting polybasic region of the C2B domain20 or the SNARE complex49 in a Ca2+-independent manner. Ca2+ can induce a re-orientation of the C2AB domain on the plasma membrane by changing the binding mode with the SNARE complex49 or PIP245. This change in orientation may act as a switch to trigger synaptotagmin-1-dependent vesicle fusion in neurons and neuroendocrine cells. Our results do not rule out the possibility for Ca2+-independent interactions of synaptotagmin-1 with the SNARE complex despite extremely weak interaction49 and it remains a topic of further study to include Ca2+-independent interactions of synaptotagmin-1 in our system for physiological relevance.
Data availability
The datasets generated during the current study are available from the corresponding author on reasonable requests.
References
Jahn, R. & Scheller, R. H. SNAREs—engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7, 631–643 (2006).
Brunger, A. T., Choi, U. B., Lai, Y., Leitz, J. & Zhou, Q. Molecular mechanisms of fast neurotransmitter release. Annu. Rev. Biophys. 47, 469–497 (2018).
Ramakrishnan, N. A., Drescher, M. J. & Drescher, D. G. The SNARE complex in neuronal and sensory cells. Mol. Cell Neurosci. 50, 58–69 (2012).
Shao, X. et al. Synaptotagmin-syntaxin interaction: The C2 domain as a Ca2+-dependent electrostatic switch. Neuron 18, 133–142 (1997).
Park, Y. & Ryu, J. K. Models of synaptotagmin-1 to trigger Ca(2+) -dependent vesicle fusion. FEBS Lett. 592, 3480–3492 (2018).
Takamori, S. et al. Molecular anatomy of a trafficking organelle. Cell 127, 831–846 (2006).
Stein, A., Radhakrishnan, A., Riedel, D., Fasshauer, D. & Jahn, R. Synaptotagmin activates membrane fusion through a Ca2+-dependent trans interaction with phospholipids. Nat. Struct. Mol. Biol. 14, 904–911 (2007).
Vennekate, W. et al. Cis- and trans-membrane interactions of synaptotagmin-1. Proc. Natl. Acad. Sci. U S A 109, 11037–11042 (2012).
Holt, M., Riedel, D., Stein, A., Schuette, C. & Jahn, R. Synaptic vesicles are constitutively active fusion machines that function independently of Ca2+. Curr. Biol. 18, 715–722 (2008).
Park, Y. et al. Controlling synaptotagmin activity by electrostatic screening. Nat. Struct. Mol. Biol. 19, 991–997 (2012).
Nyenhuis, S. B., Thapa, A. & Cafiso, D. S. Phosphatidylinositol 4,5 Bisphosphate Controls the cis and trans Interactions of Synaptotagmin 1. Biophys. J. 117, 247–257 (2019).
Dietz, J. et al. Forces, kinetics, and fusion efficiency altered by the full-length synaptotagmin-1-PI(4,5)P2 interaction in constrained geometries. Nano Lett. 22, 1449–1455 (2022).
Sun, J. et al. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature 450, 676–682 (2007).
Lou, X., Scheuss, V. & Schneggenburger, R. Allosteric modulation of the presynaptic Ca2+ sensor for vesicle fusion. Nature 435, 497–501 (2005).
Schneggenburger, R. & Neher, E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406, 889–893 (2000).
Dodge, F. A. Jr. & Rahamimoff, R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J. Physiol. 193, 419–432 (1967).
Heidelberger, R., Heinemann, C., Neher, E. & Matthews, G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature 371, 513–515 (1994).
Thomas, P., Wong, J. G., Lee, A. K. & Almers, W. A low affinity Ca2+ receptor controls the final steps in peptide secretion from pituitary melanotrophs. Neuron 11, 93–104 (1993).
Augustine, G. J. & Neher, E. Calcium requirements for secretion in bovine chromaffin cells. J. Physiol. 450, 247–271 (1992).
Park, Y. et al. Synaptotagmin-1 binds to PIP(2)-containing membrane but not to SNAREs at physiological ionic strength. Nat. Struct. Mol. Biol. 22, 815–823 (2015).
Perez-Lara, A. et al. PtdInsP2 and PtdSer cooperate to trap synaptotagmin-1 to the plasma membrane in the presence of calcium. Elife 5, 25 (2016).
Fernandez, I. et al. Three-dimensional structure of the synaptotagmin 1 C2B-domain: Synaptotagmin 1 as a phospholipid binding machine. Neuron 32, 1057–1069 (2001).
Ubach, J., Zhang, X., Shao, X., Sudhof, T. C. & Rizo, J. Ca2+ binding to synaptotagmin: How many Ca2+ ions bind to the tip of a C2-domain?. EMBO J. 17, 3921–3930 (1998).
Birinci, Y., Preobraschenski, J., Ganzella, M., Jahn, R. & Park, Y. Isolation of large dense-core vesicles from bovine adrenal medulla for functional studies. Sci. Rep. 10, 7540 (2020).
Pobbati, A. V., Stein, A. & Fasshauer, D. N- to C-terminal SNARE complex assembly promotes rapid membrane fusion. Science 313, 673–676 (2006).
Radhakrishnan, A., Stein, A., Jahn, R. & Fasshauer, D. The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 284, 25749–25760 (2009).
Wilson, J. E. & Chin, A. Chelation of divalent cations by ATP, studied by titration calorimetry. Anal. Biochem. 193, 16–19 (1991).
Llinas, R., Sugimori, M. & Silver, R. B. Microdomains of high calcium concentration in a presynaptic terminal. Science 256, 677–679 (1992).
Parekh, A. B. Ca2+ microdomains near plasma membrane Ca2+ channels: Impact on cell function. J. Physiol. 586, 3043–3054 (2008).
Park, Y. et al. alpha-SNAP interferes with the zippering of the SNARE protein membrane fusion machinery. J. Biol. Chem. 289, 16326–16335 (2014).
Gumurdu, A. et al. MicroRNA exocytosis by large dense-core vesicle fusion. Sci. Rep. 7, 45661 (2017).
Schoenmakers, T. J., Visser, G. J., Flik, G. & Theuvenet, A. P. CHELATOR: An improved method for computing metal ion concentrations in physiological solutions. Biotechniques 12(870–874), 876–879 (1992).
Patton, C., Thompson, S. & Epel, D. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium 35, 427–431 (2004).
Kabbara, A. A. & Allen, D. G. The use of the indicator fluo-5N to measure sarcoplasmic reticulum calcium in single muscle fibres of the cane toad. J. Physiol. 534, 87–97 (2001).
Bers, D. M., Patton, C. W. & Nuccitelli, R. A practical guide to the preparation of Ca(2+) buffers. Methods Cell Biol. 99, 1–26 (2010).
Murray, D. & Honig, B. Electrostatic control of the membrane targeting of C2 domains. Mol. Cell 9, 145–154 (2002).
Zanetti, M. N. et al. Ring-like oligomers of Synaptotagmins and related C2 domain proteins. Elife 5, 25 (2016).
Kobbersmed, J. R. L., Berns, M. M. M., Ditlevsen, S., Sorensen, J. B. & Walter, A. M. Allosteric stabilization of calcium and phosphoinositide dual binding engages several synaptotagmins in fast exocytosis. Elife 11, 20 (2022).
Bers, D. M. A simple method for the accurate determination of free [Ca] in Ca-EGTA solutions. Am. J. Physiol. 242, C404-408 (1982).
Tsien, R. Y. New calcium indicators and buffers with high selectivity against magnesium and protons: Design, synthesis, and properties of prototype structures. Biochemistry 19, 2396–2404 (1980).
Woehler, A., Lin, K. H. & Neher, E. Calcium-buffering effects of gluconate and nucleotides, as determined by a novel fluorimetric titration method. J. Physiol. 592, 4863–4875 (2014).
Chapman, E. R. How does synaptotagmin trigger neurotransmitter release?. Annu. Rev. Biochem. 77, 615–641 (2008).
Davletov, B. A. & Sudhof, T. C. A single C2 domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid binding. J. Biol. Chem. 268, 26386–26390 (1993).
Tran, H. T., Anderson, L. H. & Knight, J. D. Membrane-binding cooperativity and coinsertion by C2AB tandem domains of synaptotagmins 1 and 7. Biophys. J. 116, 1025–1036 (2019).
Katti, S., Nyenhuis, S. B., Her, B., Cafiso, D. S. & Igumenova, T. I. Partial metal ion saturation of C2 domains primes synaptotagmin 1-membrane interactions. Biophys. J. 118, 1409–1423 (2020).
Kuo, W., Herrick, D. Z., Ellena, J. F. & Cafiso, D. S. The calcium-dependent and calcium-independent membrane binding of synaptotagmin 1: Two modes of C2B binding. J. Mol. Biol. 387, 284–294 (2009).
Vrljic, M. et al. Post-translational modifications and lipid binding profile of insect cell-expressed full-length mammalian synaptotagmin 1. Biochemistry 50, 9998–10012 (2011).
Li, L. et al. Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J. Biol. Chem. 281, 15845–15852 (2006).
Rizo, J., David, G., Fealey, M. E. & Jaczynska, K. On the difficulties of characterizing weak protein interactions that are critical for neurotransmitter release. FEBS Open Bio 12, 1912–1938 (2022).
Acknowledgements
This work was supported by the grant from Qatar Biomedical Research Institute (Project Number SF 2019 004 to Y.P.).
Funding
Open Access funding was provided by the Qatar National Library.
Author information
Authors and Affiliations
Contributions
Y.P. and H.Y.A.M. purified vesicles and performed experiments. Y.P. collected and analyzed data. Y.P. wrote the manuscript and all authors read and provided their comments.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ali Moussa, H.Y., Park, Y. Electrostatic regulation of the cis- and trans-membrane interactions of synaptotagmin-1. Sci Rep 12, 22407 (2022). https://doi.org/10.1038/s41598-022-26723-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-26723-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
