
Abstract
This study was to understand the genetic structure and diversity of the Korean Malus species. We used genotyping-by-sequencing (GBS) technology to analyze samples of 112 individuals belonging to 18 populations of wild Malus spp. Using GBS, we identified thousands of single nucleotide polymorphisms in the species analyzed. M. baccata and M. toringo, two dominant mainland species of the Korean Peninsula, were distinguishable based on their genetic structure. However, M. toringo collected from Jeju Island exhibited a different genetic profile than that from the mainland. We identified M. cf. micromalus as a hybrid resulting from the Jeju Island M. toringo (pollen donor) and the mainland M. baccata, (pollen recipient). Putative M. mandshurica distributed on the Korean Peninsula showed a high structural and genetic similarity with M. baccata, indicating that it might be an ecotype. Overall, this study contributes to the understanding of the population history and genetic structure of Malus in the Korean Peninsula.
Similar content being viewed by others


Introduction
Malus Mill. (tribe Maleae, Rosaceae) is an economically important genus comprising 25–55 taxa and is widely distributed in the temperate regions of the northern hemisphere1,2,3. Malus domestica Borkh., a representative crop of apples, underwent hybridization and gene introgression with various wild Malus species over decades4. Interspecific hybridization in Malus is well documented5,6,7,8,9. Although several taxonomists proposed reasonable classification systems based on its morphology10,11,12,13,14,15,16,17,18,19, reticulated processes of Malus have hindered the accuracy of its phylogenetic inferences5,6,7. The concept of hybridization leading to the formation of new species has been proposed in the 1950s20. Hybrids usually possess an intermediate phenotype of the two parental species; describing their morphology can be subjective and difficult to predict owing to the combined expression of parental genes21. Some hybrids are identified based on their morphological properties, but their origins remain unknown (e.g., Malus × robusta Rehd. and Malus × micromalus Mak.; assessed at 30th April 2022 https://www.treesandshrubsonline.org/). Similarly, distinguishing interspecific hybrids of Malus among species of the Korean Peninsula can be difficult. Since the identification of two subspecies by Nakai10 (Pyrus baccata var. sibirica Maxim. [synonym of Malus rockii Rehder] and Pyrus baccata var. manshurica Maxim. [synonym of Malus manshurica (Maxim.) Kom. ex Skvortsov]), up to eight taxa including cultivar have been reported in the Korean Peninsula over the last 20 years (Table S1). M. baccata is primarily distributed in northern Asia (Bhutan, China, India, Kashmir, Korea, Mongolia, Nepal, and Russia) and has been widely used as a rootstock for breeding because it is disease-free and cold-resistant22,23,24.
M. baccata and M. toringo (Siebold) Siebold ex de Vriese are the most widely distributed representative taxa in the Korean Peninsula10,11,12,13,14,15,16,17,18,19. M. baccata is predominantly distributed along the main mountain-range connecting the north and the south (called Baekdu-daegan) up to the central region and is vertically distributed from the lowlands to about 1500 m above sea level (Fig. 1). M. toringo, a 2–6 m small ornamental tree or shrub, is distributed in East Asian countries including China, Japan, Korea, and Russia25. In the Korean Peninsula, M. toringo is distributed in the central and central-southern regions and in the Jeju Island mainly growing in lowland areas (Fig. 1)11,12.

Heat map of 833 specimens deposited in both the Korea National Arboretum Herbarium (KH) and the National Institute of Biological Resources Herbarium (KB) were visualized using Qgis v3.26.1. Red color indicates M. toringo and green represents M. baccata. The black square indicates the sampling sites information for this study. Map source: https://www.gisdeveloper.co.kr/.
M. baccata and M. toringo can be morphologically differentiated based on their leaf lobes (M. baccata: not lobed and M. toringo: 3–5 lobed leaf), petiole (M. baccata: 2–5 cm and M. toringo: 1.5–2.5 cm), pedicel length (M. baccata: 4–7 cm and M. toringo: 1.2–4 cm), bud characteristics (M. baccata lineage: convoluted bud and M. toringo lineage: folded bud), and fruit size (M. baccata: 6–9 mm and M. toringo: 4–6 mm)11,12,13,14,25,26. However, individuals with intermediate morphology between M. baccata and M. toringo possess the following characteristics27: (1) ovate leaf but slightly lobed, short petiole and pedicel; (2) slightly lobed leaf, long petiole and pedicel; and (3) variations in fruit sizes.
Previous research has not deduced consistent results with regard to the relationship between the two species. Although phylogenetic studies performed using sequences of partial chloroplast and nuclear regions have confirmed that M. baccata and M. toringo are distinguished based on their morphological characters28,29, maximum likelihood (ML) tree constructed using 79 protein-coding chloroplast genes clustered the sequences based on geographic distribution, and not the species monophyletic group30. In addition, the unweighted pair group method with arithmetic mean (UPGMA), based on morphological characters and random amplified polymorphic DNA (RAPD) analysis, revealed that the individuals formed two distinct clusters based on species data31,32. Intermediate morphological features14,27 and inconsistent molecular evidence30 have raised doubts over the natural hybridization of the two species as suggested by Chang et al.14; however, no assessment has been conducted at the population level.
M. micromalus Makino is another putative hybrid species, which was first described by Makino in 1908, and introduced in South Korea from China33. This species is presumed to have resulted from a cross between M. baccata and M. spectabilis (Sol.) Borkh25,34. M. micromalus is distributed at high elevations in areas such as the Hallasan Mountain (> 1500 m) on Jeju Island, which is approximately 140 km south of the Korean Peninsula mainland33. The species has the following morphological characteristics: tomentose in the midrib, pubescent petiole when young, pedicels tomentose-pubescent, and persistent calyx at pomes10,11,12,13,14,15,16,17,18,33. Although this taxon has been described in various studies, there are discrepancies in its classification12,13. Moreover, literature on whether calyx is caducous or persistent is inconsistent11,15,18,33, making it difficult to identify distinct morphological differences between M. micromalus and other related species.
Another relevant species is M. mandshurica (Maxim.) Kom. ex Juz., which is distributed in China, Russia, Japan, North Korea, and South Korea25. Although M. mandshurica possess similar characteristics as M. baccata, it exhibits sparse pubescence on the petiole and abaxial pubescence on leaves, or subglabrous along midvein and lateral veins25,26. However, preliminary investigation of specimen and image data of both herbariums (Herbarium of Korea National Arboretum: KH; National Institute of Biological Resources: KB) revealed that it was difficult to differentiate M. mandshurica from M. baccata. Moreover, there have been discrepancies in the ranking of this taxon, with some studies classifying it as a separate species and others classifying it as a variety of M. baccata16,19,25,26. Additionally, M. mandshurica has also been classified as an ecotype and treated as a synonym of M. baccata14,35. Previous studies that performed simple sequence repeat (SSR) analyses using several markers could not differentiate the two species because the genetic distance within M. baccata taxa was scattered across other Malus species36. Additionally, the low resolution of a phylogenetic approach cannot efficiently represent the relationship between the two species29,32,37. Species boundaries are often difficult to identify morphologically; therefore, integrated taxonomy which includes additional information, such as molecular data, biogeography, and ecology is required38,39. When several lines of evidence (e.g., morphology, molecules, ecology, or distribution) independently indicate a certain species boundary, discretely evolving lineages can be identified40,41,42.
Recently, next-generation sequencing (NGS) has identified sufficient single nucleotide polymorphisms (SNPs) from population genetic studies to facilitate the study of genetic diversity in Malus43,44,45. Molecular diagnosis of genetic diversity within the nuclear genome using GBS has the potential to examine hybrid signals, introgressions, and the population–species boundary46,47,48. This is the first study to explore the possibility of hybridization and evolutionary relationship of Malus across the Korean Peninsula. The aim of this study was to identify Malus-specific SNPs in the Korean Peninsula and establish their phylogenetic relationships by GBS approaches. The objectives of this study were to (1) verify whether there is a hybrid or genetic exchange between M. baccata and M. toringo; (2) identify the entity of M. cf. micromalus distributed in Jeju Island; (3) compare the genetic structure of M. toringo distributed in the mainland and Jeju Island; and (4) verify the entity of M. cf. madshurica collected in the Korean Peninsula.
Methods
Sample collection and DNA extraction
In this study, we collected a total of 112 individuals (5 taxa 18 populations) from natural habitats (Table S2) as well as Sequence Read Archive (SRA) data (Table S3) from the National Center for Biotechnology Information (NCBI). A total of 25 accessions of SRA data (10 accessions of M. baccata, three accessions of M. toringo, four accessions of M. mandshurica, four accessions of M. micromlaus, and three accessions of M. spectabilis) were obtained from a previously conducted GBS study49. During the field survey, young leaves were collected from 112 individuals belonging to 18 populations (six populations of M. baccata, seven populations of M. toringo, one population of M. mandshurica, one population of M. cf. micromalus, and three populations of Malus sp. cultivar) in South Korea (Table 1, Fig. 1). The location of the collected samples was 30 km away from each other in the mainland and 10 km away from each other in the Jeju Island (73 km east–west and 31 km north–south). Total genomic DNA was extracted from silica-dried plant material using a DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA), following the manufacturer’s instructions. All voucher specimens were deposited in the KH (detail of voucher number is listed in S2).
GBS library construction and NGS
The GBS libraries of 112 individuals were constructed as previously described50, with minor modifications. Briefly, DNA samples were digested with ApeKI (New England Biolabs, Ipswich, MA, USA). Adapters were subsequently ligated to the sticky ends by adding T4 DNA ligase (200 U; MGMED, Korea) to each well. Digested DNA samples, each with a different barcode adapter, were combined and purified using a commercial kit (QIAquick PCR Purification Kit; Qiagen, USA), according to the manufacturer’s instructions. The libraries were sequenced using Illumina NextSeq500 for 95 samples and Hi-seqX for 20 samples.
Mapping to reference genome and SNP calling
After sequencing, raw reads were de-multiplexed according to the barcode sequences using the “process_radtags” function in STACKS v2.60 with default parameters: “—inline-null” for barcode option and “-e apeKI” for enzymes option51. Reads were trimmed, and adaptors were removed using cutadapt 3.552. In this study, two data matrices were used: (1) data set A: SRA data (25 accessions) + 112 individuals of Malus species collected from the Korean Peninsula; (2) data set B: 112 individuals of Malus species. A total of 137 individual reads were aligned to the Malus domestica chromosome sequence retrieved from NCBI (ASM211411v1) to generate the BAM files using BWA v0.7.17 and SAMtools v1.9 with the default parameters53,54,55 (Tables S2 and S3). The generated BAM files were input into the gstacks, a core program included in STACKS v2.60, under default parameters.
Two major output files were generated (catalog.fa.gz and catalog.calls) which were subsequently input into the “Populations” program of STACKS v2.60 that utilize SNP calling. The ‘-p’ option, which indicates the minimum number of populations required to process a locus, was set to 23 for 137 accessions (data set A) and 18 for 112 accessions (data set B). The designation of parameters 23 and 18 in two data sets for SNPs calling is a strict strategy to extract only the SNPs observed in all groups. The minimum percentage of individuals in a population required to process a locus for that population (-r) was set to 1 for 137 accessions of data set A because for the maximum value (1) is to minimize the error of SNPs generated due to external data. And we set the 0.8 for 112 accessions of data set B. The options listed were later equally applied to both datasets. The minimum percentage of individuals in/across a population required to process a locus for that population were increased (--min-samples-per-pop 1, --min-samples-overall 1). Additionally, we set the minimum minor allele frequency to 0.05 (--min_maf 0.05), maximum observed heterozygosity to 0.95 (--max_obs_het 0.95) and restricted the study to only the first SNP per locus (--write-single-snp).
Distribution map
Qgis v3.26.1 desktop application (https://qgis.org/en/site/) was used for visualizing the heat map and the source of GPS coordinates was collected from 833 specimens deposited in KH and KB herbariums.
SplitsTree analysis vs minimum spanning network
To explore the genetic distance of data set A and B, we constructed a SplitsTree network using strictly filtered 563 and 613 SNPs, respectively. The vcf file generated from “populations” in STACKS v2.60 was converted into the FASTA format from the vcf2phylip (https://github.com/edgardomortiz/vcf2phylip.git), with the value for minimum samples per locus (MIN_SAMPLES_LOCUS) set to 137 in data set A and 112 in data set B. The network was created and visualized using SplitsTree4 software56.
Population genetic analysis
To investigate the population structure, we used VCF file generated using the “populations” function in STACKS v2.60. To illustrate the relationship among various individuals, principal component analysis (PCA) was conducted based on 563 SNPs (137 accession) in data set A and 8426 SNPs (112 accession) in data set B, using graph Laplacian PCA (gLPCA)57 and plotted using R studio58. To investigate the population structure, plink files were converted into BED files, using Plink v1.0759, which were then used as input files to determine cross validation (CV) of K = 1–10 values, using admixture_ linux-1.3.060. The graphical display of the population structure was generated using DISTRUCT61.
Genetic diversity and differentiation
Common measures of genetic diversity, including private allele number (AP), percentage of polymorphic loci (%Poly), observed and expected heterozygosity (Ho and He), nucleotide diversity (π), inbreeding coefficient (FIS), and population differentiation (pairwise FST), were calculated using the “populations” function in STACKS v2.60.
An analysis of molecular variance (AMOVA) was performed to estimate genetic variation among and within populations, using the adegenet62 and poppr63 package in R studio64. The AMOVA analysis was performed for four categories: (1) all samples (C1–C7; 112 individuals); (2) M. toringo, including C3 and C5 (60 individuals); (3) M. baccata (C1 and C2) and M. mandshurica (C7; 39 individuals); (4) M. baccata (C1 and C2), M. mandshurica (C7), and M. micromalus (C6; 52 species), as well as M. toringo (C3 and C5) and M. micromalus (C6; 73 individuals).
Migration rates calculation
To investigate the ancient gene flow, we utilized a coalescent approach implemented in MIGRATE- N 3.6.11 to calculate the migration rates between groups representing populations65,66. Structure file produced from the “populations” program of STACKS v2.60 was converted into the SNP model (heat map data) using the “vcfR2migrate” plug in vcfR package67. We redefined groups based on the genetic clusters inferred from STRUCTURE (Table 1). We set starting values of θ and M with an “Estimate with FST measure.” The gene flow parameter M was used (M = m/μ; m, immigration rate per generation; μ, mutation rate), with the mutation rate as a constant68. A more permissive acceptance criteria was applied (“heating”) with four chains set at different temperatures (1.0, 1.5, 3.0, and 100,000). The run used 100 long-samples with an increment of 10,000 (1,000,000 iterations) after a burn-in of 100,000. The number of migrants per generation (Nm) was calculated using the following equation: Nm = [(θ x * M y → x)/4])68.
Ethics declarations
The materials used in this study are not included IUCN red list. Sample collections was conducted in compliance with the regulations of the Act on the creation and furtherance of arboretums and gardens.
Results
SNP discovery using data set A and B
A total of 397,876,068 raw reads (average of 3.5 million reads per sample) were generated from data set B (Table S2). After quality filtering, a total of 368,008,854 reads from 112 accessions and 46,503,805 reads from 25 accessions (five taxa, NCBI SRA data) were aligned to the Malus domestica reference genome. Approximately 83.2% of the 112 accessions and 92.3% of the 25 SRA data were mapped to the genome (Table S3). Subsequent analysis was performed using the generated BAM file, and SNP calling was performed with two separate data consisting of 112 accessions (data set B) collected from the Korean Peninsula and 137 accessions from data set A, including SRA data (Tables S2 and S3). Finally, a total of 562 SNPs were identified from 23 populations in data set A and 8426 SNPs were identified in data set B representing 18 Korean populations.
Phylogenetic network relationship and genetic structure of data set A and B
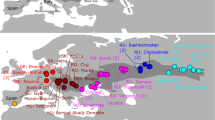
Phylogenetic networks were constructed to determine the relationships among Malus and its relatives (Fig. 2A,B). Both of the phylogenetic networks generated using data sets A and B were divided into two clusters (Fig. 2A,B). Cluster I in Fig. 2A included all accession of M. baccata, M. micromalus (including M. cf. micromalus in JEJU type population), M. mandshurica (including M. cf. mandshurica in Mand_GW population), and M. spectabilis. Within cluster I, comparisons between collected samples and SRA data formed a subgroup based on their original collection site. Particularly, four SRA-M. baccata (SRR12446151: Korea_Wild_Apple; SRR12446074 and SRR12383338: China; SRR12446158: unkown49) were closely related to the Korean population (Fig. 2A). However, most of the SRA data of M. mandshurica, M. micromalus, M. spectabilis, and M. baccata (SRR12383267, 12383311, 12446095, 12446077, 12446141, and 12446144) were found to be of a different lineage, a long branch that was separately from the Korean population. Nevertheless, no clear distinction at the species level could be observed in the SRA branch. Putative M. micromalus collected from Jeju Island (hyh381-398; Table S2) formed separate clusters from SRA-M. micromalus, with some individuals related to M. baccata, while more than half were positioned at the border with cluster II. Similarly, M. mandsurica did not form a cluster with SRA-M. mandsurica data and M. cf. mandsurica collected from the Korean Peninsula. Furthermore, cluster II included all M. toringo accessions and three cultivar that maintained an independent group. Two separate groups were identified within the M. toringo accessions in SplitsTree. SRA-M. toringo was more similar to the cultivar than the Korean M. toringo. Additionally, PCA divided M. toringo into two groups: PC1 and PC2 (Fig. 3).

Unrooted network of 137 species of the Malus genus. (A) Neighbor-Net constructed by SplitsTree using 563 SNP markers of data set A. (B) Neighbor-Net constructed by SplitsTree using 612 SNP markers of data set B. Red dotted line indicates the division of 137 species based on the different leaf morphological characters (Cluster I: Leaf blade not lobed, and Cluster II: Leaf blade usually lobed). Capital in samples name indicate the abbreviation of scientific name (B: M. baccata, C: Malus cultivar, T: M. toringo, Ma: M. mandshurica, Mi: M. micromalus).

PCA of 137 Malus species was conducted with the top two components.
Population genetic structure and relationship analysis using data set B
Population genetic structure analysis (PCA and STRUCTURE) was conducted based on 8426 SNPs, using the STACKS v.2.60 software. The eigenvalues of PC1 (47.7%) and PC2 (11.35%) explained approximately 59.1% of the total variance (Fig. 4). PC1 separated the two major populations of wild Malus (excluding cultivar) from M. toringo, M. baccata, M. cf. mandshurica, and M. cf. micromalus. PC2 separated M. toringo into two groups, according to the geographic distribution. Three populations of cultivar were located in the center of M. toringo in PC2 (Fig. 4). From the assigned group, all samples clustered with each other except for M. cf. micromalus on JEJUtype population. JEJUtype accessions (hyh382, hyh386, hyh387, and hyh392) formed a group which was distinct from the other accessions in quadrant 4 (Fig. 4). This phenomenon was also observed in the STRUCTURE data (Fig. 5). We set the range of delta K as 2–10 for ancestral populations. Although K = 5 was determined as the optimal value from the CV error, the difference in CV values was insignificant; therefore, the results of K = 2–10 are presented (Fig. 5). M. toringo was divided into two distinct genotypic groups, one group with five populations (T_JB1, T_JB2, T_JB, T_GN, and T_JN) and the other with two populations (T_JJ2 and T_JJ3). The distinct groups remained consistent across the entire range of K values (Fig. 5). Two populations (T_JJ2 and T_JJ3) sampled from the southern Jeju Island of the Korean Peninsula showed a different genetic profile compared to that of the mainland M. toringo group (Fig. 5). Three accessions, including three cultivar populations (C_JJ1, T_GW, and T_IC) contained some of the mixed genetic compositions of M. toringo. Among the M. baccata populations, eight were divided into two clusters. The composition of the M. cf. mandshurica population revealed geographical similarities rather than species entity. The M. cf. micromalus (JEJUtype) population collected from Jeju Island shared both M. toringo and M. baccata genetic composition. Specifically, the M. baccata genotype was mixed with geographically more distant populations (B_GW1, B_GW2, and B_GB1) than closer populations (B_GB2, B_JB, and B_GN). The highly polymorphic and diverse JEJUtype population exhibited mixed components of M. baccata (B_GW1, B_GW2, and B_GB1) and M. toringo (T_JJ2 and T_JJ3).

PCA of 112 Malus species was conducted with the top two components.

Genetic population structure prediction (K = 2–10). The distribution of the accessions to different populations is indicated by the color code.
Population genetic diversity
The mean expected heterozygosity (He) of the 18 Malus populations varied from 0.074 in T_GW to 0.203 in the island JEJUtype. The observed heterozygosity (Ho) of the 18 Malus populations varied from 0.074 in T_GW to 0.260 in B_GN. Wright’s F-statistic (FIS) of the 18 Malus populations ranged from -0.170 in B_JB to 0.042 in Mand_GW (Table 1). When the Ho value is higher than He, FIS has a negative value, with most Ho values being higher than He (Table 1). The average nucleotide diversity (π) ranged from 0.083 in T_GW to 0.211 in the JEJUtype. High π was observed in three M. baccata (B_GB2, B_JB, and B_GN) groups, which were geographically distributed close to the M. toringo population (Fig. 6). The highest π was recorded in the M. cf. micromalus population from Jeju Island (Table 1).

Pie charts of the location map used for the genetic population structure analysis of 18 Malus populations. The pie charts show the frequency of each cluster in a population based on STRUCTURE results (K = 5). Map source: https://www.gisdeveloper.co.kr/.
Genetic differentiation
Pairwise FST (genetic differentiation index) between populations ranged from 0.011–0.47 (Table 2); a similar trend was observed after the genetic structure analysis (Figs. 5 and 6). The lowest FST value was identified among M. toringo populations, specifically among five mainland groups (T_JB, T_JB1, T_JB2, T_GN, and T_JN) with similar geographical distributions. The lowest FST value was identified among the three M. baccata populations (B_GB2, B_GN, and B_JB). These findings indicated that the genetic difference within populations was greater than those between populations as well as the occurrence of gene flow between populations. Most FST values between the M. baccata and M. mandshurica groups as well as M. toringo were more than 0.3 (Table 2). FST values between JEJUtype, three M. baccata populations (B_GB1, B_GW1, and B_GW2), and three M. toringo populations (T_IC, T_JJ1, and T_JJ2) were extremely low. Additionally, the FST values between M. mandsurica and M. baccata (0.074–0.181) were lower or similar to the FST values measured between M. baccata populations, indicating the occurrence of gene flow between populations69,70.
Molecular variance and migration rates
AMOVA results (Table 3) revealed that 52% of the genetic variation occurred between groups and 40% occurred within samples. Within samples, variation from the three measurements (M. toringo; M. baccata, and M. mandshurica; and M. baccata, M. mandshurica, and M. micromalus) was extremely high (84%, 96%, and 94%, respectively). Our analysis further revealed that between the two groups of M. toringo and M. micromalus, 52% of the total variations occurred within samples, while the remaining 37.8% and 19.2% of variations occurred between groups and between populations/within group, respectively. The results of the migration rate analysis revealed that the Nm between all populations without model specification was 0.373, with the highest value obtained (0.748) at M3 → 5 (Table S4).
Discussion
Although previous studies have examined the genetic diversity of Malus in Europe71,72,73 and central Asia74,75, with several reports on cultivar76,77, studies on Malus in the Korean Peninsula are limited. In this study, we evaluated the genetic diversity and structure of Malus species in the East Asia, using a GBS-based analysis. The molecular diagnosis used in this study provides extensive information on variations of the Korean Malus, which could facilitate the tracking of geneflow, and deciphering of taxonomic delimitation, and historical evolution of divergence.
The level of heterozygosity was significantly lower within the Korean Malus population (mean He = 0.07–0.2; mean Ho = 0.07–0.26; Table 1) compared with that reported in previous studies (He > 0.6; Ho > 0.7)71,72,78. However, this discrepancy could be attributed to differences in research methodology79. Moreover, the findings of the present study are in agreement with those of previous GBS-based studies80. However, the low diversity observed in most Korean Malus populations is unusual for Malus (Table 1), which is self-incompatible and relies on cross pollination81. Other evolutionary forces affecting genetic diversity include mode of reproduction, geographical distribution, and population size, among which the mode of reproduction is highly relevant as it involves direct genetic exchange82.
The FIS values obtained in this study were close to zero in six populations and negative in the remaining ones (Table 1). Also, the FIS values show regional differences even within the same species (Table 1 and Fig. 6). For instance, the FIS value of M. baccata converges to 0 and − 0.1 in C1 and C2, respectively (Table 1). Generally, outcrossing species are known to exhibit a higher genetic diversity than that of selfing species83; nevertheless, it is surprising that the genetic diversity in our results is low in Malus (He, Ho, and Pi; Table 1). It is possible that the low genetic diversity is a result of spatial isolation80, or that Malus, specifically in the Korean Peninsula, has high homozygosity. Geographical barriers and small population sizes facilitate genetic drift and bottlenecks84,85. Geographically, the Korean Peninsula is surrounded by the sea along the east, west, and south coasts which can limit gene flow into the Korean Malus population from marginal states. Such spatial or landscape structure barriers can increase genetic isolation80.
Molecular diagnosis of M. baccata and M. toringo distributed in the Korean Peninsula
M. baccata and M. toringo from the Korean Peninsula can be differentiated and described based on distinct morphological features, such as leaf lobes, length of petioles, pedicel, fruit size, and bud character11,12,13,14,25,26,27,34. However, morphologically intermediate species from geographically overlapping regions have been reported27. Contrary to our hypothesis that hybridization would be observed, our results indicated the presence of two genetically distinct groups, M. baccata and M. toringo, without hybrid signals (Figs. 2, 3, 4, and 5). From K = 4–7, the genetic structure between M. baccata and M. toringo was clearly distinguished, and no indication of hybridization between the two species was detected in the mainland population (Figs. 5 and 6). In B_GN, some individuals showed mixed profiles that were affected by cultivar genotypes of individuals planted in the recreation forest (Fig. 5). Additionally, traces of genetic exchange between JB and GN have not been identified in this study despite the similar forest distribution of the two species, implying reproductive isolation between the two species (Fig. 6). PCA indicated that M. baccata and M. toringo were clearly distinguished based on the PC1 component (Fig. 4). Moreover, most M. baccata populations share similar genetic profiles; the mainland M. toringo was clearly distinguishable from the Jeju Island group based on the PC2 component (Fig. 4).
Notably, it is important to understand the distribution and spatial pattern of living organisms to interpret their biological differentiation and ecological evolutionary history86. The segregation of biogeographic regions is based on geography, geology, and climate87. Several studies attempted to subdivide the biogeographic region of the Korean Peninsula88,89,90,91. Jung and Cho91 segregated four biogeographic regions based on the coordinates of 310,000 vascular plant specimens collected from the Korean Peninsula. In the population groups redefined in Table 1, C1 and C7, which share similar genetic profiles (Fig. 6) belonged to the central Korean Peninsula, named Zone I (Cold floristic zone) and Zone II (Cool floristic zone)91. C3, which included five populations of M. toringo with similar genetic profiles belonged to Zone III (Warm floristic zone) and Zone IV (Miratic zone 2)91. C2 belonged to the boundary between Zone II and Zone III in the low-altitude regions of the Korean Peninsula91, which is similar to the biogeographic regional boundary proposed by Lee and Yim90. Nevertheless, the possibility that the distance between populations, altitude, and climatic zone changes affect the genetic composition of Malus cannot be ruled out (Table 2).
PCA and STRUCTURE analysis results indicated the segregation of M. toringo into two distinct groups: mainland and Jeju Island populations (Figs. 4, 5, 6). Additionally, the 35.65% difference “between pop” of M. toringo populations in the molecular variation analysis suggests that the analyzed individuals could belong to more than one group (Table 3). With an increase in the K value, the difference between the two groups remained distinct, suggesting that gene flow was not a recent occurrence (Fig. 5). A historical migration rate of 0.748 was observed between the mainland M. toringo (C3 in Table 1) and Jeju Island M. toringo (C5 in Table 1 and Table S4). Genetic variations resulting from mutation, recombination, and division of basic gene pool can cause the emergence of a morphologically or genetically evolutionarily significant units82,92. Our results imply M. toringo is genetically difference groups between mainland (C3 in Table 1) and Jeju Island (C5). However, our data have limitation for verifying the evolutionarily significant units as species. Therefore, further studies in aspect of morphological and molecular methods are necessary with extensive sampling.
M. cf. micromalus on Jeju Island
Interspecific hybridization of Malus has been inferred in the past7 and confirmed by a recently conducted phylogenomic analysis of Malus hybridization93. The genotypes of symmetric hybrids are composed of half of each putative parent genotype94 and share similar structural patterns with the cultivar cluster identified in this study (Fig. 5). It has been hypothesized that M. micromalus is a hybrid of M. baccata and M. spectabilis (as detailed in the “Introduction”); however, this has never been evaluated. The M. cf. micromalus population (JEJUtype) collected from Jeju Island did not exhibit a close relationship with SRA-M. micromalus, nor did it show a close relationship with M. spectabilis (Fig. 2A). Some individuals within the JEJUtype clustered with M. baccata while others were positioned between the M. baccata and M. toringo lineage (Fig. 2A).
The present study indicated that the JEJUtype did not cluster with previously reported targets (M. micromalus; Fig. 2) and should be treated as a separate lineages generated by hybridization between C5 (JJ2 and JJ3) and M. baccata (Fig. 5)95,96. Additionally, one-sided genetic invasion from C5 to M. baccata is presumed to be the result of asymmetric hybridization, where C5 acts as a pollen donor and M. baccata as a pollen acceptor (Fig. 5)97,98,99. Only a few JEJUtype individuals were identified as F1 descendants with half the genotypes of both parental species (Fig. 5). A similar case has been reported in association with Prunus yedoensis in Rosaceae100. JEJUtype species are characterized by lanceolate-ovate leaves similar to those of M. baccata and short peduncles similar to those of M. toringo; the species are also characteristic of small-sized, narrow, and unlobed leaves (Fig. 7). The major taxonomical key character of M. micromalus, the persistence of sepal in fruit, was not identified in this population. Based on these findings, we suggest that JEJUtype (Malus cf.), which has been the center of debate for nearly 100 years since Makino described a new species (M. micromalus) in 190833, is a different entity from M. micromalus. To better understand the evolutionary history of this hybrid, extensive sampling is necessary to identify genetic clues in their natural habitat and assess their phenetic characters, particularly in the species-rich neighboring countries (China and Japan).

Photos of habitat, leaf, inflorescence, and fruits of Malus in the Korea Peninsula: A–D: M. micromalus (in Jeju Island). E–G: M. Baccata (in GW). H–J: M. Toringo (in JB). All photograph was taken by Y.-H.H.
Taxonomical decision of M. mandshurica
The M. cf. mandshurica (C7) analyzed in this study exhibited similar morphological characteristics with previously identified M. mandshurica, such as tomentose hair on petiole and adaxial leaf pubescence25,26. However, SplitsTree analysis indicated that there was no close relationship between M. cf. mandshurica accessions (population: Mand_GW) and SRA-M. mandshurica, which is a remarkable finding (Fig. 2). Additionally, the analyses conducted in this study indicated that C7 shares a similar genetic profile with M. baccata (Figs. 4 and 5; Tables 2 and 3). However, previous studies have reported inconsistencies in the identification/characterization of M. mandshurica, with some studies reporting a close relationship between M. mandshurica and M. baccata or M. micromalus28,29,31,37. Physiologically, studies have reported that the leaves of plants become smaller and thicker, and the amount of hair increases as the altitude increases101,102; thus, further studies are needed to analyze the status of M. mandshurica at the species level.
Conclusion
The current study primarily explored the genetic structure and geneflow of Malus in the Korean Peninsula using GBS analysis. Following sequencing, we identified high quality SNPs (8426) using the reference mapping method. Notably, majority of the Malus populations distributed in the Korean Peninsula form geographically distinct groups that coincide with those in the floristic zones91. Contrary to our prediction that hybridization would occur between M. baccata and M. toringo, we observed that the two species were genetically differentiated. Putative M. micromalus from the Jeju Island might represent a new hybrid. Our findings provide valuable insights into the genetic profile of Korean Malus. However, thorough morphological studies with extensive sampling are needed to clarify these species attributes.
Data availability
All sequencing data analyzed in this study are publicly available from the National center of Biotechnology Information (https://www.ncbi.nlm.nih.gov/) under the BioProject ID: PRJNA826537.
References
Phipps, J. B., Robertson, K. R., Smith, P. G. & Rohrer, J. R. A checklist of the subfamily Maloideae (Rosaceae). Can. J. Bot. 68, 2209–2269 (1990).
Korban, S. S. & Skirvin, R. M. Nomenclature of the cultivated apple. Hortic. Sci. 19, 177–180 (1984).
Morgan, J. & Richards, A. The New Book of Apples: The Definitive Guide to Over 2,000 Varieties (Ebury Press, 2002).
Sun, X. et al. Phased diploid genome assemblies and pan-genomes provide insights into the genetic history of apple domestication. Nat. Genet. 52, 1423–1432 (2020).
Janick, J. Horticultural Reviews, Volume 29: Wild Apple and Fruit Trees of Central Asia (Wiley Online Library, 2002).
Potter, D. Flora of North America: North of Mexico in Magnoliophyta: Picramniaceae to Rosaceae. Q. Rev. Biol. 91, 103 (2016).
Robertson, K. R., Phipps, J. B., Rohrer, J. R. & Smith, P. G. A synopsis of genera in Maloideae (Rosaceae). Syst. Bot. 16, 376 (1991).
Kron, P. & Husband, B. C. Hybridization and the reproductive pathways mediating gene flow between native Malus coronaria and domestic apple M. domestica. Botany 87, 864–874 (2009).
Korban, S. S. Interspecific hybridization in Malus. Hortic. Sci. 21, 41–48 (1986).
Nakai, T. Flora Koreana Vol. 26 (Imperial University of Tokyo, 1909).
Lee, T. B. Coloured Flora of Korea Vol. I, II (Hayangmunsa, 2003) (in Korean).
Lee, Y. N. New Flora of Korea (Kyohaksa Publishing Co, 2006).
Im, R. J. Flora Coreana, Sci. Technol (Publ House, 1997).
Chang, C. S., Kim, H. & Chang, K.-S. Illustrated Encyclopedia of Fauna and Flora of Korea, Vol. 43, Woody Plants (Minist. Educ., 2011).
Nakai, T. Flora Sylvatica Koreana Vol. 7 (Forest Experiment Station Government General of Chosen, 1917).
Park, C.-W. (ed.) The Genera of Vascular Plants of Korea (Academy Publishing Co., 2007) (in Korean).
Chung, T. H. Korean Flora 1025 (Shinjisa, 1957).
Lee, W. T. Lineamenta Florae Koreae (Academy Publ, 1996) (in Korean).
Korea National Arboretum. Checklist of Vascular Plants in Korea (Korea National Arboretum, 2017).
Stebbins, G. L. The role of hybridization in evolution. Proc. Am. Philos. Soc. 103, 231–251 (1959).
Bensmihen, S. et al. Mutational spaces for leaf shape and size. HFSP J. 2, 110–120 (2008).
Volk, G. M. et al. The vulnerability of US apple (Malus) genetic resources. Genet. Resour. Crop Evol. 62, 765–794 (2015).
Fiala, J. L. Flowering crabapples: The genus Malus. Choice Rev. Online 32, 32–5077 (1995).
Kew Royal Botanic Garden. Plants World Online. https://powo.science.kew.org/ (2022).
Gu, C. et al. Rosaceae. In Flora of China (Pittosporaceae through Connaraceae) (eds Wu, Z. Y. et al.) 46–434 (Science Press, 2003).
Iwatsuki, K., Yamazaki, T., Boufford, D. E. & Ohba, H. Angiospermae, Dicotyledoneae, Archichlamydeae. In Flora of Japan (eds Iwatsuki, K. et al.) (Kodansha, 2006).
Chang, K.-S. & Chang, C.-S. The distribution of the woody plants of South Korea based on herbarium (SNUA) material of the Arboretum (XII). Bull. Arbor. 26, 1–46 (2007).
Robinson, J. P., Harris, S. A. & Juniper, B. E. Taxonomy of the genus Malus Mill (Rosaceae) with emphasis on the cultivated apple, Malus domestica Borkh.. Plant Syst. Evol. 226, 35–58 (2001).
Lo, E. Y. Y. & Donoghue, M. J. Expanded phylogenetic and dating analyses of the apples and their relatives (Pyreae, Rosaceae). Mol. Phylogenet. Evol. 63, 230–243 (2012).
Cho, M.-S., Kim, J. H., Yamada, T., Maki, M. & Kim, S.-C. Plastome characterization and comparative analyses of wild crabapples (Malus baccata and M. toringo): Insights into infraspecific plastome variation and phylogenetic relationships. Tree Genet. Genomes 17, 41 (2021).
Kumar, C. et al. Genetic diversity and population structure studies of the wild apple genotypes using RAPD markers. Indian J. Hortic. 75, 546 (2018).
Forte, A. V., Ignatov, A. N., Ponomarenko, V. V., Dorokhov, D. B. & Savelyev, N. I. Phylogeny of the Malus (apple tree) species, inferred from the morphological traits and molecular DNA analysis. Russ. J. Genet. 38, 1150–1161 (2002).
Makino, T. Observations on the Flora of Japan. Bot. Mag. XXII (The Tokyo Botanical Society, 1908).
Ohashi, H., Kadota, Y., Murata, J., Yonekura, K. & Kihara, H. Wild Flowers of Japan Vol 3 Rosaceae-Meliaceae (Heibonsha, 2016).
Chang, C. S., Kim, H. & Gil, H. Y. A field guide to Korean woody plants. Designpost Paju 180–191 (2012).
Hokanson, S. C., Lamboy, W. F., Szewc-McFadden, A. K. & McFerson, J. R. Microsatellite (SSR) variation in a collection of Malus (apple) species and hybrids. Euphytica 118, 281–294 (2001).
Nikiforova, S. V., Cavalieri, D., Velasco, R. & Goremykin, V. Phylogenetic analysis of 47 chloroplast genomes clarifies the contribution of wild species to the domesticated apple maternal line. Mol. Biol. Evol. 30, 1751–1760 (2013).
Dayrat, B. Towards integrative taxonomy. Biol. J. Linn. Soc. 85, 407–415 (2005).
Padial, J. M., Miralles, A., De la Riva, I. & Vences, M. The integrative future of taxonomy. Front. Zool. 7, 16 (2010).
Padial, J. M. & De La Riva, I. A response to recent proposals for integrative taxonomy. Biol. J. Linn. Soc. 101, 747–756 (2010).
De Queiroz, K. Species concepts and species delimitation. Syst. Biol. 56, 879–886 (2007).
Valdecasas, A. G., Williams, D. & Wheeler, Q. D. ‘Integrative taxonomy’ then and now: A response to Dayrat (2005). Biol. J. Linn. Soc. 93, 211–216 (2007).
Boucher, F. C. et al. Discovery of cryptic plant diversity on the rooftops of the Alps. Sci. Rep. 11, 11128 (2021).
Marchán, D. F., Fernández, R., Domínguez, J., Díaz Cosín, D. J. & Novo, M. Genome-informed integrative taxonomic description of three cryptic species in the earthworm genus Carpetania (Oligochaeta, Hormogastridae). Syst. Biodivers. 18, 203–215 (2020).
Brandrud, M. K., Paun, O., Lorenzo, M. T., Nordal, I. & Brysting, A. K. RADseq provides evidence for parallel ecotypic divergence in the autotetraploid Cochlearia officinalis in Northern Norway. Sci. Rep. 7, 5573 (2017).
Lioi, L., Zuluaga, D. L., Pavan, S. & Sonnante, G. Genotyping-by-sequencing reveals molecular genetic diversity in Italian common bean landraces. Diversity 11, 154 (2019).
Zhao, P. et al. Population genetics, phylogenomics and hybrid speciation of Juglans in China determined from whole chloroplast genomes, transcriptomes, and genotyping-by-sequencing (GBS). Mol. Phylogenet. Evol. 126, 250–265 (2018).
Hong, U. V. T. et al. Insights into opium poppy (Papaver spp.) genetic diversity from genotyping-by-sequencing analysis. Sci. Rep. 12, 111 (2022).
Migicovsky, Z. et al. Genomic consequences of apple improvement. Hortic. Res. 8, 9 (2021).
Elshire, R. J. et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6, e19379 (2011).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10 (2011).
Daccord, N. et al. High-quality de novo assembly of the apple genome and methylome dynamics of early fruit development. Nat. Genet. 49, 1099–1106 (2017).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Jiang, B., Ding, C., Luo, B. & Tang, J. Graph-Laplacian PCA: Closed-form solution and robustness. In 2013 IEEE Conference on Computer Vision and Pattern Recognition 3492–3498. https://doi.org/10.1109/CVPR.2013.448 (2013).
R. C. Team. R: A Language and Environment for Statistical Computing v Vol. 3 (R Foundation for Statistical Computing, 2019).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Zhou, H., Alexander, D. H. & Lange, K. A quasi-Newton method for accelerating the convergence of iterative optimization algorithms. Stat. Comput. (2009).
Rosenberg, N. A. distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138 (2003).
Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Kamvar, Z. N., Tabima, J. F. & Grünwald, N. J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2, e281 (2014).
RStudio Team. RStudio: Integrated development environment for R (Version 1.4.1106). J. Wildl. Manag. 75 (2021).
Beerli, P. Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 22, 341–345 (2006).
Beerli, P. & Palczewski, M. Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 185, 313–326 (2010).
Knaus, B. J. et al. Package ‘vcfR’ (2020).
Hodel, R. G. J., de Cortez, M. B. S., Soltis, P. S. & Soltis, D. E. Comparative phylogeography of black mangroves (Avicennia germinans) and red mangroves (Rhizophora mangle) in Florida: Testing the maritime discontinuity in coastal plants. Am. J. Bot. 103, 730–739 (2016).
Pereira-Dias, L., Vilanova, S., Fita, A., Prohens, J. & Rodríguez-Burruezo, A. Genetic diversity, population structure, and relationships in a collection of pepper (Capsicum spp.) landraces from the Spanish centre of diversity revealed by genotyping-by-sequencing (GBS). Hortic. Res. 6, 54 (2019).
Holsinger, K. E. & Weir, B. S. Genetics in geographically structured populations: Defining, estimating and interpreting FST. Nat. Rev. Genet. 10, 639–650 (2009).
Cornille, A. et al. Anthropogenic and natural drivers of gene flow in a temperate wild fruit tree: A basis for conservation and breeding programs in apples. Evol. Appl. 8, 373–384 (2015).
Schnitzler, A., Arnold, C., Cornille, A., Bachmann, O. & Schnitzler, C. Wild European apple (Malus sylvestris (L.) Mill.) population dynamics: Insight from genetics and ecology in the Rhine valley. Priorities for a future conservation programme. PLoS One 9, e96596 (2014).
Denoirjean, T. et al. Genetic structure of Malus sylvestris and potential link with preference/performance by the rosy apple aphid pest Dysaphis plantaginea. Sci. Rep. 11, 5732 (2021).
Ha, Y.-H., Oh, S.-H. & Lee, S.-R. Genetic admixture in the population of wild Apple (Malus sieversii) from the Tien Shan Mountains, Kazakhstan. Genes (Basel) 12, 104 (2021).
Richards, C. M. et al. Genetic diversity and population structure in Malus sieversii, a wild progenitor species of domesticated apple. Tree Genet. Genomes 5, 339–347 (2009).
Chang, Y. et al. Application of high-throughput sequencing to evaluate the genetic diversity among wild apple species indigenous to Shandong, China, and introduced cultivars. Plant Mol. Biol. Rep. 37, 63–73 (2019).
Hamid, B. et al. An additional area of apple domestication with crop-wild gene flow, and also cultivation of the local wild apple, in the Caucasus. bioRxiv (2021).
Cornille, A., Gladieux, P. & Giraud, T. Crop-to-wild gene flow and spatial genetic structure in the closest wild relatives of the cultivated apple. Evol. Appl. 6, 737–748 (2013).
Qin, A. et al. Low genetic diversity and population differentiation in Thuja sutchuenensis Franch., an extremely endangered rediscovered conifer species in southwestern China. Glob. Ecol. Conserv. 25, e01430 (2021).
Wang, X. et al. Application of genome-wide insertion/deletion markers on genetic structure analysis and identity signature of Malus accessions. BMC Plant Biol. 20, 540 (2020).
Degrandi-Hoffman, G., Hoopingarner, R. & Klomparens, K. Influence of honey bee (Hymenoptera: Apidae) in-hive pollen transfer on cross-pollination and fruit set in apple. Environ. Entomol. 15, 723–725 (1986).
Hao, R. M., Zhao, H. B., Wang, J. H. & Zhou, L. H. Observation and study on breeding system of wild Osmanthus fragrans. J. Plant Resour. Environ. 20, 17–24 (2011).
Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 13, 1143–1155 (2004).
Barrett, S. C. H. Genetic and evolutionary consequences of small population size in plants: Implications for conservation. Genet. Conserv. Rare Plants (1991).
Hilfiker, K., Gugerli, F., Schütz, J.-P., Rotach, P. & Holderegger, R. Low RAPD variation and female-biased sex ratio indicate genetic drift in small populations of the dioecious conifer Taxus baccata in Switzerland. Conserv. Genet. 5, 357–365 (2004).
Ricklefs, R. E. A comprehensive framework for global patterns in biodiversity. Ecol. Lett. 7, 1–15 (2004).
Abellán, P. & Ribera, I. Geographic location and phylogeny are the main determinants of the size of the geographical range in aquatic beetles. BMC Evol. Biol. 11, 344 (2011).
Takhtadzhian, A. L. The Floristic Regions of the World (Nauka, 1988).
Nakai, T. Report on the Vegetation of the Island Ooryongto or Dagelet Island, Corea, February 1818 [I.E. 1918] (2011).
Lee, W.-T. & Yang, J. Y. Studies on the distribution of vascular plants in the Korean Peninsula. Korean J. Plant Taxon. 8, 1–8 (1978).
Jung, S. & Cho, Y. Redefining floristic zones in the Korean Peninsula using high-resolution georeferenced specimen data and self-organizing maps. Ecol. Evol. 10, 11549–11564 (2020).
Stuessy, T. F. et al. Anagenetic evolution in island plants. J. Biogeogr. 33, 1259–1265 (2006).
Liu, B. et al. Phylogenomic conflict analyses in the apple genus Malus s.l. reveal widespread hybridization and allopolyploidy driving diversification, with insights into the complex biogeographic history in the Northern Hemisphere. J. Integr. Plant Biol. 64, 1020–1043 (2022).
Nishio, S. et al. Genetic evidence that Chinese chestnut cultivars in Japan are derived from two divergent genetic structures that originated in China. PLoS One 15, e0235354 (2020).
Wu, C.-C. et al. Identification of hybridization and introgression between Cinnamomum kanehirae Hayata and C. camphora (L.) Presl using genotyping-by-sequencing. Sci. Rep. 10, 15995 (2020).
Li, X., Wei, G., El-Kassaby, Y. A. & Fang, Y. Hybridization and introgression in sympatric and allopatric populations of four oak species. BMC Plant Biol. 21, 266 (2021).
Pickup, M. et al. Mating system variation in hybrid zones: Facilitation, barriers and asymmetries to gene flow. New Phytol. 224, 1035–1047 (2019).
Abdelaziz, M. et al. Asymmetric reproductive barriers and gene flow promote the rise of a stable hybrid zone in the Mediterranean high mountain. Front. Plant Sci. 12, 687094 (2021).
Schulke, B. & Waser, N. M. Long-distance pollinator flights and pollen dispersal between populations of Delphinium nuttallianum. Oecologia 127, 239–245 (2001).
Cho, M.-S. & Kim, S.-C. Multiple lines of evidence for independent origin of wild and cultivated flowering cherry (Prunus yedoensis). Front. Plant Sci. 10, 1555 (2019).
Liu, W., Zheng, L. & Qi, D. Variation in leaf traits at different altitudes reflects the adaptive strategy of plants to environmental changes. Ecol. Evol. 10, 8166–8175 (2020).
Kofidis, G. Contemporary seasonal and altitudinal variations of leaf structural features in oregano (Origanum vulgare L.). Ann. Bot. 92, 635–645 (2003).
Acknowledgements
We thank Sa-Bum Jang and Eun-Ho Lee for sampling and laboratory assistance throughout the project. We appreciate the reviewers for reviewing our manuscript and for providing supportive comments. The study was supported by the research project of Korea National Arboretum (KNA1-1-13, 14-1).
Author information
Authors and Affiliations
Contributions
J.-H.K. and K.C. designed the project and obtained funding. Y.-H.H. and H.-Y.G. collected samples. Y.-H.H. conducted identification of the plant material. Y.-H.H., H.-Y.G. and S.-C.K. performed the genetic analyses. Y.-H.H. wrote the manuscript. All authors edited the manuscript and agreed with the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ha, YH., Gil, HY., Kim, SC. et al. Genetic structure and geneflow of Malus across the Korean Peninsula using genotyping-by-sequencing. Sci Rep 12, 16262 (2022). https://doi.org/10.1038/s41598-022-20513-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-20513-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
