
Abstract
Base treatment and metal doping were evaluated as means of enhancing the photocatalytic activity of ZrO2 nanoparticles (NPs) via the generation of oxygen vacancies (OvS), and the sites responsible for this enhancement were identified and characterized by spectroscopic and microscopic techniques. We confirmed that OvS produced by base treatment engaged in photocatalytic activity for organic pollutant degradation, whereas surface defects introduced by Cr-ion doping engaged in oxidative catalysis of molecules. Moreover, we verified that base-treated ZrO2 NPs outperformed their Cr-ion doped counterparts as photocatalysts using in situ X-ray photoelectron spectroscopy and scanning transmission electron microscopy coupled with electron energy loss spectroscopy (STEM-EELS). Thus, our study provides valuable information on the origin of the enhanced photocatalytic activity of modified ZrO2 NPs and demonstrates the practicality of in situ spectroscopy and STEM-EELS for the evaluation of highly efficient metal oxide photocatalysts.
Similar content being viewed by others

Introduction
In view of their photocatalytic activity, metal oxide nanoparticles (MO NPs; e.g., TiO2, CeO2, and ZnO) find numerous applications1,2,3,4,5. This activity comes from the occurrence of oxygen vacancies (OvS) on the NP surface and can therefore be enhanced by increasing the amount of OvS, ideally without inducing structural changes. Although the involvement of versatile defect sites in photocatalysis is widely accepted, the distinguishable mechanisms of their actions are not fully understood, largely because direct monitoring of these sites is hindered by their inherent instability6,7,8,9,10,11. The precise control of OvS characteristics through defect structure engineering is widely used to tailor the intrinsic properties of photocatalysts and thus rationally control their photocatalytic degradation (PCD) activity and selectivity12,13,14.
A common method of generating defect sites in MO NPs relies on surface modification via metal doping or base treatment15,16,17,18. During metal doping, surface defects are formed through charge transfer due to the combination of oxygen in MO NPs and doped transition metal ions. Meanwhile, many OH- groups formed on the surface during base treatment of NP affect the formation of hydroxyl radicals (·OH) as a formation of OvS which is directly related to the photocatalytic activity2. As it is currently unclear which of the two methods is more efficient for photocatalytic activity enhancement, there is a need to identify and characterize photocatalytic-related defect structures directly affecting photocatalytic properties. The generated positive holes in the valence band (VB) react with water and produce ·OH. As soon as ·OH is produced, they react with organic compounds and oxidize them, and eventually, CO2 and H2O as end products are produced19.
Contrary to other MOs, ZrO2 NPs cannot efficiently utilize visible light because of their wide bandgap (~ 5.0 eV) but show promising physicochemical properties, namely high thermal and chemical stability, low thermal conductivity, high corrosion resistance, and high strength. Accordingly, ZrO2 NPs have attracted much attention as multifunctional materials for catalysis, dye-sensitized solar cells, fuel cells, and gas sensors20,21,22. Therefore, this research is also meaningful and can be worth maximizing the photocatalytic activity of ZrO2 NPs by generating defect sites on their surface via metal doping or base treatment.
Herein, we verified base treatment and Cr-ion doping in terms of their ability to enhance the photocatalytic activity of ZrO2 NPs and determined which method affords defect structures directly responsible for this activity. The defect and electronic structures of three types (pristine, base-treated, and Cr-doped) of ZrO2 NPs were probed using high-resolution X-ray photoelectron spectroscopy (HRXPS), X-ray absorption spectroscopy (XAS), and scanning transmission electron microscopy coupled with electron energy loss spectroscopy (STEM-EELS), while photocatalytic activity was assessed by monitoring the degradation of 4-chlorophenol (4-CP) and phenol in aqueous solutions. In addition, the generation of ·OH over the above photocatalysts was assessed by following the formation of p-hydroxybenzoic acid (p-HBA) from benzoic acid (BA). For abundant OvS, charge carrier activity increases in the external supply of additional energy (i.e., photons with energies exceeding that of the bandgap) during spectroscopic measurements23,24,25. Therefore, in situ XPS measurements were herein performed under the conditions of the PCD reaction to characterize the change of electronic structures produced upon the irradiation of modified ZrO2 NPs and probe the effects of OvS relied on photocatalytic properties26,27,28,29.
Results and discussion
Photocatalyst characterization
The role of ZrO2 NPs surfaces is an important discrepancy that distinguishes photocatalytic activity from catalytic properties. The HR-TEM images of all ZrO2 NPs (Fig. 1a–c) featured the typical lattice fringes (111) and (− 111) of monoclinic ZrO2 NPs (0.315 and 0.28 nm), which indicated that defect generation did not destroy the crystal structure.

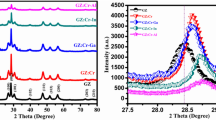
High-resolution TEM images and electron diffraction patterns of (a) ZrO2 (b) ZrO2-B, and (c) Cr@ZrO2 NPs. (d) XRD patterns and (e) Raman spectra of ZrO2, ZrO2-B, and Cr@ZrO2 NPs. The inset of (e) shows the Raman shift of ZrO2-B NPs toward the lower energy region. # and * indicate the Ag and Bg Raman vibrational modes, respectively.
The XRD patterns of all samples (Fig. 1d) exhibited characteristic peaks at 2θ = 23.9°, 28.1°, 31.6°, 34.4°, 35.4°, 40.9°, and 45.1°, which corresponded to reflections from the (110), (111), (11−1), (002), (200), (−102), and (−211) planes of monoclinic ZrO2 (m-ZrO2), respectively30,31. Therefore, neither treatment-induced phase transitions. The intensity and width of XRD peaks provide data on crystallite size and structure. Herein, the (111) peak was used to calculate the crystallite sizes of ZrO2, ZrO2-B, and Cr@ZrO2 NPs according to the Scherrer formula as 27.6 ± 0.5, 28.5 ± 0.5, and 26.6 ± 0.5 nm, respectively, which indicated that neither treatment-induced noticeable changes in the particle size distribution32.
The Raman spectra of all samples (Fig. 1e) featured characteristic peaks at 178.1 (Ag vibrational mode), 190.3 (Bg vibrational mode), 221 (Bg), 309 (Bg), 332 (Bg), 345 (Ag), 380 (Ag), 478 (Ag), 503 (Bg), 539 (Bg), 558 (Ag), 613 (Bg), and 635 cm−1 (Ag)33,34. These peaks indicated the presence of m-ZrO2 as the dominant phase, in agreement with the results of XRD analysis. We confirm that for base-treated ZrO2 NPs (marked as ZrO2-B NPs) there was a red-shift of the Raman peaks as shown in the inset of Fig. 1e. This observation clearly supports that the defects were formed because it is relatively weak as the bond length increases. Subsequently, we aimed to determine and compare whether the defect structure formed by base treatment or Cr-ion doping was OvS affecting photocatalytic properties or a simple defect structure. The combined results of HR-TEM, XRD, and Raman spectroscopic analyses revealed that modification did not result in significant structural changes.
UV light–induced PCD and hydroxyl radical formation
PCD studies were carried out using 4-CP, phenol, and BA as target pollutants to compare the effects of defects on the photocatalytic activity of ZrO2 NPs35,36. As the large bandgap of ZrO2 NPs (5.0 eV) does not allow them to exhibit photocatalytic properties under irradiation with visible light (Fig. S1), the PCD activities of modified ZrO2 NPs were assessed using UV light at a wavelength (λ ≥ 225 nm).
As shown in Fig. 2 and Table 1, PCD efficiency strongly depended on the modification method. ZrO2-B NPs were more efficient at degrading 4-CP and phenol (Fig. 2a,b) than Cr@ZrO2 NPs, which indicated that the abundance of OvS in the former catalyst significantly contributed to its enhanced activity. Additionally, by following the production of p-HBA from BA, we evaluated the formation of ·OH over the tested NPs and elucidated the influence of these radicals on the photocatalytic reaction37. As shown in Fig. 2c, the ability to produce ·OH was highest for ZrO2-B NPs, which is consistent with the results of the 4-CP and phenol degradation experiments. Hence, base treatment was concluded to be more effective than Cr-ion doping in increasing the amount of OvS, which is critical for the improvement of PCD efficiency. Moreover, we could evaluate that Cr2O3 formed by Cr-ion doping only slightly improved PCD efficiency at wavelengths above 225 nm (see Fig. S1) as we confirm a little effect of PCD activity. Therefore, the doped Cr ions were concluded to act as co-catalysts rather than directly enhancing photocatalytic activity (Fig. S2).

PCD activity of (a) 4-CP, (b) phenol, and (c) BA over ZrO2, ZrO2-B, or Cr@ZrO2 NPs under UV light irradiation (λ ≥ 225 nm). The PCD activities extracted from the plots are listed in Table 1.
Catalyst reusability was tested by using recovered ZrO2-B NPs to promote the PCD of 4-CP (Fig. S3). As the photocatalytic activity of ZrO2-B NPs was maintained for up to five consecutive cycles (15 h in total), the introduced defects were concluded to be stable. In addition, to test the stability of samples after PCD experiments, we measured XPS after the five consecutive photocatalytic cycles. (Fig. S4) After confirming the stability tests, we correlated it with electronic structure using HRXPS and STEM-EELS to explain the high photocatalytic activity of ZrO2-B NPs.
Investigation of defect structures using HRXPS and XAS
HRXPS and XAS were used to analyze the bonding configurations of Zr and O atoms on the surface of NPs and thus evaluate their electronic structures concerning defects.
The three types of ZrO2 NPs exhibited similar core-level spectra that featured two characteristic bonding configurations with differing intensities and thus indicated the varying presence of defect structures. Zr 3d core-level spectra (Fig. 3a) featured two peaks at 182.8 and 180.2 eV corresponding to the Zr 3d5/2 transitions of pristine ZrO2 and ZrOx with defects, respectively38,39. Remarkably, more defects were present in ZrO2-B NPs than in Cr@ZrO2 NPs, which was in line with the higher PCD activity of the former. After the deconvolution procedure of O 1 s peaks (Fig. 3b), we exhibit the three distinct components clearly at the binding energy of 530.1 eV (ZrO2), 531.6 eV (oxygen vacancy; Ov), and 532.7 eV (-OH), respectively40,41. Focusing on the intensity ratio of –OH and Ov peaks, we can identify that the amounts of defect sites of ZrO2-B NPs are larger than others, which indicates that treatment with base resulted in surface modification42,43. Meanwhile, the intensity change of the ZrOx peak is a good indicator for the quantification of defects such as surface hydroxyl-related defects and oxygen vacancies (Ovs). For ZrO2, ZrO2-B, and Cr@ZrO2 NPs, the ZrOx/ZrO2 (Zr 3d) peak intensity ratio equaled ~ 0.08, 0.175, and 0.097, respectively. The intensity ratio difference between the samples prepared by the two modification methods confirmed that base treatment is more effective at forming OvS than Cr-ion doping.

(a) Zr 3d and (b) O 1 s core-level HR-XP spectra and (c) Zr M-edge and (d) O K-edge X-ray absorption spectra of ZrO2 (bottom), ZrO2-B (middle), and Cr@ZrO2 (top) NPs.
Figure 3c,d show the X-ray absorption spectra of ZrO2 NPs. Based on dipole selection rules, signals in the Zr M-edge spectra (Fig. 3c) were assigned to transitions between the M2,3 p-core states of Zr atoms and the conduction band states derived from the 4d (A and B and A’ and B’) and the 5 s (C and C’) atomic states of Zr44,45. As in the case of XPS analysis, the spectrum of ZrO2-B NPs was markedly different from those of the other two samples, featuring a strongly attenuated peak A, which may indicate an increased defect content. A similar trend was observed for O K-edge spectra (Fig. 3d), in which case the decrease in the intensity of the t2g peak observed for ZrO2-B NPs was explained by a change in defect structure. Thus, the intensities of defect-attributable peaks (ZrOx) in the spectra of the base-treated sample exceeded those in the spectra of the Cr-ion-doped sample. Consequently, the apparent difference between the X-ray absorption spectrum of ZrO2-B NPs and those of the other two samples was attributed to remarkable changes in defect sites due to base treatment, which agreed with the results of HRXPS analysis. The enhanced photocatalytic activity of the ZrO2-B NPs shows that OH- and OvS formed around the defect structure can form a relatively large amount of ·OH affecting the photocatalytic reaction of the ZrO2 NPs46,47. Moreover, to verify the oxidation states of the doped Cr ions in Cr@ZrO2 NPs, we investigate the electronic states of Cr by using HRXPS and XAS as shown in Fig. S5.
STEM-EELS and in situ XPS during UV irradiation
As the difference in the defect site ratio determined using HRXPS represented the average over many NPs, further analysis was required to precisely compare the number of defect sites for single NPs. Thus, to obtain site-specific defects data for the surface and core of a single NP, we used high-resolution STEM-EELS to detect changes in the spectral profile of the O K-edge, including those in the defect-induced peak (surface hydroxyl-related defects or Ovs)48,49.
Figure 4 shows the O K-edge energy-loss near-edge structure (ELNES) spectra obtained for the surface and core sites of each NP. In the energy loss range of 525–545 eV, two peaks corresponding to eg (~ 533.2 eV) and t2g (~ 536.4 eV) were observed, corresponding to the hybridization of O 2p and Zr 4d states, respectively. The eg/t2g peak intensity ratio changes with the amounts of OvS because of the formation of deep donor states due to oxygen deficiency and is, therefore, a sensitive indicator of the relative concentration of OvS within the investigated spatial region50. The O-K ELNES spectra of a single pristine ZrO2 NP (Fig. 4a) exhibited the features typical of ZrO2 NPs with no changes in the above intensity ratio (which equaled 0.89 ± 0.03 and 0.96 ± 0.02 for the surface and the core, respectively) or the spectral profile51. A prominent change was observed for ZrO2-B NPs (eg/t2g = 0.78 ± 0.04 (surface) and 0.92 ± 0.03 (core)), suggesting a significant alteration of electronic structure and indicating that base treatment significantly affected the amounts of OvS and thus increased PCD activity. Conversely, no significant difference in the amounts of OvS was observed between Cr-doped (eg/t2g = 0.87 ± 0.05 (surface) and 0.95 ± 0.02 (core)) and pristine samples. This confirms that base treatment had a significantly larger effect on OvS than Cr-ion doping, which is directly related to the photocatalytic activity.

(a–c) STEM–EELS images and O K-edge ELNES spectra of (a) ZrO2, (b) ZrO2-B, and (c) Cr@ZrO2 NPs. Red and blue cones indicate the probing sites at the surface and the core of each NP, respectively.
Furthermore, the distribution of OvS within a single NP can be evaluated by comparing the eg/t2g peak intensity ratios of the surface and core regions of each particle. Herein, the ZrO2-B NPs exhibited an eg/t2g peak intensity ratio ⁓11.5% lower than that of Cr@ZrO2 NPs in all particle regions.
As shown in Fig. 2, ZrO2-B NPs exhibited higher PCD activity than other samples. HRXPS (Fig. 3) also allowed us to distinguish the hydroxyl-induced oxygen vacancy of ZrO2-B NPs from the defect structures of Cr@ZrO2 NPs. To investigate differences in the PCD activities of the three samples due to defect formation, we used XPS under irradiation with UV light of the same wavelength (λ ≥ 225 nm) as that used for the PCD reaction28,52.
Figure 5 shows changes in the in situ X-ray photoelectron spectra (Zr 3d and O 1 s) of the three samples recorded with and without UV irradiation (λ ≥ 225 nm). As expected, a large change was observed for ZrO2-B NPs (Fig. 5b). Regarding Zr 3d spectra, no shift of the ZrO2 peak was observed for any sample upon light on/off. However, the ZrOx peak due to photocatalytic-related OvS, exhibited a large shift, particularly in the case of ZrO2-B (0.18 eV). To explain this behavior, we focused on the intensity change of the ZrOx peak in ZrO2-B NPs. Upon irradiation with additional 225-nm light, the intensity of the ZrOx peak of ZrO2-B NPs increased by ~ 13.5%, whereas no such increase was meaningfully observed for other samples. This finding is consistent with the results of PCD activity evaluation, according to which only ZrO2-B NPs exhibit enhanced photocatalytic activity. Therefore, the increased OvS of ZrO2-B NPs underwent a large change due to UV light irradiation, which explains the difference in the results of PCD activity evaluation.

In situ X-ray photoelectron spectra of (a) ZrO2, (b) ZrO2-B, and (c) Cr@ZrO2 NPs recorded with (red) and without (black) UV light irradiation (λ ≥ 225 nm).
The same trend was observed for O 1s core-level spectra. Specifically, in the spectrum of ZrO2-B NPs, the Zr–OH peak corresponding to surface hydroxyl-related defects exhibited a shift of ~ 0.24 eV and an intensity decrease of 13.0%, and Ov peaks show an increase of approximately 13.0% at the same time, which was attributed to the enhanced photocatalytic properties of this sample. Therefore, the presence of many -OH groups on the surface of ZrO2-B NPs was indicative of a more OvS structure, which is closely related to PCD activity. Conversely, the intensity changes of the ZrOx peak in the X-ray photoelectron spectra of ZrO2 and Cr@ZrO2 NPs were not vivid and were almost equivalent to those observed for the ZrO2 peak during UV light irradiation, i.e., UV light irradiation induced only a small change in the oxygen-deficient structure. Therefore, unlike that of ZrO2-B NPs, the photocatalytic activity of these two samples did not markedly improve. The results of in situ XPS measurements precisely confirmed the existence of many –OH groups can be an indicator of the formation of abundant OvS upon irradiation and demonstrated that the extent of this transfer was enhanced in ZrO2-B NPs. The peak shift values of ZrOx in Zr 3d and –OH on O 1s core-level spectra of the three samples are listed in Table 2.
Notably, although defects were observed in both ZrO2-B and Cr@ZrO2 NPs, they exhibited markedly different photocatalytic properties, which was correlated with the presence/absence of ·OH on the NP surface, which originated from OvS. Among these two catalysts, ZrO2-B NPs featured –OH peak larger than others in their high-resolution X-ray photoelectron spectrum, thus forming relatively larger amounts of OvS. Even for similar oxygen-deficient structures, photocatalytic activity considerably increased in the presence of many -OH groups. To investigate the improvement of the photocatalytic properties of ZrO2-B NPs with many OvS, we additionally conducted an experiment on free radical trapping using radical scavenger (5,5-Dimethyl-1-pyrroline N-oxide; DMPO). As expected, when the radical scavenger (DMPO; 20 µM) was added together with 4-CP (10 µM) to cause the PCD reaction, it was confirmed that DMPO reacted preferentially with the ·OH radical generated by the photoreaction and that it interrupt the PCD reaction of the ZrO2–B NPs. (Fig. S6).
As is evidently in Fig. S2, it confirms that the PCD extent of 4-CP and thiophenol depended on the concentration of doping Cr ions in Cr@ZrO2 NPs, which indicates that Cr2O3 facilitates oxidation rather than acts as a photocatalyst.
Lastly, we found that the base treated ZrO2 shows the enhanced PCD activity due to many OH- at the surface of ZrO2 NPs. To clarify this effect, we also did pH dependent PCD test while changing the pH solution from 7.0 to 13.0. As expected, we confirmed the larger the pH value, the greater the change was due to the formation of many OH- ions under basic conditions. However, the change in PCD activity according to the change in the pH was not shown proportionally though as shown in Fig. S7.
Consequently, compared to Cr-ion doping, base treatment was better at improving the photocatalytic properties of ZrO2 NPs, increasing the number of ·OH near the -OH groups (Scheme 1).

The behavior of Ov sites on ZrO2 NPs modified using two different methods during PCD.
Conclusions
Modified ZrO2 NPs were prepared to use base treatment and Cr-ion doping, and the former treatment was shown to have a larger effect on photocatalytic properties than the latter. Mesoscale HRXPS, XAS, and atomic-scale STEM-EELS measurements showed that the enhanced PCD activity of the base-treated sample was due to an increase in the content of defect sites without a marked alteration of atomic structure. The rates of the ZrO2 NPs-catalysed PCD of organic pollutants were strongly affected by modification type, as it influenced the defect sites and, hence, oxygen-storage properties and the adsorption and reduction of dissolved O2. Using in situ XPS, we demonstrated that defect sites (Ov) formed in the presence of many –OH groups at the ZrO2 NP surfaces directly affect photocatalytic activity. Thus, our study provides a novel approach for the development of highly stable defect-engineered photocatalysts.
Material and methods
Synthesis of pristine ZrO2 NPs
A solution of zirconium isopropoxide (12 g) in ethanol (80 mL) was added to distilled deionized water (DDW; 500 mL) over 1 h upon gentle stirring, and the reaction mixture was heated in an autoclave at 220 °C for 10 h. After cooling to 25 °C, the produced ZrO2 NPs were selectively precipitated, dried at 90 °C for 48 h, and used for the synthesis of base-treated ZrO2 NPs (ZrO2-B NPs) or Cr-doped ZrO2 NPs (Cr@ZrO2 NPs)53,54.
Synthesis of ZrO2-B NPs
An aqueous solution with a KOH-adjusted pH of 13 was allowed to stand for 2 h and then supplemented with pristine ZrO2 NPs (3.6 g) upon stirring. The gel solution obtained after ~ 30 min was transferred to an autoclave and heated at 220 °C for 8 h in a convection oven. The reaction mixture was cooled to 25 °C, and the produced ZrO2-B NPs were precipitated, washed with DDW, and dried at 90 °C for 48 h.
Synthesis of Cr@ZrO2 NPs
Pristine ZrO2 NPs (3.0 g) were added to a solution of Cr(NO3)3·9H2O (0.612 g) in DDW (100 mL). After 30-min stirring at 90 °C, the heterogeneous mixture was transferred to an autoclave and heated at 220 °C for 8 h in a convection oven. The obtained Cr@ZrO2 NPs were precipitated, washed with DDW, and dried at 90 °C for 48 h.
Evaluation of photocatalytic performance
The catalyst of choice (0.015 g) was dispersed in distilled water (30 mL). The resulting suspension was stirred for 30 min to allow the adsorption of 4-CP, phenol, or BA on the NPs to reach equilibrium and then irradiated with a 300-W Xe arc lamp (Oriel Lighting, Darra, QLD, Australia) equipped with a cut-off filter (λ ≥ 225 nm). Aliquots were intermittently withdrawn using a 1-mL syringe and filtered through a 0.45-μm polytetrafluoroethylene filter (Millipore Sigma, Burlington, MA, USA) to remove the suspended ZrO2 NPs. The filtrates were analyzed by high-performance liquid chromatography (LC-20AD Pump, Shimadzu, Kyoto, Japan) to quantify the residual 4-CP, phenol, or BA and thus determine photocatalytic activity.
Instrumentation
The phase composition was probed by X-ray diffraction (XRD; M18XHF, MAC Science Co., Yokohama, Japan), while surface functional groups were probed by Raman spectroscopy (Labram ARAMIS instrument with an Ar+-ion continuous-wave (514.5 nm) laser, Horiba Ltd., Kyoto, Japan). Schottky field-emission STEM measurements were performed at 200 kV using an instrument equipped with a probe-forming spherical aberration (Cs) corrector (JEM-2100F, JEOL Ltd., Tokyo, Japan). Bright-field high-resolution transmission electron microscopy (HR-TEM) images were captured by a charge-coupled device camera (OneView, Gatan, Inc., Pleasanton, CA, USA) at full 4 k × 4 k resolution using an acquisition time of 16 s. Chemical composition was probed by STEM coupled with energy-dispersive X-ray spectroscopy (STEM-EDS; JED-2300T, JEOL Ltd., Tokyo, Japan), while O K-edge STEM-EELS measurements (GIF Quantum ER, Gatan, Inc., Pleasanton, CA, USA) were carried out at an energy resolution of 0.8 eV and a dispersion of 0.1 eV/pixel for an exposure time of 1.0 s. Electronic structures were probed by HRXPS and XAS at the 10A2 beamline of the Pohang Accelerator Laboratory. During in situ XPS measurements, which were performed at the above beamline, a low-power 225 nm light-emitting diode (Solis High-Power LED, Thorlabs, Inc., Newton, NJ, USA) was placed at ~ 25 cm from the samples to investigate electron density changes under light irradiation.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Zhang, J. & Medlin, J. W. Catalyst design using an inverse strategy: From mechanistic studies on inverted model catalysts to applications of oxide-coated metal nanoparticles. Surf. Sci. Rep. 73, 117–152 (2018).
Tran, B. et al. Oxygen vacancy engineering of cerium oxide for the selective photocatalytic oxidation of aromatic pollutants. J. Hazard. Mater. 404, 123976 (2021).
Ong, C. B., Ng, L. Y. & Mohammad, A. W. A review of ZnO nanoparticles as solar photocatalysts: Synthesis, mechanisms and applications. Renew. Sustain. Energy Rev. 81, 536–551 (2018).
Hwang, Y., Yang, S. & Lee, H. Surface analysis of N-doped TiO2 nanorods and their enhanced photocatalytic oxidation activity. Appl. Catal. B 204, 209–215 (2017).
Theerthagiri, J. et al. A review on ZnO nanostructured materials: Energy, environmental and biological applications. Nanotechnology 30, 392001 (2019).
Yu, H. et al. Three-in-One oxygen vacancies: Whole visible-spectrum absorption, efficient charge separation, and surface site activation for robust CO2 photoreduction. Angew. Chem. Int. Ed. 58, 3880–3884 (2019).
Zhang, X., Wang, Y., Liu, B., Sang, Y. & Liu, H. Heterostructures construction on TiO2 nanobelts: A Powerful tool for building high-performance photocatalysts. Appl. Catal. B 202, 620–641 (2017).
Ji, Y. & Luo, Y. New Mechanism for Photocatalytic Reduction of CO2 on the Anatase TiO2 (101) surface: The essential role of oxygen vacancy. J. Am. Chem. Soc. 138, 15896–15902 (2016).
Yang, Y. et al. An Unusual strong visible-light absorption band in red anatase TiO2 photocatalyst induced by atomic hydrogen-occupied oxygen vacancies. Adv. Mater. 30, 1704479 (2018).
Xu, W. et al. Porous cobalt oxide nanoplates enriched with oxygen vacancies for oxygen evolution reaction. Nano Energy 43, 110–116 (2018).
Jang, W.-S. et al. Direct probing of atomic site selectivity of dopants in superior photocatalytic tin oxide nanoparticles. Appl. Catal. B 305, 121083 (2022).
Tran, B., Im, S., Kim, K., Kim, W. & Lee, H. Photocatalytic degradation of phenolic compounds of defect engineered Fe3O4: An alternative approach to solar activation via ligand-to-metal charge transfer. Appl. Surf. Sci. 509, 144853 (2020).
Kim, W., Tachikawa, T., Moon, G. H., Majima, T. & Choi, W. Molecular-level understanding of the photocatalytic activity difference between anatase and rutile nanoparticles. Angew. Chem. Int. Ed. 53, 14036–14041 (2014).
Kim, W., Tachikawa, T., Moon, G. H., Majima, T. & Choi, W. Photocatalytic degradation of recalcitrant organic pollutants in water using a novel cylindrical multi-column photoreactor packed with TiO2-coated silica gel beads. J. Hazard. Mater. 285, 398–408 (2015).
Puigdollers, A. R., Schlexer, P., Tosoni, S. & Pacchioni, G. Increasing oxide reducibility: The role of metal/oxide interfaces in the formation of oxygen vacancies. ACS Catal. 7, 6493–6513 (2017).
Zhao, Y. et al. Tuning oxygen vacancies in ultrathin TiO2 nanosheets to boost photocatalytic nitrogen fixation up to 700 nm. Adv. Mater. 31, 1806482 (2019).
Yan, L. et al. Electronic modulation of cobalt phosphide nanosheet arrays via copper doping for highly efficient neutral-pH overall water splitting. Appl. Catal. B 265, 118555 (2020).
Li, L. et al. Molecularly imprinted carbon nanosheets supported TiO2: Strong selectivity and synergic adsorption-photocatalysis for antibiotics removal. J. Hazard. Mater. 383, 121211 (2020).
Wang, R. et al. Graphdiyne-modified TiO2 nanofibers with osteoinductive and enhanced photocatalytic antibacterial activities to prevent implant infection. Nat. Commun. 11, 4465 (2020).
Yamada, T. et al. Single crystal ZrO2 nanosheets formed by thermal transformation for solid oxide fuel cells and oxygen sensors. ACS Appl. Nano Mater. 2, 6866–6873 (2019).
Zhang, Y. et al. Oxygen vacancy enhancing Fenton-like catalytic oxidation of norfloxacin over prussian blue modified CeO2: Performance and mechanism. J. Hazard. Mater. 398, 122863 (2020).
Li, W. et al. ZrO2 support imparts superior activity and stability of Co catalysts for CO2 methanation. Appl. Catal. B 220, 397–408 (2018).
Tahir, M. N. et al. Facile synthesis and characterization of monocrystalline cubic ZrO2 nanoparticles. Solid State Sci. 9, 1105–1109 (2007).
Bai, X., Pucci, A., Freitas, V. T., Ferreira, R. A. S. & Pinna, N. One-step synthesis and optical properties of benzoate- and biphenolate-capped ZrO2 nanoparticles. Adv. Funct. Mater. 22, 4275–4283 (2012).
Dong, X. et al. CO2 hydrogenation to methanol over Cu/ZnO/ZrO2 catalysts prepared by precipitation-reduction method. Appl. Catal. B 191, 8–17 (2016).
Kou, J. et al. Selectivity enhancement in heterogeneous photocatalytic transformations. Chem. Rev. 117, 1445–1514 (2017).
Yang, T. et al. Enhanced photocatalytic ozonation degradation of organic pollutants by ZnO modified TiO2 nanocomposites. Appl. Catal. B 221, 223–234 (2018).
Low, J., Dai, B., Tong, T., Jiang, C. & Yu, J. In situ irradiated X-Ray photoelectron spectroscopy investigation on a direct Z-scheme TiO2/CdS composite film photocatalyst. Adv. Mater. 31, 1802981 (2019).
Wang, S. et al. Direct Z-scheme ZnO/CdS hierarchical photocatalyst for enhanced photocatalytic H2-production activity. Appl. Catal. B 243, 19–26 (2019).
Ni, J. et al. Tuning electron density of metal nickel by support defects in Ni/ZrO2 for selective hydrogenation of fatty acids to alkanes and alcohols. Appl. Catal. B 253, 170–178 (2019).
Wang, Y. et al. Low-temperature catalytic CO2 dry reforming of methane on Ni-Si/ZrO2 catalyst. ACS Catal. 8, 6495–6506 (2018).
Martínez, A., Prieto, G., Arribas, M. A., Concepción, P. & Sánchez-Royo, J. F. Influence of the preparative route on the properties of WOx–ZrO2 catalysts: A detailed structural, spectroscopic, and catalytic study. J. Catal. 248, 288–302 (2007).
Gomes, A. S. O., Yaghini, N., Martinelli, A. & Ahlberg, E. A Micro-raman spectroscopic study of Cr(OH)3 and Cr2O3 nanoparticles obtained by the hydrothermal method. J. Raman Spectrosc. 48, 1256–1263 (2017).
Zou, W. et al. Engineering the Cu2O–reduced graphene oxide interface to enhance photocatalytic degradation of organic pollutants under visible light. Appl. Catal. B 181, 495–503 (2016).
Chen, L. et al. Production of hydroxyl radical via the activation of hydrogen peroxide by hydroxylamine. Environ. Sci. Technol. 49, 10373–10379 (2015).
Matolín, V. et al. Activation of binary Zr–V non-evaporable Getters: Synchrotron radiation photoemission study. Appl. Surf. Sci. 243, 106–112 (2005).
Fernández-García, M. et al. Ca doping of nanosize Ce−Zr and Ce−Tb solid solutions: Structural and electronic effects. Chem. Mater. 17, 4181–4193 (2005).
Seo, J., Cha, D., Takanabe, K., Kubota, J. & Domen, K. Electrodeposited ultrafine NbOx, ZrOx, and TaOx nanoparticles on carbon black supports for oxygen reduction electrocatalysts in acidic media. ACS Catal. 3, 2181–2189 (2013).
Chang, C.-Y. et al. Room-temperature solution-processed n-doped zirconium oxide cathode buffer layer for efficient and stable organic and hybrid perovskite solar cells. Chem. Mater. 28, 242–251 (2016).
Wang, J. et al. Oxygen vacancy induced band-gap narrowing and enhanced visible light photocatalytic activity of ZnO. ACS Appl. Mater. Interfaces 4, 4024–4030 (2012).
Baek, M., Kim, D. & Yong, K. Simple but effective way to enhance photoelectrochemical solar water-splitting performance of ZnO nanorod arrays: Charge trapping Zn(OH)2 annihilation and oxygen vacancy generation by vacuum annealing. ACS Appl. Mater. Interfaces 9, 2317–2325 (2017).
Weissbach, T., Peckham, T. J. & Holdcroft, S. CeO2, ZrO2 and YSZ as mitigating additives against degradation of proton exchange membranes by free radicals. J. Membr. Sci. 498, 94–104 (2016).
Ouyang, J., Zhao, Z., Suib, S. L. & Yang, H. Degradation of congo red dye by a Fe2O3@CeO2-ZrO2/Palygorskite composite catalyst: Synergetic effects of Fe2O3. J. Colloid Interface Sci. 539, 135–145 (2019).
Lucovsky, G. et al. Electronic structure of noncrystalline transition metal silicate and aluminate alloys. Appl. Phys. Lett. 79, 1775 (2001).
Rawat, M. et al. Micro-raman and electronic structure study on kinetics of electronic excitations induced monoclinic-to-tetragonal phase transition in zirconium oxide films. RSC Adv. 6, 104425–104432 (2016).
Piao, Y. et al. Identify Zr promotion effects in atomic scale for Co-based catalysts in Fischer-Tropsch synthesis. ACS Catal. 10, 7894–7906 (2020).
Dong, C. et al. Supported metal clusters: Fabrication and application in heterogeneous catalysis. ACS Catal. 10, 11011–11045 (2020).
Timoshenko, J. & Frenkel, A. I. “Inverting” X-ray absorption spectra of catalysts by machine learning in search for activity descriptors. ACS Catal. 9, 10192–10211 (2019).
Frati, F., Hunault, M. O. J. Y. & de Groot, F. M. F. Oxygen K-edge x-ray absorption spectra. Chem. Rev. 120, 4056–4110 (2020).
Toyao, T. et al. Machine learning for catalysis informatics: Recent applications and prospects. ACS Catal. 10, 2260–2297 (2020).
Filippov, T. N. et al. Photocatalytic and photochemical processes on the surface of uranyl-modified oxides: An in situ XPS study. Appl. Catal. A 558, 81–90 (2018).
Feng, N. et al. Understanding the high photocatalytic activity of (B, Ag)-codoped TiO2 under solar-light irradiation with XPS, solid-state NMR, and DFT calculations. J. Am. Chem. Soc. 135, 1607–1616 (2013).
Witoon, T., Chalorngtham, J., Dumrongbunditkul, P., Chareonpanich, M. & Limtrakul, J. CO2 hydrogenation to methanol over Cu/ZrO2 catalysts: Effects of zirconia phases. Chem. Eng. J. 293, 327–336 (2016).
Yang, X. et al. Interface effect of mixed phase Pt/ZrO2 catalysts for HCHO oxidation at ambient temperature. J. Mater. Chem. A. 5, 13799–13806 (2017).
Acknowledgements
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIT) (2021R1A2C2007992, NRF-2020R1A2C1014203, and NRF-2020R1A2C1010725). We also acknowledge support from MSIT and PAL.
Author information
Authors and Affiliations
Contributions
All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, H.S., Kim, YJ., Son, Y.R. et al. Verifying the relationships of defect site and enhanced photocatalytic properties of modified ZrO2 nanoparticles evaluated by in-situ spectroscopy and STEM-EELS. Sci Rep 12, 11295 (2022). https://doi.org/10.1038/s41598-022-15557-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-15557-0
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
