
Abstract
The interest in chemical RNA modifications is rapidly growing in the field of molecular biology. Dynamic and reversible alterations of N6-methyladenosine (m6A) RNA modification are responsible for a platter of structural and functional changes in healthy and cancerous cell states. Although many studies reported the link between tumor initiation/progression and m6A modulators, there are few studies exploring transcriptome-wide m6A profile of non-coding RNAs. The aim of current study was to identify glioma stem cell (GSC) specific m6A landscape of long non-coding RNAs (lncRNAs) applying MeRIP-seq approach. MeRIP-seq analysis assigned 77.9% of m6A peaks to mRNAs and 8.16% to lncRNAs. GSCs and differentiated cells showed 76.4% conservation of m6A peaks, while 19.4% were unique to GSCs. Seven novel GSC-specific m6A modified lncRNAs were identified: HRAT92, SLCO4A1-AS1, CEROX1, PVT1, AGAP2-AS1, MIAT, and novel lncRNA gene ENSG00000262223. Analysis disclosed a strong negative correlation between lncRNAs m6A modification rate and expression. MeRIP-seq analysis revealed m6A modifications on previously reported glioma-associated lncRNAs: LINC000461, HOTTIP, CRNDE, TUG1, and XIST. Moreover, current study disclosed that most highly m6A modified lncRNAs primarily contain m6A modifications close to 3′ and 5′ ends. Our results provide basis and insight for further studies of m6A modifications in non-coding transcriptome of GSCs.
Similar content being viewed by others
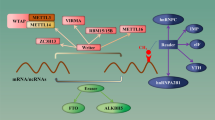


Introduction
More than 150 distinct post-transcriptional RNA modifications were identified in eukaryotes1. Next generation sequencing revealed widespread epigenetic RNA modifications of N6-methyladenosine (m6A), 5-methylcytosine (5mC), and pseudouridine (Ψ)2,3,4. m6A is the most prevalent epigenetic RNA modification5,6 affecting RNA stability, shape, and translation7,8,9,10,11. Approximately 0.1–0.4% of all adenines are methylated at the nitrogen-6 position of total RNA which translates 3–5 m6A modifications per mRNA1,12,13.
Most of m6A modifications are post-transcriptionally installed onto RNA by two independent writer complexes: multicomponent METTL3/METTL14 complex9,14,15 and METTL16 complex16. m6A mark can be removed by erasers–demethylases: fat obesity protein (FTO)17,18 and ALKBH519. m6A modifications are recognised by reader proteins, which contain a conserved YTH domain20,21 and affect RNA charge, base pairing, location, secondary structures12,22,23,24. Epigenetic m6A RNA machinery provides a new level of gene expression control and highlights the complexity and importance of the epitranscriptome. Recent development of methylated RNA immunoprecipitation followed by sequencing (MeRIP-seq) enabled transcriptome-wide m6A detection. MeRIP-seq detects methylated regions by m6A specific antibody immunoprecipitation of RNA, relative to input RNA25. Such discoveries have promoted the research of novel epigenetic markers for disease prognostics and therapeutics.
Glioblastoma (GBM) is one of the deadliest brain tumors. Even combination of various treatments including surgical resection, radiation therapy, and chemotherapy results in 5-year survival estimate of 5% for GBM patients26,27,28,29. Low efficiency of treatments is due to heterogeneity and high resistance of glioma stem cells (GSCs)30. Reduction of m6A RNA levels lead to increased GSCs growth, self-renewal, and tumorigenesis31. Therefore, novel therapies are required to combat potent GSCs and one of the possibilities could be targeting epitranscriptome marks like m6A. Majority of m6A epitranscriptomic studies focus on mRNAs and little is known on how long non-coding RNAs (lncRNAs) are affected by this modification. mRNA mimicking lncRNA molecules are usually modified near or at the 3′ end (MALAT132) and the 5′ end regions (HOTAIR33). m6A modifications in lncRNAs affect secondary structure, charge, binding capabilities, and binding targets3. Although many studies have identified the link between tumor initiation/progression with dynamic m6A modifications, there are only few studies exploring the m6A landscape of non-coding RNAs, including lncRNAs. Recently, Liu et al. demonstrated that m6A modifications regulate the stability of the lncRNA THOR, thus can maintain the oncogenic role of the lncRNA THOR34. Nevertheless, there are no transcriptome-wide studies about m6A modified lncRNAs and their potential role on GSC driven glioma development and progression.
Here we provide, first to date lncRNA m6A landscape of GSC. Our results highlight the importance of m6A modification for GSC specific lncRNA functionality and provide basis for further research of GSC epitranscriptome.
Results
Differentially modified m6A peaks in lncRNAs
To assess differentially modified lncRNAs between glioblastoma tumor mass-forming differentiated cells and glioma stem cells (both derived from human glioblastoma tumors), we performed m6A methylated RNA immunoprecipitation followed by high-throughput sequencing (MeRIP-seq) of U87-MG and NCH421K cells (see Supplementary Fig. S1 for experimental design). MeRIP-seq analysis resulted in 33,986 identified m6A peaks: 6579 specific to NCH421K, 1443 specific to U87-MG, and 25,964 shared between cell lines (Fig. 1A, B). Peaks in protein coding transcripts composed a major part of data—77.91% (26,478), peaks in lncRNA transcripts composed 8.16% (2773); in pseudogenes 3.8%; in miRNAs—0.15% (52); in snoRNAs 0.18% (61); in miscRNAs—0.1% (31); in snRNAs—0.11% (37), and 0.1% and 9.57% of total sequenced peaks were composed of other type of molecules and unassigned transcripts, respectively (Fig. 1C). Next, DESeq2 analysis was applied to select the most differentially m6A modified lncRNAs between cell lines. Analysis incorporated NCH421K and U87-MG m6A-IP data previously normalized to the input control. FDR threshold (q < 0.01) adjustment resulted in 659 remaining lncRNA peaks out of 2773, of which 423 were specific to GSCs (NCH421K) and 236 peaks specific to U87-MG cells (Fig. 1D, E). More than two-thirds of the m6A peaks in the identified lncRNAs (n = 425) were localized in 5′ or 3′ end positions of transcripts (Fig. 1D). Log2fold change (log2FC) method was applied to select the most differentially modified targets between cell lines with a Benjamini–Hochberg adjusted p-value < 1 × 10−10 (FDR q < 0.01) and log2FC≥ 4 after the Wald test. Analysis resulted in 61 differentially modified m6A peak. Among them, 19 peaks were specific to GSC (NCH421K) and 42 to U87-MG cells (Fig. 1E, F; Supplementary Table S1). Even though FDR threshold adjustment resulted in 64% of the peaks specific to NCH421K cells, most of the peaks stayed under the threshold after the application of differentially modified peak analysis.

Distribution of MeRIP-seq data (A) Volcano-plot of detected m6A peaks in U87-MG (left) and NCH421K (right) cell lines applying MeRIP-seq. (B) Venn-diagram of detected cell line specific and common m6A peaks after MeRIP-seq. (C) The percentage of detected m6A peaks by annotation type. (D) Pie chart of the percentage of detected m6A peaks in lncRNA transcripts (n = 659) by peak localization. (E) Volcano-plot of all detected lncRNA m6A peaks. (F) Volcano-plot of differentially modified lncRNAs m6A peaks between U87-MG and NCH421K cell lines after FDR, log2FC and p-value cut-off. Dot-lines indicate the selected threshold of p-value < 1 × 10−10 (y-axis) and selected threshold of log2FC ≥ 4 (16-fold) (x-axis); blue and red colored dots respectively indicate U87-MG and NCH421K specific lncRNA m6A peaks that were selected for the further analysis.
GSC specific m6A modified lncRNAs
Next, we revised all 19 m6A peaks which were specifically expressed in GSCs. Using Integrative Genomic Viewer (IGV) tool we assessed the following parameters: peak position and peak annotation. Peak position assessment—only peaks that were annotated mostly to lncRNA exon zones (± 30 bases) were selected. Peak annotation assessment: if the peaks were annotated to the two gene loci (plus and minus strand), only peaks which originated from the correct strand (the strand where lncRNA was presented) based on gene expression data output information, were selected. Only peaks that matched selection criteria were further analyzed. Peak revision resulted in 7 novel GSC specific targets: HRAT92, SLCO4A1-AS1, CEROX1, PVT1; AGAP2-AS1; MIAT and novel lncRNA gene entrez ID: ENSG00000262223, see Table 1.
Highly m6A enriched GSC specific lncRNA peaks were predominantly distributed in the first (close to the 5′ end of the RNA molecule) or in the last (close to the 3′ end of the RNA molecule) exons of selected targets. Slightly lower peaks or no peaks were detected within internal exons. The major m6A peak of HRAT92 lncRNA was identified in the 1st out of 3 exons. Moreover, two more GSC specific lower peaks were observed in the second and the third exons of the target, (Supplementary Fig. S2). The main SLCO4A1-AS1 peak was in the 3rd out of 4 exons, furthermore peaks of lower signal were identified in the 1st and 4th exon.
The last (3rd) exon of CEROX1 was the most differentially m6A methylated between cell lines, while the 1st exon showed lower m6A methylation level. The 1st exon of PVT1 in NCH421K cells was highly m6A enriched while the remaining 8 exons did not reveal any differences between cell lines. The most prominent m6A peaks in MIAT lncRNA were located at the beginning of the last (4th) exon, (Supplementary Fig. S2). Considerably lower m6A peaks were detected in other MIAT exons. However, there were no differences in peaks enrichment between cell lines. Highly enriched m6A peak at the 1st exon of AGAP2-AS1 was detected in glioma stem cells as compared to U87-MG cell line. Single m6A peak of lncRNA ENSG00000262223 was located in the 1st out of 3 exons (Supplementary Fig. S2).
Analysis of m6A pattern revealed that identified GSC specific and highly m6A modified lncRNAs primarily contain m6A modifications close to 3′ and/ or 5′ ends. Markedly lower m6A modification level was observed in the middle part of the lncRNA molecules. Similar m6A modification pattern was observed in previously identified and tumor associated lncRNAs like MALAT131 and HOTAIR32.
GSC specific m6A modified lncRNAs gene expression
Next, we checked the expression level of selected GSC specific lncRNAs. Interestingly, we found that the expression level of GSC specifically modified lncRNAs was higher in U87-MG cell line (Fig. 2A, B). We analyzed expression of selected lncRNAs at both—gene type level (Fig. 2B) and transcript type level (Fig. 2C). LncRNA expression data analysis using either gene type or transcript type data revealed similar results. Correlation analysis showed strong negative (r = −0.744, q = 3.56 × 10−4) association between m6A modification rate and expression level (Fig. 2D, E).

Visualization of screened-out lncRNAs (A) Volcano-plot of seven selected highly in NCH421K cells modified lncRNAs (black dots with yellow edging) in the context of all detected lncRNAs (FDR q < 0.01). (B) Volcano-plot of gene expression. Screened out lncRNAs indicated in black dots with yellow edging. (C) Volcano-plot transcripts expression. (D) The heat-map of gene expression (Log2FC) and m6A modification (Log2FC) of screened out lncRNAs. (E) The correlation scatterplot of screened out lncRNAs expression and m6A modification.
GSC specific m6A modified lncRNAs gene expression and prognostic impact in glioma tumors
Differentially m6A methylated GSC associated genes were selected for further analysis on web-based visualization tool Gene Expression Profiling Interactive Analysis (GEPIA) database35. The present study employed AGAP2-AS1, SLCO4A1-AS1, HRAT92, PVT1, MIAT, CEROX1 and ENSG00000262223 data in GEPIA dataset to explore its differential expression and effect on GBM prognosis.
GEPIA data analysis revealed that AGAP2-AS1 and PVT1 gene expression was higher in GBM than in normal tissue (N) (log2FC ≥ 1, FDR p-adj. = 3.5 × 10−60 and p-adj. = 2.33 × 10−53, respectively) (Fig. 3A). Significant downregulation of CEROX1, MIAT and ENSG00000262223 was observed in GBM as compared to normal tissue (log2FC ≥ 1, FDR p-adj. = 1.75 × 10−27; p-adj. = 7 × 10−104; p-adj. = 1.4 × 10−25, respectively). Several lncRNAs were associated with GBM molecular subtype; CEROX1 and novel lncRNA ENSG00000262223 expression was upregulated in proneural as compared to mesenchymal glioblastoma subtype (data not shown). Gene expression-based survival analysis showed that six from seven detected highly m6A modified lncRNAs (PVT1, SLCO4A1, HRAT92, AGAP2-AS1, CEROX1 and ENSG00000262223 had an impact on the prognosis of low-grade glioma (LGG) and GBM (FDR adj. p < 0.01) (Fig. 3B).

lncRNAs analysis on Gepia platform. (A) lncRNAs gene expression (log2(TPM + 1)) in low grade glioma (LGG, n = 518), glioblastoma (GBM, n = 163) and normal tissue (N, n = 207) for AGAP2-AS1, SLCO4A1-AS1, HRAT92, PVT1, MIAT, ENSG00000262223 and CEROX1. Significant difference shown as asterisk: * = log2FC = 1, p-adj. < 0.01. (B) Prognostic impact of lncRNAs expression level based on the survival heat map in LGG and GBM. Heat map shows hazard ratios (HR) in log10 for different genes. The red and blue blocks denote higher and lower risks, respectively. The rectangles with frames mean the significant (FDR p-adj. > 0.01) unfavorable and favorable results in prognostic analyses.
m6A modifications of glioma associated lncRNAs
Numerous lncRNAs are shown to be deregulated in glioblastoma and associated with glioma pathogenesis and GSCs properties36,37. Nevertheless, m6A RNA modification profile of deregulated lncRNAs requires further exploration. Thus, here we checked m6A modifications of previously reported glioma associated lncRNAs: MALAT1, NEAT1, XIST1, TUG1, CRNDE, LINC00461, and HOTTIP.
MeRIP-Seq analysis detected three m6A peaks in MALAT1 lncRNA that previously was shown to play an important role in regulating the expression of stemness markers and GSCs proliferation37,38 (Supplementary Fig. S3). The peak localized at the 5′ end of MALAT1 transcript revealed slightly higher m6A enrichment (1.4-fold, p = 2.8 × 10−6) in U87-MG cells as compared to NCH421K. The remaining two peaks were similar between cell lines. NEAT1 was also highly modified at the 5′ end in both cell lines, nevertheless the peak was 3.1-fold more (p = 3.9 × 10−17) enriched in U87-MG as compared to NCH421K (Fig. 4A; Supplementary Fig. S3). Five relatively low m6A peak enrichment signals were detected in XIST of which only one was significantly enriched in NCH421K cells (p = 0.002, 27.6-fold). m6A signals were localized at the 1st and the last 6th exon of XIST (Supplementary Fig. S3). TUG1 previously associated with self-renewal of GSCs and tumor development39,40 in present study was found to be highly m6A methylated in both cell lines. In total 9 m6A peaks were detected that were localized in all TUG1 exons (Supplementary Fig. S3). The analysis revealed single differentially methylated m6A peak (localized in the last exon) to be more (1.37-fold, p = 4.1 × 10−3) enriched in NCH421K cells as compared to U87-MG. Next lncRNA that was shown to be deregulated in GSCs—CRNDE in the present study was found to be lowly m6A methylated at two sites of which one peak (localized at the 5′ end) showed different enrichment between cell lines (3.9-fold, p = 3.7 × 10−4) with the higher enrichment in NCH421K cells (Supplementary Fig. S3). Seven m6A peaks were detected in LINC00461 of which 5 were localized in RefSeq exons of the lncRNA and two in intronic zone. Three peaks localized in LINC00461 exons revealed significantly higher enrichment in NCH421K cells as compared to U87-MG (19.6-fold, p = 0.0124; 16.6-fold, p = 6.9 × 10−3;16.1-fold, p = 2.5 × 10−5). Single m6A peak was found in HOTTIP third exon that was more enriched in NCH421K cells when compared to U87-MG cells (18.5-fold, p = 2.86 × 10−3) (Supplementary Fig. S3). Different modification levels (IP enrichment scores) between previously described lncRNAs were detected, therefore different scales were applied for peak visualization using IGV (Supplementary Fig. S3). m6A modification enrichment of most of the above-described glioma associated lncRNAs was higher in NCH421K as compared to U87-MG (Fig. 4A, B). Interestingly, the overall expression level of these lncRNAs was higher in U87-MG as compared to NCH421K (Fig. 4C).

Glioma associated lncRNAs m6A modification (A) Bar plot of glioblastoma associated lncRNAs m6A peaks log2FC detected by MeRIP-seq in NCH421K and U87-MG. Note that multiple peaks map to the same gene. Bars with solid black border represent significantly enriched peaks (Benjamini–Hochberg adjusted p < 0.01). (B) Volcano-plot showing m6A peaks in glioma associated lncRNAs (colored dots) in the context of all detected lncRNAs . (C) Volcano-plot of glioma associated lncRNAs expression (calculated at gene level) in the context of all detected lncRNAs. Screened out lncRNAs indicated in black dots with yellow edging.
Discussion
Here we investigated m6A modifications of lncRNAs by MeRIP-seq in NCH421K and U87-MG cells. Based on strict criteria we identified 19 GSC specific m6A peaks, of which 7 specifically corresponded to lncRNAs: HRAT92, SLCO4A1-AS1, CEROX1, PVT1; AGAP2-AS1; MIAT and an uncharacterized lncRNA transcript ENSG00000262223. We also found that m6A modification rate negatively correlated with lncRNAs expression level. GEPIA gene expression and survival analysis confirmed involvement of all 7 lncRNAs (in present study identified as GSC specific and highly m6A modified) in glioma pathogenesis. Furthermore, we assessed m6A modifications in previously glioma associated lncRNAs: LINC00461, HOTTIP, CRNDE, TUG1, XIST, MALAT1 and NEAT1. To our knowledge we are first to report m6A modifications in LINC00461, HOTTIP, and CRNDE lncRNAs in GSCs.
In accordance with Patil et al.9, we found high m6A enrichment in XIST lncRNA. We also observed possible m6A peak conservation between different cell types as indicated by identification of already reported and m6A modified lncRNAs XIST, TUG1, MALAT1 and NEAT19,41,42,43. Furthermore, highly m6A methylated lncRNAs were lowly expressed and vice versa between two cell types. m6A modification affects RNA secondary structure, charge, and base pairing12,22,23,24. Therefore, m6A induced structural RNA changes can affect RNA half-life and/or functionality. Primary functions of lncRNA include sponging various RNA molecules and interacting with proteins. It is known that m6A modifications affect mRNA half-life44. mRNA’s m6A is recognized by readers, YTH proteins which promote and induce mRNA degradation5,45,46 while IGF2BP protects mRNA from degradation47, however it is not known if that is the case for lncRNA. lncRNA m6A modifications predominantly localizes in 3′ and 5′ end of the molecule32,33,42. Moreover, m6A induced structural changes in lncRNAs may not only affect sponging efficiency but also change the profile of RNA it can interact with. Therefore, it is possible that (1) significant lncRNA structural changes occur only when m6A marks are abundantly installed; (2) m6A methylation might work as a “switch”, which either promotes or inhibits lncRNA functionality as indicated by XIST9.
As Patil et al.9 suggests that m6A modulates lncRNA functional efficiency, it is possible to hypothesize further that m6A serves as an alternative path for stimuli response. Various stimuli’s result in changes in gene expression by interacting with DNA for expression of novel RNAs or promoting translation of current RNA molecules. We propose that m6A offers an alternative path for stimuli response. Instead of transcribing new RNAs, the present RNAs can be m6A modified (or m6A removed), which would result in required response whether its changed lncRNA binding efficiency and profile or changed half-life and translation efficiency of mRNAs. An alternative approach focuses on efficiency rather than the abundance of the molecule.
Even though, MeRIP-seq provided intriguing results it may not be the best approach to study m6A’s in lncRNAs. We and others hypothesise that lncRNAs contain more m6A marks than mRNA, however comparison of a single lncRNA versus all mRNA could be biased. One of the main limitations for m6A research is its detection methodology. While MeRIP-seq is great at detecting molecules containing m6A it lacks resolution to accurately measure m6A quantity48. Methods like individual-nucleotide resolution Cross-Linking and Immuno Precipitation (iCLIP) offers single nucleotide resolution49 but does not allow investigation of all m6A epi-transcriptome. The lack of bioinformatic tools for m6A analysis preclude complete understanding of m6A biology, which is of particular importance for lncRNA not only because of possibly containing more m6A modifications than mRNA but also because of lncRNA annotation. Therefore, future studies should look for more sensitive m6A detection methods at higher resolution, and bioinformatic tools specific for lncRNA to provide further insight into lncRNA m6A biology. Furthermore, 5′ and 3′ regions of non-coding RNAs should receive accurate nomenclature to enable continuous growth of the research field and more detailed analysis of functionality of specific m6A peaks.
Taken all together, our results indicate that m6A in lncRNA may overcome the need for abundance in molecular numbers. Highlight an additional level of lncRNA tissue specificity and functionality. Provide first insight into GSCs epi-transcriptome and illustrate the importance of epi-transcriptomic studies.
Material and methods
Cell culture
Glioblastoma stem-like cell line NCH421K (CLS Cell Lines Service GmbH, Eppelheim, Germany, cat. no. 300118) was a kind gift of Dr. A. Jekabsone from the Faculty of Pharmacy of Lithuanian University of Health Sciences, Kaunas, Lithuania. NCH421K represents glioma stem cells from primary glioblastoma without EGFR amplification, is enriched with neural stem cell markers NESTIN and CD133 and is characterized by glioma-typical amplifications of Cdk4, MDM2 and PDGFRA50.
NCH421K cells were grown as spheroid suspension in complete DMEM/Ham F-12 media (Sigma-Aldrich, cat. no. D8437), supplemented with 100 IU/mL of penicillin, 100 µg/mL of streptomycin (Gibco, cat. no. 15140122), 0.12% bovine albumin fraction V (Gibco, cat. no. 15260037), 1 × Minimum essential media (Gibco, cat. no. 11140035), 0.8 g/L D-Gliucose solution (Sigma-Aldrich, cat. no. G8644), 0.5 × B-27 (Gibco, cat. no. 17504044), 0.5 × N-2 (Gibco, cat. no. 17502048), 20 ng/mL bFGF and EGF (Gibco, cat. no. PHG0261 and PHG0311). Upon treatment with serum-containing media, neurospheres of GSCs quickly attached to cell cultivation flask, branched out, and lost their neurosphere-like shape (Supplementary Fig. S4A, B). Stemness was verified by assessing gene expression levels of SOX2, POU5F1, MYC, PROM1, KLF4, NANOG, and GFAP by RT-qPCR (Supplementary Fig. S4C, D; Table S2).
Glioblastoma (U87-MG) cells were obtained from the European Collection of Cell Cultures (ECACC, cat. no. 89081402) and cultivated in high glucose DMEM solution media (Gibco, cat. no. 10566016) with 10% fetal bovine serum (Gibco, cat. no. 10500064) and p/s. Both cell lines were incubated in a humidified atmosphere with 5% CO2 at 37 °C. Mycoplasma contamination was checked by a PCR assay designed to detect the 16S ribosomal DNA coding region51.
RNA extraction
Trizol reagent (Invitrogen, cat. no 15596026) was used to isolate total RNA from U87-MG and NCH421K cells. Cell culture media was replaced with 1 ml of ice-cold PBS. Cells were scraped from the culture dish. Obtained cell pallets were lysed with TRIzol (1 mL TRIzol per 5 × 106 cells). The procedure was performed according to the manufacturer’s recommendations. Extracted RNA was eluted in RNase free water. RNA quantity and quality was assessed with NanoDrop 2000 spectrophotometer (Thermo Scientific), agarose gel electrophoresis, and Agilent 2000 Bioanalyzer with “Agilent RNA 6000 Pico kit” (Agilent, cat. no. 5067-1513). RNA of quality no less than 1.9 at 260/280 and 2.2 at 260/230 and with RIN no less than 9.6 was used for further analysis.
N6-methyladenine immunoprecipitation following by next-generation sequencing (MeRIP-seq)
Poly-A RNA enrichment and fragmentation
“Dynabeads mRNA DIRECT kit” (Invitrogen, cat. no. 61012) was used for the enrichment of polyA RNA. The procedure was performed according to the manufacturer’s instructions. 600–700 µg of total RNA was used to get 15–25 µg of polyA enriched RNA. 250 µL of beads were used per 40 µg of total RNA. PolyA enriched RNA was precipitated overnight (O/N) at −80 °C with 1/10 volume of 3 M sodium acetate (pH 5.52), 100 µg/mL (final) glycogen (Thermofisher, cat. no. R0551), and 2.5 volumes of 100% ethanol, and resuspended in RNase free water. PolyA RNA was shredded into ~ 100 nt length fragments. 18 µg polyA RNA in a total volume of 20 µL with 10 × fragmentation buffer (H2O, TRIS–HCl, pH 7.0; ZnCl2), at 94 °C for 3 min. in AB Veriti thermal cycler (Applied Biosystems). PolyA RNA fragmentation efficiency was checked on a 1.5% agarose gel and by Agilent Bioanalyzer.
m6A Immunoprecipitation
For N6-methyladenine immunoprecipitation (MeRIP) we followed protocols of “Magna MeRIP m6A kit” (Sigma-Aldrich, cat. No. 1710499), Dominissini and Meyer group33,52. 0.125 mg of Pierce Protein A/G Magnetic Beads (Thermofisher, cat. no. 88803) were coupled with 5 µg of m6A antibody (Synaptic Systems, cat. no. 202–003) in 500 μL IP buffer (50 mM Tris–HCl 7.5 pH, 150 mM NaCl, 0.1% (vol/vol) Igepal) for 6 h at 4 °C. Bead-antibody complex was then washed twice with 1 mL IP buffer for 10 min. 5 µg of fragmented polyA RNA was mixed with 500 µL IP buffer (supplemented with 0.3 U/µL SUPERase-In RNase inhibitor (Invitrogen, cat. no. AM2694) and 2 mM RVC) and placed on washed bead-antibody complex for overnight incubation at 4 °C. Mixture was washed twice for 10 min. with 1 mL IP buffer (supplemented with 0.1 U/µL SUPERase-In RNase inhibitor) and once with 1 mL high salt buffer (50 mM Tris–HCl 7.5 pH, 300 mM NaCl, 0.1% (vol/vol) Igepal, 0.1 U/µL SUPERase-In RNase inhibitor). Bead-antibody-RNA complex was treated twice with 100 µL elution buffer (IP buffer supplemented with m6A salt at 6.7 mM (Sigma-Aldrich, cat. no. M2780)) for 1 h at 4 °C. RNA was recovered by overnight ethanol precipitation (2.5 vol 100% ethanol and 1/10 volume of 3 M sodium acetate: pH 5.2) at −80 °C.
MeRIP-seq
Both m6A immunoprecipitated (IP) and RNA input (as control) samples were sequenced in triplicates. The RNA amount used for sequencing varied in a range of 0.2–7 ng, since the yield of IP samples was extremely low (0.016–0.479 ng/µl). Library preparation and sequencing service was done applying RIP-seq protocol (Novogene, Europe). In short, fragmented RNA (80–120 nt) was first synthesized to double-stranded cDNA using random hexamer primers, adapter ligated on both 5′ and 3′ ends, and PCR amplified prior sequencing (single-end 50 bp (SE50), 25 M reads/sample).
m6A peak calling
All sequenced samples overwent a quality control check, after which the 3rd replicate of U87-MG was indicated as an extreme outlier (Supplementary Fig. S5), thus, this sample was eliminated from the downstream analysis.
Sequencing reads were processed using “nf-core’s”53 “chipseq” pipeline (ver.: 1.2.2) run within Singularity54 container through Nextflow process scheduler55. In short, raw reads were evaluated with FastQC56. Fifty-eight and 63 nucleotides of 5′ and 3′ adapters (respectively) were trimmed using Trim Galore57. Then, trimmed reads were aligned against Ensembl’s GRCh37 reference genome from Amazon’s iGenomes database58 with BWA59 and filtered out from ambiguous reads using Picard60, SAMtools61, BAMtools62 and Pysam63. Afterwards, the peaks were called with MACS264, providing input samples from the MeRIP experiment as a control, and m6A enrichments were identified using HOMER65. Finally, m6A peaks were compared between cell lines using DESeq266.
Gene expression analysis
Input samples, from the MeRIP experiments, were used to measure gene expression level of NCH421K and U87-MG cells. To do so, “nf-core’s” “rnaseq” pipeline (ver. 3.1)53 was applied. In brief, quality of the reads was estimated with FastQC, adapters were trimmed using Trim Galore, genome/ribosomal RNA sequences were discarded with BBSplit67/SortMeRNA68. Filtered reads were aligned to Ensembl’s GRCh37 reference genome using STAR69 sort, indexed with SAMtools and gene/transcript quantified applying Salmon70/StringTie71. Finally, gene expression levels were compared between cell lines using DESeq2. All the computational analysis were performed on the GenomeDK cluster.
Data visualization, targets selection, m6A modification and expression level comparison were done using machine learning and data visualization toolkit “Orange” (ver.3.28, University of Ljubljana). Integrative Genomics Viewer (IGV ver.2.9.2, University of California) was applied for visual exploration of transcriptomic data.
Clinical relevance verification of expression of lncRNAs in GEPIA
The relative expression of selected genes was verified in database GEPIA2 (Gene Expression Profiling Interactive Analysis). GEPIA2 is an online tool providing data analysis concerning gene expression, tumor stage and survival (http://gepia.cancer-pku.cn/), and is widely used to compare gene expression between tumor and normal tissue exploring RNA-seq expression data from the TCGA and the GTEx projects35. In our study, relative gene expression was detected using log2 fold change (log2FC = 1), and FDR adjusted p < 0.01. On survival analysis pane, survival contribution of genes between different tumors (GBM vs LGG) was evaluated using Mantel-Cox test with FDR adjusted p < 0.01. Median gene expression values were used as group cut-off.
References
Boccaletto, P. et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx1030 (2018).
Gilbert, W., Bell, T. A. & Schaening, C. Messenger RNA modifications: Form, distribution, and function. Science (New York, NY) 352, 1408–1412 (2016).
Esteller, M. & Pandolfi, P. P. The epitranscriptome of noncoding RNAs in cancer. Cancer Discov. 7, 359–368 (2017).
Dinescu, S. et al. Epitranscriptomic signatures in lncRNAs and their possible roles in cancer. Genes 10, 52 (2019).
Shi, H. et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 27, 315–328 (2017).
Chen, T. et al. m6A RNA methylation is regulated by MicroRNAs and promotes reprogramming to pluripotency. Cell Stem Cell 16, 289–301 (2015).
Dominissini, D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).
Alarcón, C. R., Lee, H., Goodarzi, H., Halberg, N. & Tavazoie, S. F. N6-methyladenosine marks primary microRNAs for processing. Nature 519, 482–485 (2015).
Patil, D. P. et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373 (2016).
Barbieri, I. et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 552, 126–131 (2017).
Lin, S., Choe, J., Du, P., Triboulet, R. & Gregory, R. I. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 62, 335–345 (2016).
Roundtree, I. A., Evans, M. E., Pan, T. & He, C. Dynamic RNA modifications in gene expression regulation. Cell 169, 1187–1200 (2017).
Thapar, R. et al. RNA modifications: Reversal mechanisms and cancer. Biochemistry 58, 312–329 (2019).
Agarwala, S. D., Blitzblau, H. G., Hochwagen, A. & Fink, G. R. RNA methylation by the MIS complex regulates a cell fate decision in yeast. PLoS Genet. 8, e1002732 (2012).
Schwartz, S. et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 8, 284–296 (2014).
Nance, D. J. et al. Characterization of METTL16 as a cytoplasmic RNA binding protein. PLoS ONE 15, e0227647 (2020).
Fu, Y. et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat. Commun. 4, 1798 (2013).
Mauer, J. et al. Reversible methylation of m6 Am in the 5′ cap controls mRNA stability. Nature 541, 371–375 (2017).
Zheng, G. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013).
Xu, C. et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat. Chem. Biol. 10, 927–929 (2014).
Zhang, B. et al. Alternative splicing-related factor YT521: An independent prognostic factor in endometrial cancer. Int. J. Gynecol. Cancer 20, 492–499 (2010).
Zhao, B. S., Roundtree, I. A. & He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 18, 31–42 (2016).
Nachtergaele, S. & He, C. The emerging biology of RNA post-transcriptional modifications. RNA Biol. 14, 156–163 (2017).
Liu, N. & Pan, T. N6-methyladenosine-encoded epitranscriptomics. Nat. Struct. Mol. Biol. 23, 98–102 (2016).
McIntyre, A. B. R. et al. Limits in the detection of m6A changes using MeRIP/m6A-seq. Sci. Rep. 10, 1–15 (2020).
Poon, M. T. C., Sudlow, C. L. M., Figueroa, J. D. & Brennan, P. M. Longer-term (≥ 2 years) survival in patients with glioblastoma in population-based studies pre- and post-2005: A systematic review and meta-analysis. Sci. Rep. 10, 11622 (2020).
Dressler, E. et al. Patterns and disparities of care in glioblastoma. Neuro-Oncol. Pract. 6, 37–46 (2019).
Omuro, A. & DeAngelis, L. M. Glioblastoma and other malignant gliomas: A clinical review. J. Am. Med. Assoc. 310, 1842–1850 (2013).
Ostrom, Q. T., Gittleman, H., Stetson, L., Virk, S. & Barnholtz-Sloan, J. S. Epidemiology of Intracranial Gliomas. Prog. Neurol. Surg. 30, 1–11 (2017).
Singh, S. K. et al. Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004).
Cui, Q. et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 18, 2622–2634 (2017).
Brown, J. A., Kinzig, C. G., DeGregorio, S. J. & Steitz, J. A. Methyltransferase-like protein 16 binds the 3’-terminal triple helix of MALAT1 long noncoding RNA. Proc. Natl. Acad. Sci. U.S.A. 113, 14013–14018 (2016).
Meyer, K. D. et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646 (2012).
Liu, H. et al. A novel N6-methyladenosine (m6A)-dependent fate decision for the lncRNA THOR. Cell Death Dis. 11, 1–15 (2020).
Tang, Z., Kang, B., Li, C., Chen, T. & Zhang, Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 47, W556–W560 (2019).
Peng, Z., Liu, C. & Wu, M. New insights into long noncoding RNAs and their roles in glioma. Mol. Cancer 17, 1 (2018).
Han, Y. et al. Downregulation of lncRNA-MALAT1 Affects proliferation and the expression of stemness markers in glioma stem cell line SHG139S. Cell. Mol. Neurobiol. 36, 1097–1107 (2016).
Ghafouri-Fard, S. et al. Role of long non-coding RNAs in conferring resistance in tumors of the nervous system. Front. Oncol. 11, 2054 (2021).
Katsushima, K. et al. Targeting the Notch-regulated non-coding RNA TUG1 for glioma treatment. Nat. Commun. 7, 13616 (2016).
Cai, H. et al. Long non-coding RNA taurine upregulated 1 enhances tumor-induced angiogenesis through inhibiting microRNA-299 in human glioblastoma. Oncogene 36, 318–331 (2017).
Wang, X. et al. N(6)-methyladenosine modification of MALAT1 promotes metastasis via reshaping nuclear speckles. Dev. Cell 56, 702-715.e8 (2021).
Liu, N. et al. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA (New York, NY) 19, 1848–1856 (2013).
Guo, T., Liu, D.-F., Peng, S.-H. & Xu, A.-M. ALKBH5 promotes colon cancer progression by decreasing methylation of the lncRNA NEAT1. Am. J. Transl. Res. 12, 4542–4549 (2020).
Lee, Y., Choe, J., Park, O. H. & Kim, Y. K. Molecular mechanisms driving mRNA degradation by m(6)A modification. Trends Genet. 36, 177–188 (2020).
Lu, W. et al. N(6)-Methyladenosine-binding proteins suppress HIV-1 infectivity and viral production. J. Biol. Chem. 293, 12992–13005 (2018).
Tirumuru, N. et al. N(6)-methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. Elife 5, 15528 (2016).
Huang, H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295 (2018).
Linder, B. et al. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772 (2015).
König, J. et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 17, 909–915 (2010).
Campos, B. et al. Differentiation therapy exerts antitumor effects on stem-like glioma cells. Clin. Cancer Res. 16, 2715–2728 (2010).
Uphoff, C. C. & Drexler, H. G. Comparative PCR analysis for detection of mycoplasma infections in continuous cell lines. In Vitro Cell. Dev. Biol. Anim. 38, 79–85 (2002).
Dominissini, D., Moshitch-Moshkovitz, S., Salmon-Divon, M., Amariglio, N. & Rechavi, G. Transcriptome-wide mapping of N6-methyladenosine by m 6A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 8, 176–189 (2013).
Ewels, P. A. et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 38, 276–278 (2020).
Kurtzer, G. M. et al. hpcng/singularity: Singularity 3.7.3. (2021) https://doi.org/10.5281/ZENODO.4667718.
di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 35, 316–319 (2017).
Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Babraham Bioinformatics - Trim Galore! https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
AWS iGenomes. https://ewels.github.io/AWS-iGenomes/.
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 26, 589–595 (2010).
Picard Tools - By Broad Institute. https://broadinstitute.github.io/picard/.
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, 1–4 (2021).
Barnett, D. W., Garrison, E. K., Quinlan, A. R., Stromberg, M. P., Marth, G. T. BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27(12), 1691–1692 (2011). https://doi.org/10.1093/bioinformatics/btr174.
GitHub - pysam-developers/pysam: Pysam is a Python module for reading and manipulating SAM/BAM/VCF/BCF files. It’s a lightweight wrapper of the htslib C-API, the same one that powers samtools, bcftools, and tabix. https://github.com/pysam-developers/pysam.
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Homer Software and Data Download. http://homer.ucsd.edu/homer/download.html.
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
GitHub - BioInfoTools/BBMap: (Not Offical) BBMap short read aligner, and other bioinformatic tools. https://github.com/BioInfoTools/BBMap.
Kopylova, E., Noé, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics (Oxford, England) 28, 3211–3217 (2012).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Acknowledgements
This project is funded by Lithuanian Research Council, project No S-SEN-20-7.
Author information
Authors and Affiliations
Contributions
D.S., G.S., A.T. generated an idea, and planned experiments. I.V., R.U. cultivated cells and extracted RNA. G.S., R.K., R.U., R.D. performed poly-A enrichment, m6A-IP. R.S. performed bioinformatic analysis and MeRIP-seq pipeline. G.S., R.S. analyzed MeRIP-seq data. G.S. performed data visualization. D.S., G.S., R.K. prepared the manuscript. All the authors have read the manuscript and approved the last version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Steponaitis, G., Stakaitis, R., Valiulyte, I. et al. Transcriptome-wide analysis of glioma stem cell specific m6A modifications in long-non-coding RNAs. Sci Rep 12, 5431 (2022). https://doi.org/10.1038/s41598-022-08616-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-08616-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
