
Abstract
A subset of essential thrombocythemia (ET) cases are negative for disease-defining mutations on JAK2, MPL, and CALR and defined as triple negative (TN). The lack of recurrent mutations in TN-ET patients makes its pathogenesis ambiguous. Here, we screened 483 patients with suspected ET in a single institution, centrally reviewed bone marrow specimens, and identified 23 TN-ET patients. Analysis of clinical records revealed that TN-ET patients were mostly young female, without a history of thrombosis or progression to secondary myelofibrosis and leukemia. Sequencing analysis and human androgen receptor assays revealed that the majority of TN-ET patients exhibited polyclonal hematopoiesis, suggesting a possibility of reactive thrombocytosis in TN-ET. However, the serum levels of thrombopoietin (TPO) and interleukin-6 in TN-ET patients were not significantly different from those in ET patients with canonical mutations and healthy individuals. Rather, CD34-positive cells from TN-ET patients showed a capacity to form megakaryocytic colonies, even in the absence of TPO. No signs of thrombocytosis were observed before TN-ET development, denying the possibility of hereditary thrombocytosis in TN-ET. Overall, these findings indicate that TN-ET is a distinctive disease entity associated with polyclonal hematopoiesis and is paradoxically caused by hematopoietic stem cells harboring a capacity for cell-autonomous megakaryopoiesis.
Similar content being viewed by others

Introduction
In approximately 80% of patients with essential thrombocythemia (ET), disease-defining mutations such as JAK2 V617F, MPL exon 10, and CALR exon 9 are found in a mutually exclusive manner1. Any of these mutant gene products induces the constitutive activation of MPL, the thrombopoietin (TPO) receptor, and its downstream molecules, leading to the clonal expansion of hematopoietic stem cells and the cell-autonomous expansion of megakaryocytes, thus causing thrombocytosis1. However, approximately 20% of ET cases do not harbor any of these mutations and thus are called triple-negative ET (TN-ET)2,3,4,5,6,7,8. Studies have shown the dominance of female patients in the TN-ET patient population, while other clinical parameters, such as age, platelet count, and frequency of thrombosis, vary between studies7,9,10, perhaps due to the diversity in ethnicities and medical practices in different institutions. Studies have also shown noncanonical mutations in JAK2 or MPL in a subset of patients with TN-ET. However, noncanonical mutants exhibit a subtle capacity to activate MPL signaling11,12, leaving the pathogenesis of TN-ET ambiguous. Here, we performed a single institution study to investigate the clinical and biological features of TN-ET.
Results
Younger females are dominant in the TN-ET patient population with no incidence of thrombotic events or progression to fibrosis
Based on WHO 2016 criteria, 178 patients were defined as having ET, and 13% (n = 23) of them were defined as having TN-ET. Statistical analysis was performed based on the clinical data of the patients grouped by their driver mutation status (Fig. 1A,B; Table 1). The TN-ET patients were mostly females (78.3%) and younger (median age of 36.0 years) than those in the other groups (Table 1). These results suggest that TN-ET is biologically different from ET harboring driver mutations (hereinafter referred to as mutated ET).

Comparison of clinical parameters between the TN-ET patients and mutated ET patients. (A) Frequencies of driver mutations in the ET patients in our cohort. (B) A diagram presenting the mutation profiles of the TN-ET patients. NC-JAK2/MPL: noncanonical JAK2 and MPL mutation. The WBC count (C), Hb value (D), platelet count (E), and LDH level (F) for the patients classified based on driver mutation status are shown. Gray highlight shows normal range. * < 0.05, ** < 0.01, *** < 0.001, ns: not significant.
The blood count data of the TN-ET patients resembled those of the ET patients harboring MPL exon 10 or CALR exon 9 mutations (Fig. 1C–E). However, a significant decrease in WBC count and hemoglobin (Hb) value and an increase in platelet count were observed in the TN-ET patients compared to the ET patients harboring the JAK2 V617F mutation (Fig. 1C–E). The serum LDH level was significantly reduced in the TN-ET patients compared to the ET patients harboring JAK2 V617F and CALR exon 9 mutations (Fig. 1F). All TN-ET patients displayed a normal or favorable risk karyotype except for one patient with an unfavorable risk karyotype (Table 1; Table S1)13.
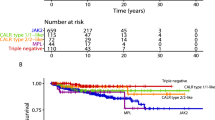
The analysis of clinical events such as thrombosis, progression to fibrosis, and leukemia transformation revealed that the TN-ET patients exhibited none of these events (Table 1; Fig. 2A,B). No significant differences were observed in fibrosis-free survival (FFS), leukemia-free survival (LFS), or overall survival (OS) between the ET groups (Fig. 2A–C), presumably owing to the small size of the cohort. Statistical analysis showed that the number of thrombotic events was significantly reduced in the TN-ET patients compared to the ET patients harboring the JAK2 V617F mutation (p = 0.006, Table 1). Again, these data strongly suggest that TN-ET exhibits clinical features that are different from those of mutated ET.

Survival data of the ET patients grouped by driver mutation status. The fibrosis-free survival (A), leukemia-free survival (B), and overall survival (C) of the patients stratified by driver mutation status are shown.
Noncanonical JAK2 and MPL mutant exhibits wild-type-equivalent levels of STAT5 activation
Because noncanonical mutations in JAK2 and MPL can be found in a subset of triple-negative myeloproliferative neoplasm (TN-MPN) patients11,12, we sequenced all exons of JAK2 and MPL for all the TN-ET patients (see Methods) and found 4 noncanonical JAK2 or MPL variants in 4 patients: JAK2 I724T (germline), MPL X636WX12 (germline), MPL S204F (somatic), and MPL A58V (unknown status due to a lack of germline control) (Fig. 1B; Table S1). To examine the oncogenic properties of these variant gene products, we performed a STAT5 reporter assay (see Methods). We found that, unlike canonical mutants, such as JAK2 V617F and MPL W515L, all noncanonical variants displayed wild-type-equivalent levels of STAT5 activation in the absence of TPO (Fig. 3). In agreement with this result, aside from MPL S204F, the noncanonical variants observed are rare variants in the general population (Table S2). In addition, by whole-exome sequencing (WES) analysis of 11 patients with available genomic DNA (gDNA) samples from both peripheral blood and CD3-positive cells, we found that 2 patients harbored somatic mutations (details shown in Table S1). One such patient was found to harbor MPL S204F. Nevertheless, no mutation was common in these patients and has been implicated as a driver mutation in MPN.

Noncanonical mutants of JAK2 and MPL exhibit wild-type-equivalent levels of STAT5 activation. (A,B) STAT5 reporter activity determined by luciferase reporter assay. The y-axis indicates values of STAT5 reporter activity adjusted by the internal control. Plasmids encoding JAK2 (A) or MPL (B) with the indicated mutation were used. Representative data from multiple experiments are presented. * < 0.05, ** < 0.01, ns: not significant.
Polyclonal hematopoiesis in TN-ET
Because of the considerably low frequency of somatic mutations in the TN-ET patients (Table S1), we suspected that the majority of the TN-ET patients had reactive thrombocytosis with bone marrow (BM) characteristics resembling the MPN. To this end, we performed a human androgen receptor (HUMARA) assay to examine the clonality of hematopoietic cells in the TN-ET patients by measuring the degree of skewed methylation on paternal and maternal X-chromosomes14. Seventeen of 18 female TN-ET patients had available gDNA from granulocytes (n = 12) or mononuclear cells (MNCs) (n = 5); we assessed their samples, and only one (9.1%) of 11 patients who exhibited judgeable results showed a clonal pattern, while 10 patients (90.9%) showed a polyclonal pattern (Fig. 4B). In contrast, all the patients (n = 3) harboring canonical driver mutations showed a clonal pattern as previously described15, and the patients (n = 3) with reactive thrombocytosis showed a polyclonal pattern (data not shown). The frequency (9.1%) of clonal hematopoiesis in TN-ET was within the range expected from a previous study where a higher frequency (14 of 31, 45.2%) of clonal hematopoiesis was observed in elderly females14. Nevertheless, despite the histology defining neoplastic features in the BM, most of the TN-ET patients in our cohort exhibited a polyclonal pattern according to the HUMARA assay.

Polyclonal hematopoiesis in the TN-ET patients harboring comparable serum levels of cytokines for megakaryopoiesis. (A) Typical profiles of capillary electrophoresis of HpaII-digested gDNA from granulocytes (n = 10) or MNCs (n = 5) and CD3-positive cells. Two HpaII-resistant peaks representing maternal and paternal alleles in polyclonal hematopoiesis. One of these alleles becomes HpaII-sensitive in granulocytes, representing clonal hematopoiesis (arrow). (B) A pie chart presenting the frequencies of clonal and polyclonal hematopoiesis judged by the HUMARA assay in TN-ET. Four of 15 patients who exhibited ambiguous patterns in the HUMARA assay (A) were excluded from the analysis. Comparison of TPO (C) and IL-6 (D) concentrations in the serum among the patients with TN-ET, patients with ET harboring driver mutations, and healthy controls.
The levels of serum cytokines were comparable among the TN-ET and ET patients harboring canonical mutations
To search for the cause of thrombocytosis associated with polyclonal hematopoiesis, we examined the levels of serum cytokines, such as TPO16,17,18,19 and interleukin-6 (IL-6)16,18,19, that have been shown to promote platelet production. By analyzing samples from 16 patients with TN-ET, 17 patients with mutated ET, and 8 healthy individuals, we found that the levels of TPO and IL-6 were increased in the patients with TN-ET and mutated ET compared to the healthy controls, but there was no significant difference among the groups, implying that the increases in the levels of TPO and IL-6 do not or only partly promote thrombocytosis in TN-ET (Fig. 4C,D).
Endogenous megakaryocyte (Mk) colonies were formed from the CD34-positive BM cells of TN-ET patients
To examine the capacity of megakaryopoiesis in stem/progenitor cells of the patients with TN-ET, we purified CD34-positive cells from cryopreserved BM cells and cultured them in semisolid media to form Mk colonies (see Methods). Twelve TN-ET, 7 mutated ET, and 7 control samples, including normal (n = 6) and reactive (n = 1) samples, were evaluated. Even in the absence of TPO, Mk colonies formed in TN-ET and mutated ET cell cultures but much less in control cell cultures (Fig. 5A). The capacity to form TPO-independent Mk colonies was determined by the ratio of the number of colonies formed in the absence and presence of TPO and compared between the groups. As shown in Fig. 5B, TN-ET and mutated ET exhibited equivalent capacities to form TPO-independent Mk colonies, and the capacity of these cells was significantly increased compared to that of the control cells. This indicates that despite polyclonal hematopoiesis in TN-ET, CD34-positive cells in TN-ET gain the capacity to promote megakaryopoiesis even in the absence of TPO.

Cell-autonomous megakaryopoiesis in hematopoietic stem/progenitor cells in TN-ET. (A) Representative images of megakaryocytic colonies (CD42b-positive) from CD34-positive BM cells from the indicated patients and controls. Scale bar indicates 100 µm. (B) Relative ratio of the number of megakaryocytic colonies for the indicated patients and controls formed in the presence and absence of TPO. ** < 0.01, *** < 0.001, ns: not significant.
Discussion
In the present study, we performed an in-depth analysis of the clinical and biological features of TN-ET in a single institution. Statistical analysis of clinical records revealed that young females were predominant in the TN-ET patient population with no incidence of thrombosis and no disease progression to fibrosis and leukemia (Table 1; Fig. 2), strongly suggesting that TN-ET is a disease entity that is biologically distinct from mutated ET. Sequencing analysis failed to identify somatic mutations in the majority of the TN-ET patients, suggesting reactive thrombocytosis associated with polyclonal hematopoiesis in TN-ET. In contrast, no significant elevation of cytokines related to platelet production was observed (Fig. 4). Biological analyses revealed that hematopoietic stem/progenitor cells in TN-ET presented a capacity to promote megakaryopoiesis in the absence of TPO (Fig. 5). Collectively, these findings indicate that TN-ET, which may still be a heterogeneous population, is a distinctive disease entity associated with polyclonal hematopoiesis but is caused in a cell-autonomous manner.
Compared to previous studies with large cohorts, in our cohort, the TN-ET patients (median age of 36.0 years) were younger (ranging between 47 and 59 years)7,9,10. Despite such differences that may reflect racial and geographical differences, we observed biological features such as polyclonal hematopoiesis and a low frequency of somatic mutations that resembled those observed among other cohorts. However, it is notable that no thrombotic events were observed in our cohort, while 10–20% of the TN-ET patients in other studies2,7,9,10 exhibited thrombosis. While the thrombosis risk determined by the IPSET-thrombosis model20 was comparable between TN-ET and CALR or MPL mutant ET (Table 1), the lack of thrombotic events in TN-ET might indicate the specific biological feature of TN-ET in our cohort.
Consistent with a previous study11,12,21, noncanonical JAK2 and MPL mutations were detected by WES or next-generation sequencing (NGS) in a fraction of the TN-ET patients in our cohort. The STAT5 reporter assay revealed that noncanonical mutants of JAK2 and MPL did not show activity similar to that of the canonical mutant proteins but rather exhibited the same level of activity as the wild-type proteins (Fig. 3). Although the noncanonical mutant MPL S204P has been shown to possess a weak gain-of-function property12, the present and previous11 studies implied that MPL S204F may not be the case. Nevertheless, the low frequency of noncanonical mutations in JAK2 and MPL in TN-ET does not fully explain the pathogenesis of TN-ET.
Polyclonal hematopoiesis was observed in nearly all the TN-ET patients examined, and these patients lack acquired mutations (Table S1). While this suggests the possibility of hereditary thrombocytosis, the platelet count before the diagnosis of all the patients (n = 10) with available blood count data showed no sign of thrombocytosis before diagnosis. Paradoxically, hematopoietic stem/progenitor cells in TN-ET exhibited an oncogenic property in megakaryopoiesis, which may explain the observation of indistinguishable morphology of BM in TN-ET with mutated ET. Based on these facts, we proposed that an environmental cue triggers a persistent epigenetic change in megakaryocytic progenitor cells to confer a capacity to promote megakaryopoiesis in the absence of TPO and results in thrombocytosis. Aberrations in epigenomic regulation not associated with genetic alteration have been shown to promote uncontrolled cell proliferation22. Such epigenomic changes may be reversible, and in fact, we observed spontaneous regression of thrombocytosis in one TN-ET patient diagnosed with thrombocytosis 1.5 years previously. Further analyses are required to better understand the pathogenesis of TN-ET, which should lead to the establishment of an appropriate treatment strategy against TN-ET.
In conclusion, we performed in-depth analysis of TN-ET and found that most of the TN-ET patients exhibited polyclonal hematopoiesis with no acquired mutation and no sign of hereditary thrombocytosis. Despite the possibility of reactive thrombocytosis, BM specimens exhibited the features of ET, and hematopoietic stem cells from TN-ET patients showed a capacity for cell-autonomous megakaryopoiesis, the hallmark of ET23. Based on these data, we propose that TN-ET, which may still be a heterogeneous population, is a biologically distinctive disease entity; thus, a different treatment strategy may need to be considered from that for mutated ET.
Methods
Patients
A total of 483 patients in the Hematology Department, Juntendo University Hospital who were suspected to have ET were analyzed and defined based on the WHO 2016 criteria24. Those who were suspected to have familial MPNs were excluded from this study. Preparation of gDNA and analysis of JAK2 V617F, MPL exon 10 and CALR exon 9 mutations were performed as described previously6,25,26,27. Histological analysis of BM was performed as described previously28. TN-ET was defined by the bone marrow morphology and by evidence of persistent thrombocytosis (> 450 × 109/L for more than six months for all except one who was only followed up for two months) with no potential cause of reactive thrombocytosis. This study was conducted in accordance with the Helsinki Declaration of 1975 and approved by the ethics committee of the School of Medicine, Juntendo University (IRB#2013020). Written informed consent was obtained prior to the use of samples and the collection of clinical records.
Exon analysis
All exons of JAK2 and MPL were sequenced as described previously6. Whole-exome sequencing was performed as described previously29. ClinVar30, gnomAD (https://gnomad.broadinstitute.org), and ToMMo-4.7KJPN in the Japanese Multi Omics Reference Panel (jMorp) (https://jmorp.megabank.tohoku.ac.jp/202001/) were used to search the frequencies of identified JAK2 and MPL noncanonical mutations in various races (Table S2).
STAT5 reporter assay
A STAT5 reporter assay was performed as described previously31. cDNA of JAK2 and MPL variants were subcloned into pcDNA3.1 plasmids, which were transfected and expressed in HEK293T cells. MPL was coexpressed with wild-type JAK2.
Statistics
Blood cell counts and biochemical parameters at the first visit prior to the treatment were analyzed. The durations for the development of fibrosis and leukemia were defined from the date of the first visit to that of the diagnosis of grade 2 or 3 fibrosis and to that of the detection of > 20% blasts in the peripheral blood or BM, respectively. Risk stratification according to the chromosome karyotype was defined according to a previous study13. The Kruskal–Wallis test/Steel–Dwass test (Table 1; Figs. 1C,E and F, 4C,D, 5B), one-way ANOVA/Tukey’s HSD test (Table 1; Fig. 1D), chi-square test/chi-square test with Bonferroni correction (Table 1), log-rank test (Fig. 2), and Student’s t test (Fig. 3) were used for statistical analysis. P values < 0.05 were considered to indicate statistical significance.
X-chromosome inactivation analysis
Peripheral blood mononuclear cells (PB-MNCs) and granulocytes (PB-Gs) were collected with Lymphosep Lymphocyte Separation Medium (MP Biomedicals) and Lymphocyte Separation Solution (Nacalai Tesque), respectively. CD3-positive cells were collected with the EasySep™ Human CD3 Positive Selection Kit II (STEMCELL Technologies) and expanded in complete medium supplemented with GTS™ OpTmizer™, CTS™ OpTmizer™ (Gibco™), L-glutamine (Gibco™), and human IL-2 (PeproTech). Genomic DNA was purified with the Gentra Puregene Blood Kit (QIAGEN). HUMARA assays, including fragment analysis by capillary electrophoresis using GeneMapper® software, were performed as previously described14,32.
Determination of the serum concentrations of TPO and IL-6
Serum samples were prepared by centrifugation at 1000 g for 15 min at room temperature and stored in a − 80 °C freezer until examination. To determine the levels of human TPO and IL-6, ELISAs were performed with the Human Thrombopoietin Quantikine ELISA Kit (R&D Systems) and the IL-6 Human Instant ELISA Kit (Invitrogen), respectively, according to the manufacturer’s instructions.
Human megakaryocyte colony formation assay
A Mk colony formation assay was performed as described previously33,34 with minor modifications. CD34-positive cells were purified from cryopreserved bone marrow mononuclear cells (BM-MNCs) using CD34 MicroBead Kit UltraPure (MACS Miltenyi Biotec). CD34-positive cells (5000) were cultured in a chamber (two-chamber slide, Matsunami) with serum-free collagen medium in the presence of 20 ng/mL human IL-3 (hIL-3) (Miltenyi Biotec) and human IL-6 (hIL-6) (Peprotech) and in the presence or absence of 50 ng/mL human thrombopoietin (hTPO) (Kyowa Kirin). The cultures were incubated at 37 °C in a humidified atmosphere of 5% CO2 in air for 12–14 days. Fixed cells were stained with anti-CD41 antibody (Clone HIP8, STEMCELL), biotin-conjugated anti-mouse IgG (Dako), streptavidin-conjugated alkaline phosphatase (Vector Laboratory), SIGMA FAST Red TR/naphthol AS-MX alkaline phosphatase substrate (Sigma), and Evans Blue (FUJIFILM Wako). BM-MNCs collected from lymphoma patients without BM infiltration and reactive thrombocytosis patients were used as negative controls.
Data availability
Exome sequencing data is deposited in the National Bioscience Database Center (NBDC; https://biosciencedbc.jp/en/).
Change history
17 January 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41598-022-04977-7
References
Vainchenker, W. & Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 129, 667–679. https://doi.org/10.1182/blood-2016-10-695940 (2017).
Tefferi, A. et al. Calreticulin mutations and long-term survival in essential thrombocythemia. Leukemia 28, 2300–2303. https://doi.org/10.1038/leu.2014.148 (2014).
Klampfl, T. et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 369, 2379–2390. https://doi.org/10.1056/NEJMoa1311347 (2013).
Nangalia, J. et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 369, 2391–2405. https://doi.org/10.1056/NEJMoa1312542 (2013).
Lundberg, P. et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood 123, 2220–2228. https://doi.org/10.1182/blood-2013-11-537167 (2014).
Shirane, S. et al. JAK2, CALR, and MPL mutation spectrum in Japanese patients with myeloproliferative neoplasms. Haematologica 100, e46-48. https://doi.org/10.3324/haematol.2014.115113 (2015).
Tefferi, A. et al. 2014 Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 124, 2507–2513. https://doi.org/10.1182/blood-2014-05-579136 (2014) (quiz 2615).
Fu, R. et al. Analysis of calreticulin mutations in Chinese patients with essential thrombocythemia: Clinical implications in diagnosis, prognosis and treatment. Leukemia 28, 1912–1914. https://doi.org/10.1038/leu.2014.138 (2014).
Rotunno, G. et al. Impact of calreticulin mutations on clinical and hematological phenotype and outcome in essential thrombocythemia. Blood 123, 1552–1555. https://doi.org/10.1182/blood-2013-11-538983 (2014).
Angona, A. et al. Molecular characterisation of triple negative essential thrombocythaemia patients by platelet analysis and targeted sequencing. Blood Cancer J. 6, e463. https://doi.org/10.1038/bcj.2016.75 (2016).
Milosevic Feenstra, J. D. et al. Whole-exome sequencing identifies novel MPL and JAK2 mutations in triple-negative myeloproliferative neoplasms. Blood 127, 325–332. https://doi.org/10.1182/blood-2015-07-661835 (2016).
Cabagnols, X. et al. Presence of atypical thrombopoietin receptor (MPL) mutations in triple-negative essential thrombocythemia patients. Blood 127, 333–342. https://doi.org/10.1182/blood-2015-07-661983 (2016).
Tefferi, A. et al. Revised cytogenetic risk stratification in primary myelofibrosis: Analysis based on 1002 informative patients. Leukemia 32, 1189–1199. https://doi.org/10.1038/s41375-018-0018-z (2018).
Tonon, L. et al. Unbalanced X-chromosome inactivation in haemopoietic cells from normal women. Br. J. Haematol. 102, 996–1003. https://doi.org/10.1046/j.1365-2141.1998.00867.x (1998).
Allen, C., Lambert, J. R., Linch, D. C. & Gale, R. E. X chromosome inactivation analysis reveals a difference in the biology of ET patients with JAK2 and CALR mutations. Blood 124, 2091–2093. https://doi.org/10.1182/blood-2014-06-580183 (2014).
Uppenkamp, M., Makarova, E., Petrasch, S. & Brittinger, G. Thrombopoietin serum concentration in patients with reactive and myeloproliferative thrombocytosis. Ann. Hematol. 77, 217–223. https://doi.org/10.1007/s002770050446 (1998).
Cerutti, A., Custodi, P., Duranti, M., Noris, P. & Balduini, C. L. Thrombopoietin levels in patients with primary and reactive thrombocytosis. Br. J. Haematol. 99, 281–284. https://doi.org/10.1046/j.1365-2141.1997.3823196.x (1997).
Griesshammer, M. et al. High levels of thrombopoietin in sera of patients with essential thrombocythemia: Cause or consequence of abnormal platelet production?. Ann. Hematol. 77, 211–215. https://doi.org/10.1007/s002770050445 (1998).
Panteli, K. E. et al. Serum interleukin (IL)-1, IL-2, sIL-2Ra, IL-6 and thrombopoietin levels in patients with chronic myeloproliferative diseases. Br. J. Haematol. 130, 709–715. https://doi.org/10.1111/j.1365-2141.2005.05674.x (2005).
Barbui, T. et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood 120, 5128–5133. https://doi.org/10.1182/blood-2012-07-444067 (2012) (quiz 5252).
Ju, M. et al. Mutation profiling by targeted sequencing of “triple-negative” essential thrombocythaemia patients. Br. J. Haematol. 181, 857–860. https://doi.org/10.1111/bjh.14723 (2018).
Ohnishi, K. et al. Premature termination of reprogramming in vivo leads to cancer development through altered epigenetic regulation. Cell 156, 663–677. https://doi.org/10.1016/j.cell.2014.01.005 (2014).
Komatsu, N. et al. Megakaryocytopoiesis in vitro of patients with essential thrombocythaemia: Effect of plasma and serum on megakaryocytic colony formation. Br. J. Haematol. 64, 241–252. https://doi.org/10.1111/j.1365-2141.1986.tb04116.x (1986).
Arber, D. A. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405. https://doi.org/10.1182/blood-2016-03-643544 (2016).
Misawa, K. et al. Mutational subtypes of JAK2 and CALR correlate with different clinical features in Japanese patients with myeloproliferative neoplasms. Int. J. Hematol. 107, 673–680. https://doi.org/10.1007/s12185-018-2421-7 (2018).
Edahiro, Y. et al. JAK2V617F mutation status and allele burden in classical Ph-negative myeloproliferative neoplasms in Japan. Int. J. Hematol. 99, 625–634. https://doi.org/10.1007/s12185-014-1567-1 (2014).
Takei, H. et al. Detection of MPLW515L/K mutations and determination of allele frequencies with a single-tube PCR assay. PLoS ONE 9, e104958. https://doi.org/10.1371/journal.pone.0104958 (2014).
Inano, T. et al. JAK2 exon 12 mutation in myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis: Not an exclusive mutation to polycythaemia vera. Br. J. Haematol. 187, e27–e31. https://doi.org/10.1111/bjh.16146 (2019).
Kawazu, M. et al. Integrative analysis of genomic alterations in triple-negative breast cancer in association with homologous recombination deficiency. PLoS Genet. 13, e1006853. https://doi.org/10.1371/journal.pgen.1006853 (2017).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067. https://doi.org/10.1093/nar/gkx1153 (2018).
Araki, M. et al. Homomultimerization of mutant calreticulin is a prerequisite for MPL binding and activation. Leukemia 33, 122–131. https://doi.org/10.1038/s41375-018-0181-2 (2019).
Wang, Y. et al. Molecular analysis of ovarian mucinous carcinoma reveals different cell of origins. Oncotarget 6, 22949–22958. https://doi.org/10.18632/oncotarget.5146 (2015).
Lansdorp, P. M. & Dragowska, W. Long-term erythropoiesis from constant numbers of CD34+ cells in serum-free cultures initiated with highly purified progenitor cells from human bone marrow. J. Exp. Med. 175, 1501–1509. https://doi.org/10.1084/jem.175.6.1501 (1992).
Hogge, D. et al. Quantitation and characterization of human megakaryocyte colony-forming cells using a standardized serum-free agarose assay. Br. J. Haematol. 96, 790–800. https://doi.org/10.1046/j.1365-2141.1997.d01-2092.x (1997).
Acknowledgements
We thank the members of the Department of Hematology, Juntendo University Graduate School of Medicine for encouraging this study. We would like to acknowledge the Laboratory of Molecular and Biochemical Research, Research Support Center, Juntendo University Graduate School of Medicine, for technical assistance.
Funding
This work was funded in part by a research program of the Project for Development of Innovative Research on Cancer Therapeutics, The Japan Agency for Medical Research and Development, the MEXT’s Promotion Plan for the Platform of Human Resource Development for Cancer Project, and JSPS KAKENHI Grants #17K16195 and #20H03715. The funders had no role in manuscript preparation. The authors declare no conflicts of interest associated with this manuscript.
Author information
Authors and Affiliations
Contributions
T.I. designed the study, carried out the experiments, and wrote the initial draft of the manuscript. M.A. contributed to manuscript preparation and the analysis and interpretation of data. S.J. contributed to all of the exon sequencing for JAK2 and MPL. Y.K. contributed to the serum cytokine analysis. M.Imai and M.O. contributed to the human megakaryocyte colony-forming assay. Y.Y. contributed to the STAT5 reporter assay. M.Ito contributed to the histological diagnosis of clinical samples. S.O. contributed to the statistical analysis. H.M. contributed to the whole-exome sequencing analysis. T.O., K.M., and Y.F. contributed to the data collection. Y.E., J.A. and N.K. supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: Figures 1 and 4, Table 1 and Supplemental Table 1 were corrected. Results were revised.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Inano, T., Araki, M., Morishita, S. et al. Cell-autonomous megakaryopoiesis associated with polyclonal hematopoiesis in triple-negative essential thrombocythemia. Sci Rep 11, 17702 (2021). https://doi.org/10.1038/s41598-021-97106-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-97106-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
