
Abstract
Biodiversity is rapidly changing due to changes in the climate and human related activities; thus, the accurate predictions of species composition and diversity are critical to developing conservation actions and management strategies. In this paper, using satellite remote sensing products as covariates, we constructed stacked species distribution models (S-SDMs) under a Bayesian framework to build next-generation biodiversity models. Model performance of these models was assessed using oak assemblages distributed across the continental United States obtained from the National Ecological Observatory Network (NEON). This study represents an attempt to evaluate the integrated predictions of biodiversity models—including assemblage diversity and composition—obtained by stacking next-generation SDMs. We found that applying constraints to assemblage predictions, such as using the probability ranking rule, does not improve biodiversity prediction models. Furthermore, we found that independent of the stacking procedure (bS-SDM versus pS-SDM versus cS-SDM), these kinds of next-generation biodiversity models do not accurately recover the observed species composition at the plot level or ecological-community scales (NEON plots are 400 m2). However, these models do return reasonable predictions at macroecological scales, i.e., moderately to highly correct assignments of species identities at the scale of NEON sites (mean area ~ 27 km2). Our results provide insights for advancing the accuracy of prediction of assemblage diversity and composition at different spatial scales globally. An important task for future studies is to evaluate the reliability of combining S-SDMs with direct detection of species using image spectroscopy to build a new generation of biodiversity models that accurately predict and monitor ecological assemblages through time and space.
Similar content being viewed by others

Introduction
Species diversity and composition vary in space and time as a consequence of historical biogeographic and environmental factors and ongoing ecological processes. Since the last millennium, however, rising human population and activities have been major drivers of environmental change on Earth1,2,3. As a consequence, biodiversity is changing at a pace solely compared to the major extinction events recorded in the geologic history1,4,5. These rapid changes in biodiversity are impacting the capacity of ecosystems to provide services to humanity that ultimately influence our well-being6,7,8. Thus, detecting and monitoring species diversity and composition is critical to developing effective management strategies and conservation actions facing global change3,9,10 that move us towards international biodiversity goals, including those posed by the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem (IPBES) and the parallel United Nations’ (UN) Sustainable Development Goals (UN-SDGs)8 and the upcoming post-2020 Convention on Biodiversity goals.
Different approaches have been proposed for assessing and monitoring the spatial and temporal patterns of species diversity and distribution10,11,12, including biodiversity models—i.e., models that aim to predict species diversity and composition13,14 and remote sensing technologies and products15,16,17,18. Among biodiversity models, the approach of Stacked Species Distribution Models (S-SDMs) has been successfully implemented for predicting species diversity and composition19,20,21. In building S-SDMs, individual SDM are modeled as a function of environmental predictors—usually derived from interpolated climate surfaces. Subsequently, these models are stacked to produce species diversity and composition assemblage predictions13,20. However, despite successful S-SDMs predictions, most previous studies were restricted to small geographic areas (~ 700 km2)19,20,22, limiting the potential of these models for biodiversity prediction and monitoring over large geographic areas, including at continental scales. Moreover, the uncertainty associated with the underlying data (i.e., occurrence records and environmental covariates) can negatively impact species model predictions23,24 and consequently the reliability of these kinds of biodiversity models.
Remote sensing products (RS-products) have been increasingly used to derive metrics that allow tracking biodiversity from space18,25,26, monitoring the state of human impacts3,27, as predictors for describing large patterns of species diversity28,29,30 or to derive Essential Biodiversity Variables, i.e., measures that allow the detection and quantification of biodiversity changes12,31,32. Despite their high spatial and temporal resolution, quasi-global coverage and range of data products (e.g., precipitation, plant productivity, biophysical variables, land cover), RS-products have been rarely used as predictors for biodiversity models17,30,33. RS-products have been dubbed important “next-generation” environmental predictors in biodiversity models34, given that remote sensing continuously captures an increasing range of Earth’s biophysical features at global scale35,36, avoiding the uncertainty associated with environmental predictors derived from traditional climatic data. Furthermore, species models derived from RS-products perform as well as those derived from interpolated climate surfaces and have the potential to provide predictions with greater spatial resolution30.
A crucial challenge for detecting and monitoring biodiversity is the integration of modeling approaches with RS-products in order to build next-generation biodiversity models at different spatial and temporal scale and levels of organization17,34. In addition, recent progress has been made in developing methods for use in S-SDM construction as well as in their evaluations21,37. By combining advances in S-SD modeling with RS-products, these developments could overcome some of the issues faced by traditional stacked models in predicting species richness and assemblage composition at different spatial scales. Therefore, the general aim of this paper is twofold: (1) to combine S-SDMs and RS-products in order to build next-generation biodiversity models, and (2) to perform a rigorous evaluation of biodiversity model predictions. In doing so, we applied a sequential procedure that allows the prediction and evaluation of species assemblage diversity and composition at continental scale. This procedure was implemented using the oak clade (genus Quercus) as a model study within the geographical area corresponding to the conterminous United States. We focused on this clade because oaks are widely distributed forest dominants, and they have the highest species richness and highest total biomass of any woody group in the conterminous U.S.38. Nevertheless, individual species have contrasting environmental distributions and range sizes. These attributes make the oaks both an important study system and a useful one for testing next generation SDMs.
Specifically, we begin by constructing individual species distribution models for each oak species using RS-products as covariates. The models we developed are designed to be biologically meaningful—i.e., we selected environmental covariates derived from Earth observations that are relevant to explaining distributions of trees in the oak genus—and to account for the accessible area of each species. To accomplish the latter, we constrained the training and transference areas. These models were then stacked to obtain predictions of oak species assemblage diversity and composition using different stacking procedures. Next, we conducted a comprehensive evaluation of assemblage predictions using observed assemblages’ diversity and composition obtained from the National Ecological Observatory Network (NEON).
Results
Species model predictions
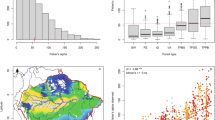
Oak species predictions were successfully calibrated for all species (Supplementary Table S1), showing high prediction accuracy as measured by the AUC (area under the receiver operating characteristic curve) and the True skill statistic (TSS) metrics (median ± SD: AUC 0.96 ± 0.03, TSS 0.82 ± 0.09; Supplementary Table S1). The number of covariates that better explain the probability of occurrence of oak species varies from 3 (Q. chapmanii; AUC = 0.99, TSS = 0.95) to 10 (Q. marilandica; AUC = 0.95, TSS = 0.76). Covariate importance evaluation (Fig. 1) indicate that precipitation seasonality (EO-BIO15 = 13.5%), annual precipitation (EO-BIO12 = 12.4%) and elevation (Elevation = 12%) were the covariates that contributed most to predict the occurrence of oak species; nonetheless, the number of covariates and their percentage of contribution varies considerably per species (Fig. 1). Notably, biophysical covariates such as LAI cumulative and LAI seasonality were selected as important covariates in predicting the occurrence for 21 and 23 oak species (Fig. 1), respectively.

Covariate importance or the relative contributions (%) of the 10 covariates used for modeling the potential distribution of oak species in the Continental United States.
Macroecological patterns of oak species richness indicate that oak species are distributed unevenly, showing high concentration in the Southeast United States, specifically in the NEON domains of the Ozark complex, the Southeast and the Mid-Atlantic (Fig. 2 top panel). Uncertainty predictions indicate that the Southeast NEON domain is the region with major uncertainty (Fig. 2 bottom panel).

Macroecological patterns of the number co-occurring oak species (top panel) and uncertainty (low panel) estimated by stacking individual species distribution model binary predictions (S-SDMs). Numbers on the legend represent the number of co-occurring within each pixel. In addition, blue and red colors mean low and high species co-occurrence (uncertainty), respectively. Star symbols (and acronyms) on the map represent the nineteen NSF-NEON sites that have data of oak species assemblages. Overlaid gray polygons represent the NSF-NEON ecoclimatic domains. Map projection: Lambert Azimuthal Equal Area. Maps were generated using ArcGis Desktop 10.6 (https://desktop.arcgis.com/).
Assemblage predictions
Mean observed richness per assemblage at the plot scale (i.e., NEON plots of 400 m2) was 3.27 (SD = 1.59, max = 10, min = 2), and mean richness prediction per assemblage for bS-SDMPLOT was 10.09 (SD = 4.19, max = 17, min = 1) and 10.54 (SD = 4.08, max = 15.80, min = 0) for pS-SDMPLOT. Mean observed richness at the site scale (i.e., NEON sites; mean area ~ 27 km2) was 6.37 (SD = 4.68, max = 17, min = 2), while the prediction for bS-SDMSITE was 8.79 (SD = 4.65, max = 17, min = 2) and for pS-SDMSITE was 9.23 (SD = 4.29, max = 15.64, min = 3.33). Bayesian correlations revealed mid positive correlations between the observed and the predicted richness derived from both the bS-SDMPLOT (ρ = 0.48 [0.38:0.57, 95% credible interval (CI)], Fig. 3B) and pS-SDMPLOT (ρ = 0.49 [0.40:0.59, 95% credible interval (CI)], Fig. 3F) at the plot scale. At the site scale the correlation increased for both bS-SDMSITE (ρ = 0.73 [0.45:0.91], Fig. 3A) and pS-SDMSITE (ρ = 0.81 [0.60:0.95], Fig. 3E). Although, richness predictions under pS-SDM correlate better with the observed richness, no evidence was found in favor of pS-SDM over bS-SDM (MAPSITE = 0.46, ROPESITE = 48%; MAPPLOT = 0, ROPEPLOT = 27%).

Predicted versus observed number of co-occurring oak species (left panels) and phylogenetic assemblage structure (right panels) at site and plot scales. Rho (ρ) values within each panel represent the median Pearson’s correlation coefficients estimated from posterior distribution and the 95% of credible intervals within brackets.
Mean absolute deviation between observed species richness and species richness predictions derived from the bS-SDM was 0.68 (SD = 0.37) and 0.16 (SD = 0.18) for the plot and site scales, respectively (Fig. 4A). The mean absolute deviation for pS-SDM was 0.73 (SD = 0.35) at the local scale and 0.18 (SD = 0.14) at the site scale. The mean species richness change (Fig. 4B) at the plot scale was 2.4 (SD = 1.69) for bS-SDM and 2.56 (SD = 1.65), while at the site scale it was 0.75 (SD = 1.18) and 0.85 (SD = 1.03) for bS-SDM and pS-SDM, respectively. Bayesian-ANOVAs reveled that bS-SDM outperformed pS-SDM, in predicting species richness, i.e., show low absolute deviation at the plot scale (MAPPLOT = 0) but not at the site scale (MAPSITE = 0.43). Similar results were found for species richness change metric (MAPPLOT = 0, MAPSITE = 0.59).

Accuracy of the number of co-occurring oak species predictions at site and plot scale. (A) Species richness deviation, measured as the absolute deviation in assemblage richness predictions divided by the maximum observed NEON plot species richness, where low and high values represent high and low accuracy respectively. (B) Species richness change, that represent the species loss or gain at plot scale. Negative values represent the number of species lost (underprediction) while positive values the gain of species (overprediction).
Mean assemblage prediction success was high for the three stacking procedures at both the site scale (bS-SDM = 0.79 ± 0.08, pS-SDM = 0.75 ± 0.07, cS-SDM = 0.88 ± 0.06; Fig. 5A) and the plot scale (bS-SDM = 0.87 ± 0.11, pS-SDM = 0.82 ± 0.10, cS-SDM = 0.88 ± 0.07; Fig. 5B). Assemblage TSS across all plots and sites ranged from low to high for the three stacking procedures (Fig. 5C,D). Mean assemblage similarity estimated using the Sørensen index were mid-low at plot scale (bS-SDM = 0.47 ± 0.17, pS-SDM = 0.39 ± 0.12, cS-SDM = 0.30 ± 0.28; Fig. 5E) while mid-high at the site scale (bS-SDM = 0.69 ± 0.17, pS-SDM = 0.59 ± 0.11, cS-SDM = 0.58 ± 0.27; Fig. 5F). Furthermore, although the mean values of the metrics used to evaluate the accuracy in assemblage predictions were relatively similar between the three stacking procedures (Fig. 5), Bayesian-ANOVAs revealed strong evidence in favor of the binary stacking (bS-SDM) in predicting assemblage composition, in other words, bS-SDM outperformed pS-SDM and cS-SDM. The higher performance of the bS-SDM procedure is consistent at both site and local scales, with the exception of prediction success metric, where cS-SDM outperformed bS-SDM at the plot scale (Fig. 5B) and no evidence was found at the site scale (Fig. 5A).

Accuracy of assemblage composition predictions at site and plot scales. Left panels (A,C,E) metrics at site scales and right panels (B,D,E) metrics at plot scale. Significant differences between stacking procedures (bS-SDM versus pS-SDM versus cS-SDM) were evaluated using Bayesian ANOVAs with NEON plot IDs as random effect (significance levels for the Maximum A Posteriori p-values are: ***p = 0, **p < 0.01, *p < 0.05, •p < 0.1, NE no evidence).
Phylogenetic assemblage structure
We were also interested in evaluating whether species composition of the assemblage predictions achieved by stacking SDMs returned similar patterns of phylogenetic structure as the observed assemblages. We found that the average SES-MPD at the plot and site scales for the observed assemblages were as follows: NEONPLOT = − 0.66 ± 1.30 and NEONSITE = − 1.04 ± 1.34; bS-SDMPLOT = − 1.12 ± 1.23 and bS-SDMSITE = − 0.71 ± 1.3; cS-SDMPLOT = − 1.42 ± 1.09 and cS-SDMSITE = − 1.02 ± 1.03. In addition, most of the predicted assemblages presented negative SES-MPD values (right panels in Fig. 3), indicating that the dominant pattern of phylogenetic structure in the predicted oak assemblages is overdispersion, a pattern that is commonly observed in natural oak assemblages39,40. Bayesian correlations showed mid to high positive associations between the phylogenetic structure obtained from the observed (NEON) and the bS-SDM at plot (ρ = 0.50 [0.41:0.59], Fig. 3D) and site (ρ = 0.75 [0.49:0.93], Fig. 3C) scales, respectively. The phylogenetic structure derived from the cS-SDM procedure, despite showing positive associations with the observed values, were mid and low for cS-SDMSITE (ρ = 0.51 [0.11:0.81], Fig. 3G) and cS-SDMPLOT (ρ = 0.20 [0.08:0.31], Fig. 3H), respectively.
Discussion
The use of RS-products in biodiversity models has been hailed as a transformative approach for providing simple and flexible biodiversity predictions17,34. In this study we evaluated the reliability of stacked species distribution models (S-SDM) to predict biodiversity in terms of species richness and composition within local assemblages13 based on individual SDMs derived only from RS-products. Our results indicate that the combination of S-SDM and RS-products perform well in predicting plot-level biodiversity as assessed by several important metrics. However, when we compared the S-SDM predictions of phylogenetic structure of oak assemblages at the plot-level with the observed structure (measured in terms of species richness and mean phylogenetic distance), the predictions tended to overestimate the diversity, even though the predicted and observed metrics were positively correlated. This result is consistent using the stacking procedure. We also found that when oak assemblages are evaluated at large scales, i.e., at the site scale, S-SDM recovered the observed pattern well, i.e., patterns of predicted diversity were highly similar to those observed at the NEON sites (Figs. 3A,C, 5A,C,E). Consequently, our results show that biodiversity models in combination with RS-products do not necessarily predict biodiversity well at the plot-level scale, but they do well at larger scales. In other words, the S-SDMs that use RS-products as environmental predictors are very useful for detecting macroecological patterns of biodiversity. These results spur new insights for biodiversity prediction and monitoring at different spatial scales.
Biodiversity models, such as the S-SDMs, depend on the reliability of individual species models (SDM)14. SDMs represent abstractions of species Hutchinsonian niches, in which species distributions are constrained by both the abiotic environment and biotic interactions with other species (Hutchinson's duality sensu41 see also Refs.30,42). Our results show that individual oak SDMs constructed using environmental covariates derived from RS-products, have good accuracy (Supplementary Table S1). However, by stacking a suite of SDMs to obtain assemblage composition predictions, no biotic constraints are considered. Consequently, S-SDM predictions tend to overestimate the number of species within local assemblages11,14. We found that species richness predictions using S-SDM overpredict the number of oak species that are observed in naturally assembled oak communities (Fig. 4A,B). This might be related to the proportion of common (high prevalence) and rare (low prevalence) species that are co-occurring in these assemblages, that in turn is affected by microhabitat variables that allow oak species to differentiate locally. Indeed, soil moisture (measured in 20 × 50 m plots on the ground) explain oak species trait distributions better than climate variables43. Local fire dynamics, not predicted by climate variables, may additionally be important in limiting and driving local species distributions44,45.
In addition, it has been suggested that incorporating some ecological assembly rules, e.g., the probability ranking rule approach (PRR), in S-SDM predictions can minimize assemblage overpredictions14,19,46. Our results corroborate this assumption; specifically, predicting species assemblages by simply overlaying individual SDMs, tends to overpredict the number of species present in local assemblages, especially at the scale of plots (Fig. 3B,F). However, through the implementation of PRR in S-SDM predictions—i.e., by constraining the number of species present within local assemblages14—we found that cS-SDM do not outperform bS-SDM assemblage predictions at both site and plot scales (Fig. 5). Rather, we found that simple stacking procedures (e.g., bS-SDM) outperform cS-SDM when the goal is to evaluate assemblage species composition (Fig. 5C–F). This conclusion is corroborated by the observation of low correlation between the phylogenetic structure of naturally assembled oaks at NEON sites and plots and assemblages predicted using cS-SDM (NEON sites = 0.51[0.11:0.81, 95% credible interval (CI)] and NEON plots = 0.20[0.08:0.31], see also Fig. 3G,H).
While applying constraints to assemblage predictions may be advantageous for predicting species richness11,14, this is not the case for predicting species identities or composition within assemblages, particularly at finer scales. Indeed, we found that no stacking procedure accurately recovered the species identities at the plot scale (Fig. 5D,F). Not surprisingly, predictions of assemblage richness and composition improved dramatically with increasing scale (Fig. 3A–E, 5C–E), a pattern observed independently of the implementation of PRR (cS-SDM) or its non-implementation (bS-SDM and pS-SDM). These contrasting results might be a consequence of the fact that S-SDMs were primarily designed for the description and assessment of macroecological patterns of species diversity11,13. This scale dependence might suggest that at larger spatial scales more uncertainty in model predictions is allowed, in other words, the probability of adding species in assemblages by increasing grain size also increases47 as well as the randomness in co-occurrence patterns40,48. This is not a small issue as no species occur within their complete geographic range, i.e., there are discontinuities in the species occurrence patterns that can be detected only at local or ecological scales. In addition, although RS-products are useful covariates for the spatial modelling of biodiversity, that allow reducing the uncertainty associated with environmental covariates in model predictions30, the coarse grain of current RS-products hampers accurate predictions of biodiversity at ecological scales30,36,49. In other words, the broad spatial resolution of current RS-products (usually with a pixel size of 30 arc-seconds or ~ 1 km) limits our ability to capture environmental features at ecological scales (e.g., microtopography, soil moisture, landscape structure) that ultimately determine the coexistence of species locally.
Furthermore, we acknowledge that we evaluated only one type of biodiversity model—S-SDM using two stacking procedures—and that other biodiversity models such as the Joint-SDM (J-SDM)50,51, could potentially improve biodiversity predictions by jointly estimating both the species-environment relationships (as in SDMs) and the species-pairwise dependencies—that reflect patterns of co-occurrence—not accounted by the covariates51,52,53. Nevertheless, recent studies have suggested that J-SDM do not improve species assemblage predictions21,53, in fact, richness predictions from S-SDMs and J-SDMs tend to return similar outcomes53, but see Ref.54. Although J-SDM represent an outstanding alternative for modeling and predicting biodiversity at assemblage level51,54, this kind of biodiversity models become untraceable when they are applied to large datasets53,55 (e.g., NEON or FIA datasets), in other words, the species-pairwise dependencies matrix or residual correlation matrix increases quadratically by adding new species to the dataset55. Further research is needed to correctly predict biodiversity at the assemblage level.
The evaluation presented here is meant to stimulate further methodological and empirical research to better predict biodiversity at different spatial and temporal scales and levels of organization. A promising approach for this purpose is the hierarchical modeling of species distributions (H-SDMs)56,57. H-SDMs allow the simultaneous modeling of spatial patterns of biodiversity at ecological and regional scales. In constructing H-SDMs, individual SDMs are first fit at regional and landscape scales, and the two predictions are fused to obtain a single species SDM, i.e., the regional model is rescaled to the landscape model56,57. This is an interesting approach because it fuses the benefits of macroecological covariates (derived from climatic surfaces or RS-products) with those derived from high-resolution remote sensing products56.
An unparalleled alternative is the direct detection of species using imaging spectroscopy58,59. For example, leaf-spectra variation among individual plants obtained using imaging spectroscopy provide sufficient information for the correct assignment of populations to species to clades60,61 and airborne imagery accurately assigns vegetation canopies to species62. Current and forthcoming hyperspectral images from DESIS sensors and forthcoming SBG and CHIMES sensors, among others18,63,64 will capture information from the Earth at fine spectral resolution, allowing the estimation of plant traits, plant nutrient content, biophysical variables (e.g., leaf area index, biomass), that can be used for direct detection of functional and perhaps community diversity from space36. Combining the detection of species using imaging spectroscopy with SDMs to build a new generation of biodiversity models, may open new avenues for the accurately assignment of species and assessment of ecological assemblages through space and time17,61,65,66, a critical hurdle to overcome in addressing the challenges posed by the global change.
Conclusion
Recent review papers17,34 proposed the applicability of remote sensing data as environmental covariates in the construction of next-generation SDMs. Here using environmental covariates derived completely from remote sensing products, we modelled the distribution of oaks (genus Quercus) and predicted the number and composition of species within assemblages at different grain sizes. Despite high variability in the predictions, modeled oak assemblages showed phylogenetic overdispersion, indicating that models recovered the observed pattern of distantly related oak species co-occurring more often than expected. Overall, we conclude that species richness can be predicted with high accuracy by applying constraints to the predictions. However, accurate predictions of species identities are still an evolving task. We suggest two alternatives (i.e., H-SDMs and the direct detection of species using imaging spectroscopy) that might increase the accuracy in assemblage composition predictions, hence, future studies should evaluate the reliability of these two alternatives using different taxa and across geographical settings.
Methods
Species occurrence dataset
The genus Quercus includes 91 species widely distributed across U.S. and showing a marked longitudinal species diversity gradient, with high species concentration in south-eastern North America. Our main occurrence dataset was assembled in a previous study67. We completed the main dataset using the Integrated Digitized Biocollections (iDigBio) and the Global Biodiversity Information Facility (GBIF) (data downloaded between 15 and 18 December, 2020) and collections from the second author (JCB)68. All occurrence data were visually examined and any localities that were outside the known geographical range of the species, in unrealistic locations (e.g., water bodies, crop fields) or in botanical gardens were discarded for accuracy. In addition, to avoid problems of spatial sampling bias and spatial autocorrelation we thinned the occurrence records of each species using a spatial thinning algorithm69 with thinning distance of 1 km for species with less than 100 occurrences up to 5 km for species with more than 10,000 records.
Species and assemblage composition evaluation data
Ecological niche models represent abstractions of the environmentally suitable areas for species to maintain long-term viable populations42. The presence of a species in a locality or a grid cell informs us about the areas that are environmentally suitable for that species42,70. Its absences inform us of the opposite pattern—namely those areas that are either not environmentally suitable for the species or are the result of historical contingences, biotic interactions that constraint the species presence even if the physical environment is suitable, recent extirpation events such as those caused by land use change, or simply because the species was not detected42,70. Here, using an independent dataset of true presences and absences from NEON71, we evaluate the assemblages’ diversity and composition predictions. NEON data follows a nested structure in which subplots are nested in larger plots and these plots in turn are nested within large areas or NEON Sites. More specifically, the presence of species is recorded at 10- and 100-m2 subplots. The recorded species within the subplots are then combined to obtain a complete species list for a plot of 400-m271. To obtain a complete list of oak species for each NEON site, we combined the species lists from the plots embedded in each site.
Here we used the species presence and absence at plot and site scale for assemblage predictions. This dataset includes a total of 277 plots of 400-m2 embedded in 19 NEON sites, spanning 8 out of the 17 NEON ecoclimatic domains in the continental United States. In addition, 36 of the 91 oak species were found in the NEON dataset; thus, we restricted all our analyses to this set of species (Fig. 1).
Environmental data derived from remote sensing
Remote sensing products used as environmental inputs to our models included a set of covariates that allow the description of distribution and environments occupied by oak species (EO-bioclimatic covariates) and covariates that allow the discrimination of local features not captured by bioclimatic information (biophysical covariates)30. Bioclimatic covariates were constructed by combining monthly Land Surface Temperature and Emissivity (LSTE) from MODIS (MOD11C3) and monthly precipitation from Climate Hazards group Infrared Precipitation with Stations (CHIRPS) following Deblauwe et al.72. Specifically, monthly LSTE and CHRIPS data were used as input to construct 19 bioclimatic covariates based on Earth observations72, similar to those of WorldClim, using the function ‘biovars’ in the R package dismo73. Prior to EO-bioclimatic construction global monthly LSTE (2001–2019) and CHIRPS (1981–2019) were downloaded using the interfaces EOSDIS Earthdata (https://earthdata.nasa.gov/) and the Climate Hazards Center (https://data.chc.ucsb.edu/products/CHIRPS-2.0/), respectively. We named each bioclimatic variable derived from Earth observation as EO-bioclimatic variables (e.g., EO-BIO1 or EO-MAT, EO-BIO12 or EO-AP) in order to avoid confusion with those bioclimatic variables derived from interpolated climate surfaces. EO-bioclimatic covariates used or modeling oak distributions include mean annual temperature (EO-BIO1), temperature seasonality (EO-BIO4), minimum temperature of coldest month (EO-BIO6), mean temperature of warmest quarter (EO-BIO10), annual precipitation (EO-BIO12), and precipitation seasonality (EO-BIO15), all critical for the distribution and differentiation of oak species67,68.
Biophysical covariates include Leaf Area Index (LAI) composites (cumulative, minimum and seasonality)28,29,30. LAI data were obtained from MODIS Terra/Aqua MOD15A2 over a 15-year period (2001–2015) using the interface EOSDIS Earthdata. The 15-year LAI composite provides a representation of the spatial variation of biophysical variables of different components of vegetation and ecosystems over the course of this time frame28,30. LAI strongly co-varies with the physical environment; for instance, higher LAI is associated with warmer, wetter and stable environments, whereas lower LAI with cooler, drier and less stable environments74,75,76. In particular, LAI has a strong relationship with climate and captures the dynamics of the growing season, including vegetation seasonality—important for characterizing plant species geographical ranges30,35. We posit, therefore, that the spatial and temporal variation in LAI—half of the total green leaf area per unit of horizontal ground surface area77—represents an important variable for determining species distributions that integrates across biotic and abiotic factors42,78. Finally, we also included mean elevation or altitude from Shuttle Radar Topography Mission (SRTM). SRTM is a high-resolution digital elevation model of the Earth that has been used for mapping and monitoring the earth’s surface79 and an important variable for predicting species distributions and community composition20,80.
Given that the original pixel size of both LSTE and CHIRPS products is 0.05° (5.6 km at the equator), we downscaled these products to 1 km to match the spatial resolution of both, biophysical covariates and SRTM. We examined the match between the original and the downscaled EO-bioclimatic covariates using Spearman rank correlations (ρ) and the Schoener’s D metric81 that is commonly used for estimating environmental similarity. D metric ranges from 0 (no similarity) to 1 (complete similarity). If the values of ρ and D are close to 1, we can hypothesize that exist a high correspondence between the two sets of EO-bioclimatic covariates, and consequently the downscaling process has null or neglected effect on the species distribution models. These analyses were performed for each species (N = 36) separately (Supplementary Table S2). We found high correspondence between the two data sets of EO-bioclimatic covariates (mean ± SD of ρ across species = 0.985 ± 0.015 and mean ± SD of D across species = 0.994 ± 0.003), thus we can conclude that our SDMs using downscaled EO-bioclimatic covariates are not compromised (Supplementary Table S2).
Modeling framework
To obtain reliable SDM predictions it is necessary to define the accessible area for a species in geographical space, i.e., the area that has been accessible to the species within a given period of time42,82. We defined the accessible area for each oak species as a bounding box around the known species occurrences, i.e., the occurrence records from the occurrence dataset, plus ~ 300 km beyond each bound. This procedure accounts for approximate species dispersal within a geographical domain and has been shown to improve model performance82,83. In other words, this procedure provides a conservative spatial representation of the environmental space in which a given species has potentially dispersed and is detectable. The individual accessible areas or species-specific accessible areas were then used to mask the covariates for each oak species and to constraint the random generation of pseudoabsences or background points before modeling species potential distributions. The number of pseudoabsences generated was similar to the number of presences84,85.
We modeled species potential distributions using Bayesian additive regression trees (BART)86. BART is a classification tree method defined by a prior distribution and a likelihood for returning occurrence predictions that enables the quantification of uncertainty around the predictions and the estimation of the marginal effects of the covariates86,87,88. BART models were run with the default parameters as implemented in dbarts89 through the R package embarcadero88. More specifically, BART models were run using 200 trees and 1000 back-fitting Marcov Chain Monte Carlo (MCMC)90 iterations, discarding 20% as burn-ins. Model performance or predictive ability was evaluated using two measures, the area under the receiver operating characteristic curve (AUC) and true skill statistics (TSS)91. To estimate the potential distribution of oak species, the resulting predictions (i.e., probability of species presences) under BART were converted to binary predictions (presence-absence maps) using TSS-maximization thresholds21,37.
To obtain assemblage composition and species richness predictions we applied three procedures for stacking species distribution models (S-SDM) (i.e., probability, binary and constrained binary). These procedures return the predicted species composition and richness within each assemblage (i.e., grid cell) across a geographical domain13,14. More specifically, probability S-SDM (pS-SDM) and binary S-SDM (bS-SDM) were obtained by stacking the probability of species presence and the binary prediction layers, respectively. Constrained binary S-SDM (cS-SDM) predictions were obtained by constraining each assemblage applying a probability ranking rule (PRR). PRR emulates ecological assembly rules by ranking the species in each assemblage based on the occurrence probability obtained from each species and the number of species per assemblage. The species with the highest probabilities in an assemblage is selected until the number of species in an assemblage, based on observed data, is reached19,37. We used the maximum number of species per assemblage from the NEON dataset as the assemblage-level constraint for cS-SDM estimations.
Evaluating species and assemblage predictions
To evaluate the performance of species assemblage predictions, we used four different metrics that are commonly used for this purpose20,21,37. The metrics used were: (a) the deviation of the predicted species richness to the observed (SR deviation); (b) species richness change (SR change); (c) the proportion of correctly predicted as present or absent (Prediction success); (d) true skill statistic (TSS) and (e) similarity between the observed and predicted community composition (Sørensen index). Assemblage metric evaluations were performed using modified functions from the R package ecospat92 using the matrices of the three assemblage predictions, i.e., probability, binary and PRR, against the observed assemblage composition from NEON. Note that the SR deviation and SR change were estimated only for the pS-SDM and bS-SDM predictions given that we used the NEON dataset in constraining the number of species per assemblage in the cS-SDM construction.
We also investigated the phylogenetic structure of oak assemblages for both the NEON dataset and the predicted assemblages, in order to explore the performance of predicted oak assemblages in recovering similar patterns of phylogenetic structure as the observed assemblages. To do so, we first obtained the latest phylogenetic hypothesis for oak lineages93. This phylogeny was constructed using restriction-site associated DNA sequencing (RAD-seq) and fossil data for node calibration and represent most highly resolved phylogenetic hypothesis for the clade globally93. We defined phylogenetic structure as the mean phylogenetic distance (MPD)94. To facilitate comparison between the two datasets, we summarized the results using standardized effects sizes (SES), which compare the observed value of an assemblage (MPD) to the mean expected value under a null model, correcting for their standard deviation. SES values > 0 and < 0 indicate phylogenetic clustering and overdispersion, respectively48,94. In SES calculations we randomized the tips of the phylogeny to generate random communities (taxon shuffle null model). All phylogenetic structure calculations were conducted using customized scripts and core functions from the picante95 package in R.
Statistical analysis
Using a Bayesian counterpart of Pearson’s correlation test, we evaluated the relationship between species richness and phylogenetic assemblage structure obtained from both NEON and predicted datasets. We chose this Bayesian alternative because it allows robust parameter estimations and accounts for outliers in the data96. We also used Bayesian-ANOVAs to test for differences in species assemblage predictions between different modelling procedures (i.e., stacking procedures), using plots as a random variable to correct for potential repeated measures. Using Maximum A Posteriori (MAP) p-values97, we then evaluated the evidence for those differences. Note that all analyses were performed for each scale separately, i.e., plot and site scales. Both Bayesian-ANOVAs and the robust Bayesian Pearson’s correlations were implemented in the probabilistic programming language Stan98 through the R packages rstanarm99 and brms100, respectively. All analyses were performed using 4 sampling chains for 10,000 generations and discarding 20% as burn-ins. MAP-based p-values was estimated as implemented in the R package bayestestR101.
Data availability
All data used in this paper are already published or publicly available. Data precipitation (CHIRPS) can be obtained from the Climate Hazards group Infrared Precipitation with Stations (CHIRPS-v2—https://www.chc.ucsb.edu/data/chirps) and Land Surface Temperature and Emissivity (LSTE) can be downloaded from EarthData (MOD11C3—https://lpdaac.usgs.gov/products/mod11c3v006/). Predicted oak species models can be found at https://doi.org/10.5281/zenodo.4611525. Code used for modeling oak species distributions is available at https://github.com/jesusNPL/BayesianSDMs_Oaks.
References
Lewis, S. L. & Maslin, M. A. Defining the anthropocene. Nature 519, 171–180 (2015).
Tilman, D. et al. Future threats to biodiversity and pathways to their prevention. Nature 546, 73–81 (2017).
Pinto-Ledezma, J. N. & Rivero Mamani, M. L. Temporal patterns of deforestation and fragmentation in lowland Bolivia: Implications for climate change. Clim. Change 127, 43–54 (2014).
Allen, J. M., Folk, R. A., Soltis, P. S., Soltis, D. E. & Guralnick, R. P. Biodiversity synthesis across the green branches of the tree of life. Nat. Plants 5, 11–13 (2019).
Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services, IPBES. Summary for policymakers of the global assessment report on biodiversity and ecosystem services. Accessed 15 Feb 2021. https://zenodo.org/record/3553579. https://doi.org/10.5281/ZENODO.3553579 (2019).
Cavender-Bares, J., Balvanera, P., King, E. & Polasky, S. Ecosystem service trade-offs across global contexts and scales. Ecol. Soc. 20, art22 (2015).
Chaplin-Kramer, R. et al. Global modeling of nature’s contributions to people. Science 366, 255–258 (2019).
Díaz, S. et al. Pervasive human-driven decline of life on Earth points to the need for transformative change. Science 366, eaax3100 (2019).
Watson, J. E. M. et al. Set a global target for ecosystems. Nature 578, 360–362 (2020).
Jetz, W. et al. Essential biodiversity variables for mapping and monitoring species populations. Nat. Ecol. Evol. 3, 539–551 (2019).
Mateo, R. G., Mokany, K. & Guisan, A. Biodiversity models: What if unsaturation is the rule?. Trends Ecol. Evol. 32, 556–566 (2017).
Pereira, H. M. et al. Essential biodiversity variables. Science 339, 277–278 (2013).
Ferrier, S. & Guisan, A. Spatial modelling of biodiversity at the community level. J. Appl. Ecol. 43, 393–404 (2006).
Guisan, A. & Rahbek, C. SESAM—A new framework integrating macroecological and species distribution models for predicting spatio-temporal patterns of species assemblages: Predicting spatio-temporal patterns of species assemblages. J. Biogeogr. 38, 1433–1444 (2011).
Cavender-Bares, J., Schweiger, A. K., Pinto-Ledezma, J. N. & Meireles, J. E. Applying remote sensing to biodiversity science. in Remote Sensing of Plant Biodiversity (eds. Cavender-Bares, J. et al.) 13–42 (Springer, 2020). https://doi.org/10.1007/978-3-030-33157-3_2.
Fawcett, D. et al. Advancing retrievals of surface reflectance and vegetation indices over forest ecosystems by combining imaging spectroscopy, digital object models, and 3D canopy modelling. Remote Sens. Environ. 204, 583–595 (2018).
Randin, C. F. et al. Monitoring biodiversity in the Anthropocene using remote sensing in species distribution models. Remote Sens. Environ. 239, 111626 (2020).
Turner, W. Sensing biodiversity. Science 346, 301–302 (2014).
D’Amen, M., Pradervand, J.-N. & Guisan, A. Predicting richness and composition in mountain insect communities at high resolution: A new test of the SESAM framework: Community-level models of insects. Glob. Ecol. Biogeogr. 24, 1443–1453 (2015).
Pottier, J. et al. The accuracy of plant assemblage prediction from species distribution models varies along environmental gradients: Climate and species assembly predictions. Glob. Ecol. Biogeogr. 22, 52–63 (2013).
Zurell, D. et al. Testing species assemblage predictions from stacked and joint species distribution models. J. Biogeogr. 47, 101–113 (2020).
D’Amen, M. et al. Improving spatial predictions of taxonomic, functional and phylogenetic diversity. J. Ecol. 106, 76–86 (2018).
Dobrowski, S. Z. et al. Modeling plant ranges over 75 years of climate change in California, USA: Temporal transferability and species traits. Ecol. Monogr. 81, 241–257 (2011).
Soria-Auza, R. W. et al. Impact of the quality of climate models for modelling species occurrences in countries with poor climatic documentation: A case study from Bolivia. Ecol. Model. 221, 1221–1229 (2010).
Rocchini, D. et al. Satellite remote sensing to monitor species diversity: Potential and pitfalls. Remote Sens. Ecol. Conserv. 2, 25–36 (2016).
Schulte to Bühne, H. & Pettorelli, N. Better together: Integrating and fusing multispectral and radar satellite imagery to inform biodiversity monitoring, ecological research and conservation science. Methods Ecol. Evol. 9, 849–865 (2018).
Hansen, M. C. et al. High-resolution global maps of 21st-century forest cover change. Science 342, 850–853 (2013).
Hobi, M. L. et al. A comparison of Dynamic Habitat Indices derived from different MODIS products as predictors of avian species richness. Remote Sens. Environ. 195, 142–152 (2017).
Radeloff, V. C. et al. The Dynamic Habitat Indices (DHIs) from MODIS and global biodiversity. Remote Sens. Environ. 222, 204–214 (2019).
Pinto-Ledezma, J. N. & Cavender-Bares, J. Using remote sensing for modeling and monitoring species distributions. In Remote Sensing of Plant Biodiversity (eds. Cavender-Bares, J., Gamon, J. A. & Townsend, P. A.) 199–223 (Springer International Publishing, 2020). https://doi.org/10.1007/978-3-030-33157-3_9.
Fernández, N., Ferrier, S., Navarro, L. M. & Pereira, H. M. Essential biodiversity variables: Integrating in-situ observations and remote sensing through modeling. In Remote Sensing of Plant Biodiversity (eds. Cavender-Bares, J., Gamon, J. A. & Townsend, P. A.) 485–501 (Springer International Publishing, 2020). https://doi.org/10.1007/978-3-030-33157-3_18.
Skidmore, A. K. et al. Priority list of biodiversity metrics to observe from space. Nat. Ecol. Evol. https://doi.org/10.1038/s41559-021-01451-x (2021).
Saatchi, S., Buermann, W., ter Steege, H., Mori, S. & Smith, T. B. Modeling distribution of Amazonian tree species and diversity using remote sensing measurements. Remote Sens. Environ. 112, 2000–2017 (2008).
He, K. S. et al. Will remote sensing shape the next generation of species distribution models?. Remote Sens. Ecol. Conserv. 1, 4–18 (2015).
Cord, A. F., Meentemeyer, R. K., Leitão, P. J. & Václavík, T. Modelling species distributions with remote sensing data: Bridging disciplinary perspectives. J. Biogeogr. 40, 2226–2227 (2013).
Jetz, W. et al. Monitoring plant functional diversity from space. Nat. Plants 2, 16024 (2016).
Scherrer, D., D’Amen, M., Fernandes, R. F., Mateo, R. G. & Guisan, A. How to best threshold and validate stacked species assemblages? Community optimisation might hold the answer. Methods Ecol. Evol. 9, 2155–2166 (2018).
Cavender-Bares, J. Diversification, adaptation, and community assembly of the American oaks (Quercus), a model clade for integrating ecology and evolution. New Phytol. 221, 669–692 (2019).
Cavender-Bares, J., Ackerly, D. D., Baum, D. A. & Bazzaz, F. A. Phylogenetic overdispersion in Floridian oak communities. Am. Nat. 163, 823–843 (2004).
Cavender-Bares, J. et al. The role of diversification in community assembly of the oaks (Quercus L.) across the continental U.S. Am. J. Bot. 105, 565–586 (2018).
Colwell, R. K. & Rangel, T. F. Hutchinson’s duality: The once and future niche. Proc. Natl. Acad. Sci. 106, 19651–19658 (2009).
Townsend Peterson, A. et al. Ecological Niches and Geographic Distributions. (Princeton University Press, 2011).
Cavender-Bares, J., Fontes, G. C. & Pinto-Ledezma, J. Open questions in understanding the adaptive significance of plant functional trait variation within a single lineage. New Phytol. https://doi.org/10.1111/nph.16652 (2020).
Cavender-Bares, J., Kitajima, K. & Bazzaz, F. A. Multiple trait associations in relation to habitat differentiation among 17 Floridian oak species. Ecol. Monogr. 74, 635–662 (2004).
Menges, E. S. & Hawkes, C. V. Interactive effects of fire and microhabitat on plants of Florida scrub. Ecol. Appl. 8, 935–946 (1998).
Calabrese, J. M., Certain, G., Kraan, C. & Dormann, C. F. Stacking species distribution models and adjusting bias by linking them to macroecological models: Stacking species distribution models. Glob. Ecol. Biogeogr. 23, 99–112 (2014).
Hurlbert, A. H. & Jetz, W. Species richness, hotspots, and the scale dependence of range maps in ecology and conservation. Proc. Natl. Acad. Sci. 104, 13384–13389 (2007).
Pinto-Ledezma, J. N., Jahn, A. E., Cueto, V. R., Diniz-Filho, J. A. F. & Villalobos, F. Drivers of phylogenetic assemblage structure of the Furnariides, a widespread clade of lowland neotropical birds. Am. Nat. 193, E41–E56 (2019).
Gamon, J. A. et al. Consideration of scale in remote sensing of biodiversity. In Remote Sensing of Plant Biodiversity (eds. Cavender-Bares, J., Gamon, J. A. & Townsend, P. A.) 425–447 (Springer International Publishing, 2020). https://doi.org/10.1007/978-3-030-33157-3_16.
Pollock, L. J. et al. Understanding co-occurrence by modelling species simultaneously with a Joint Species Distribution Model (JSDM). Methods Ecol. Evol. 5, 397–406 (2014).
Ovaskainen, O. et al. How to make more out of community data? A conceptual framework and its implementation as models and software. Ecol. Lett. 20, 561–576 (2017).
Ovaskainen, O. Joint Species Distribution Modelling: with Applications in R (Cambridge University Press, 2020).
Poggiato, G. et al. On the interpretations of joint modeling in community ecology. Trends Ecol. Evol. https://doi.org/10.1016/j.tree.2021.01.002 (2021).
Wilkinson, D. P., Golding, N., Guillera-Arroita, G., Tingley, R. & McCarthy, M. A. Defining and evaluating predictions of joint species distribution models. Methods Ecol. Evol. 12, 394–404 (2021).
Bystrova, D. et al. Clustering species with residual covariance matrix in joint species distribution models. Front. Ecol. Evol. 9, 601384 (2021).
Mateo, R. G. et al. Hierarchical species distribution models in support of vegetation conservation at the landscape scale. J. Veg. Sci. 30, 386–396 (2019).
Petitpierre, B. et al. Will climate change increase the risk of plant invasions into mountains?. Ecol. Appl. 26, 530–544 (2016).
Cavender-Bares, J. et al. Harnessing plant spectra to integrate the biodiversity sciences across biological and spatial scales. Am. J. Bot. 104, 966–969 (2017).
Schweiger, A. K. et al. Spectral Niches Reveal Taxonomic Identity and Complementarity in Plant Communities. (2020) https://doi.org/10.1101/2020.04.24.060483.
Cavender-Bares, J. et al. Associations of leaf spectra with genetic and phylogenetic variation in oaks: Prospects for remote detection of biodiversity. Remote Sens. 8, 221 (2016).
Meireles, J. E. et al. Leaf reflectance spectra capture the evolutionary history of seed plants. New Phytol. 228, 485–493 (2020).
Williams, L. J. et al. Remote spectral detection of biodiversity effects on forest biomass. Nat. Ecol. Evol. 5, 46–54 (2021).
Alonso, K. et al. Data products, quality and validation of the DLR earth sensing imaging spectrometer (DESIS). Sensors 19, 4471 (2019).
Stavros, E. N. et al. ISS observations offer insights into plant function. Nat. Ecol. Evol. 1, 0194 (2017).
Féret, J.-B. & Asner, G. P. Mapping tropical forest canopy diversity using high-fidelity imaging spectroscopy. Ecol. Appl. 24, 1289–1296 (2014).
Cavender-Bares, J. et al. BII-Implementation: The causes and consequences of plant biodiversity across scales in a rapidly changing world. Res. Ideas Outcomes 7, e63850 (2021).
Hipp, A. L. et al. Sympatric parallel diversification of major oak clades in the Americas and the origins of Mexican species diversity. New Phytol. 217, 439–452 (2018).
Cavender-Bares, J. et al. Phylogeny and biogeography of the American live oaks (Quercus subsection Virentes): A genomic and population genetics approach. Mol. Ecol. 24, 3668–3687 (2015).
Aiello-Lammens, M. E., Boria, R. A., Radosavljevic, A., Vilela, B. & Anderson, R. P. spThin: An R package for spatial thinning of species occurrence records for use in ecological niche models. Ecography 38, 541–545 (2015).
Lobo, J. M., Jiménez-Valverde, A. & Hortal, J. The uncertain nature of absences and their importance in species distribution modelling. Ecography 33, 103–114 (2010).
Barnett, D. T. et al. The plant diversity sampling design for The National Ecological Observatory Network. Ecosphere 10, e02603 (2019).
Deblauwe, V. et al. Remotely sensed temperature and precipitation data improve species distribution modelling in the tropics: Remotely sensed climate data for tropical species distribution models. Glob. Ecol. Biogeogr. 25, 443–454 (2016).
Hijmans, R. J., Phillips, S., Leathwick, J. & Elith, J. dismo: Species Distribution Modeling. R package version 1.3. https://CRAN.R-project.org/package=dismo (2020).
Myneni, R. B. et al. Global products of vegetation leaf area and fraction absorbed PAR from year one of MODIS data. Remote Sens. Environ. 83, 214–231 (2002).
Gower, S. T., Kucharik, C. J. & Norman, J. M. Direct and indirect estimation of leaf area index, fAPAR, and net primary production of terrestrial ecosystems. Remote Sens. Environ. 70, 29–51 (1999).
Reich, P. B. Key canopy traits drive forest productivity. Proc. R. Soc. B Biol. Sci. 279, 2128–2134 (2012).
Xiao, Z. et al. Use of general regression neural networks for generating the GLASS leaf area index product from time-series MODIS surface reflectance. IEEE Trans. Geosci. Remote Sens. 52, 209–223 (2014).
Soberón, J. Grinnellian and Eltonian niches and geographic distributions of species. Ecol. Lett. 10, 1115–1123 (2007).
Farr, T. G. et al. The shuttle radar topography mission. Rev. Geophys. 45, RG2004 (2007).
Dubuis, A. et al. Predicting spatial patterns of plant species richness: A comparison of direct macroecological and species stacking modelling approaches: Predicting plant species richness. Divers. Distrib. 17, 1122–1131 (2011).
Schoener, T. W. Anolis lizards of Bimini: Resource partition in a complex fauna. Ecology 49, 704–726 (1968).
Barve, N. et al. The crucial role of the accessible area in ecological niche modeling and species distribution modeling. Ecol. Model. 222, 1810–1819 (2011).
Cooper, J. C. & Soberón, J. Creating individual accessible area hypotheses improves stacked species distribution model performance. Glob. Ecol. Biogeogr. 27, 156–165 (2018).
Barbet-Massin, M., Jiguet, F., Albert, C. H. & Thuiller, W. Selecting pseudo-absences for species distribution models: How, where and how many? How to use pseudo-absences in niche modelling?. Methods Ecol. Evol. 3, 327–338 (2012).
Carlson, C. J. et al. The global distribution of Bacillus anthracis and associated anthrax risk to humans, livestock and wildlife. Nat. Microbiol. 4, 1337–1343 (2019).
Chipman, H. A., George, E. I. & McCulloch, R. E. BART: Bayesian additive regression trees. Ann. Appl. Stat. 4, 266–298 (2010).
Yen, J. D. L., Thomson, J. R., Vesk, P. A. & Mac Nally, R. To what are woodland birds responding? Inference on relative importance of in-site habitat variables using several ensemble habitat modelling techniques. Ecography 34, 946–954 (2011).
Carlson, C. J. embarcadero: Species distribution modelling with Bayesian additive regression trees in r. Methods Ecol. Evol. 11, 850–858 (2020).
Dorie, V. dbarts: Discrete Bayesian Additive Regression Trees Sampler. (2020).
Hastie, T. & Tibshirani, R. Bayesian backfitting. Stat. Sci. 15(3), 196–223 (2000).
Allouche, O., Tsoar, A. & Kadmon, R. Assessing the accuracy of species distribution models: Prevalence, kappa and the true skill statistic (TSS): Assessing the accuracy of distribution models. J. Appl. Ecol. 43, 1223–1232 (2006).
Di Cola, V. et al. ecospat: An R package to support spatial analyses and modeling of species niches and distributions. Ecography 40, 774–787 (2017).
Hipp, A. L. et al. Genomic landscape of the global oak phylogeny. New Phytol. 226, 1198–1212 (2020).
Webb, C. O., Ackerly, D. D., McPeek, M. A. & Donoghue, M. J. Phylogenies and community ecology. Annu. Rev. Ecol. Syst. 33, 475–505 (2002).
Kembel, S. W. et al. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26, 1463–1464 (2010).
Kruschke, J. Doing Bayesian Data Analysis, 2nd Ed. (2014).
Mills, J. A. & Parent, O. Bayesian MCMC estimation. In Handbook of Regional Science (eds Fischer, M. M. & Nijkamp, P.) 1571–1595 (Springer, Berlin, 2014). https://doi.org/10.1007/978-3-642-23430-9_89.
Carpenter, B. et al. Stan : A probabilistic programming language. J. Stat. Softw. 76, 1–32 (2017).
Goodrich, B., Gabry, J., Ali, I. & Brilleman, S. rstanarm: Bayesian applied regression modeling via Stan. (2020).
Bürkner, P.-C. brms: An R package for Bayesian multilevel models using Stan. J. Stat. Softw. 80, 1–28 (2017).
Makowski, D., Ben-Shachar, M. & Lüdecke, D. bayestestR: Describing effects and their uncertainty, existence and significance within the Bayesian framework. J. Open Source Softw. 4, 1541 (2019).
Acknowledgements
J.N.P.-L. was supported in part by the University of Minnesota College of Biological Sciences’ Grand Challenges in Biology Postdoctoral Program and by the National Science Foundation through the Macrosystems Biology & NEON-Enabled Science program (DEB 2017843 grant to J.N.P.-L. and J.C.-B.) with additional support provided by the National Aeronautics and Space Administration (20-BIODIV20-0048) and the National Science Foundation (DBI 2021898).
Author information
Authors and Affiliations
Contributions
J.N.P.-L. and J.C.-B. conceived the ideas presented and tested herein and contributed throughout the whole writing process.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pinto-Ledezma, J.N., Cavender-Bares, J. Predicting species distributions and community composition using satellite remote sensing predictors. Sci Rep 11, 16448 (2021). https://doi.org/10.1038/s41598-021-96047-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-96047-7
This article is cited by
-
Modeling present and future distribution of plankton populations in a coastal upwelling zone: the copepod Calanus chilensis as a study case
Scientific Reports (2023)
-
Integrating remote sensing with ecology and evolution to advance biodiversity conservation
Nature Ecology & Evolution (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
