
Abstract
Most transgenic animals are generated using a genome-modified stem cell system and genome modification directly in embryos. Although this system is well-established in the development of transgenic animals, donor cell-derived transgenic animal production is inefficient in some cases. Especially in avian models such as chickens, the efficiency of transgenic animal production through primordial germ cells (PGCs) is highly variable compared with embryonic manipulation of mammalian species. Because germ cell and germline-competent stem cell-mediated systems that contain the transgene are enriched only at the upstream level during cell cultivation, the efficiency of transgenic animal production is unreliable. Therefore, we developed an in vivo selection model to enhance the efficiency of transgenic chicken production using microsomal glutathione-S-transferase II (MGSTII)-overexpressing PGCs that are resistant to the alkylating agent busulfan, which induces germ cell-specific cytotoxicity. Under in vitro conditions, MGSTII-tg PGCs were resistant to 1 μM busulfan, which was highly toxic to wild-type PGCs. In germline chimeric roosters, transgene-expressing germ cells were dominantly colonized in the recipient testes after busulfan exposure compared with non-treated germline chimera. In validation of germline transmission, donor PGC-derived progeny production efficiency was 94.68%, and the transgene production rate of heterozygous transgenic chickens was significantly increased in chickens that received 40 mg/kg busulfan (80.33–95.23%) compared with that of non-treated germline chimeras (51.18%). This system is expected to significantly improve the efficiency of generating transgenic chickens and other animal species by increasing the distribution of donor cells in adult testes.
Similar content being viewed by others


Introduction
Transgenic animals are a powerful tool that allow functional investigations of specific genes, production of recombinant proteins, tissue implantation models in biomedical research, and comparative studies between animal models and humans. To generate transgenic models that permanently express a transgene, two major methods have been employed. Firstly, microinjection of exogenous DNA into the nuclei of embryos is used to produce genetically modified animals, including mice, rabbits, sheep, and pigs1,2,3,4. Recently, these techniques have been combined with genome editing systems, including zinc finger nucleases (ZFN), transcription activator-like effector nuclease (TALEN), and the clustered regulatory interspaced short palindromic repeats (CRISPR) system to produce model animals of various genotypes, including tissue-specific gene knockout and transgene expression5,6,7,8. Furthermore, many studies using the CRISPR/Cas9 system have demonstrated that production of genetically modified animals is quite high using this system8,9,10. On the other hand, injection of genetically modified pluripotent stem cells, including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), into the cavity of blastocysts has also been utilized. This gene transfer system holds advantages over in vitro selection strategies, as it allows development of homogenous transgene‐expressing cell lines and precise gene targeting for efficient production of transgenic animal models11,12. Despite its advantages, the production rate of transgenic animals from chimera developed using genetically modified stem cells are not consistent or guaranteed, even after drug screening13,14.
Unlike mammals, in avian systems such as the chicken, animals can be produced using primordial germ cells (PGCs), the progenitor cells of both sperm and ovum, and PGCs can be genetically modified for production of transgenic progenies15. Recent progress in long-term culture of chicken PGCs and genome modification techniques have allowed the development of numerous transgenic and genome-edited chickens16,17,18,19,20,21,22,23. At this point, the chicken is considered to be a reliable biological model representing avian species. As shown in prior studies, the chicken PGC-mediated transgenic production system can also increase production efficiency through drug selection during in vitro cultivation. Nevertheless, the production of PGC-derived genetically modified chickens is not consistent, and production efficiency is not guaranteed, similar to the stem cell-mediated system used for mammalian species. Some prior reports demonstrated that the efficiency of producing transgenic progeny derived from donor cells was consistently higher than 50%17,24. Theoretically, the efficiency of heterozygotic transgenic production is 50% at maximum because the transgene of donor PGCs is integrated into haploid germ cells. However, in most cases, germline transmission efficiency and transgenic progeny production rates are varied and low18,19,20,22,23.
The efficiencies of transgenic animal production using the mouse stem cell-mediated system and chicken PGC-mediated system are limited, as it is only possible to increase homogenous transgene‐expressing cell lines at the upstream stage of donor cell cultivation. These systems are inherently limited, as the proportion of transgene-expressing donor cells cannot be regulated, and transgene insertion is irreversible after transfer to recipient embryos. To overcome this limitation, several means to sterilize the recipients’ endogenous germ cells to increase the proportion of donor cells have been used in chickens.
Sterilization of endogenous germ cells is often used for germline chimera production. Sterilization techniques include gamma ray irradiation, X-ray irradiation, and chemical methods to eliminate endogenous germ cells25,26,27,28,29. In particular, the alkylating agent busulfan induces relatively high germ cell-specific cytotoxicity, including both PGCs and spermatogonial cells in adult testes30,31. Busulfan is also considered to be a very effective antispermatogonial agent in avian testes32,33, and allows for highly efficient germline chimera production when used to treat unhatched chicken embryos27,34. However, despite the finding that busulfan causes testicular germ cell apoptosis, a small number of spermatogonial stem cells (SSCs) survive and restore the germ cell population35. This suggests that normal spermatogenesis can be maintained after endogenous germ cells are eliminated by busulfan.
In a previous report, Harkey et al. demonstrated that overexpression of glutathione-S-transferase (GST) genes (GSTA1, GSTP1, and MGSTII) in HEK cells for hematopoietic gene therapy is significantly increased using the busulfan resistance system36. Specifically, the MGSTII gene conferred a reproducible twofold selective advantage under busulfan exposure conditions, suggesting that this approach could be used for hematopoietic gene therapy36. The GST enzymes catalyze the conjugation of a variety of small molecules, including alkylating agents such as busulfan, with glutathione (GSH), targeting these molecules for export36,37. This property enables detoxification of alkyl agents in GST-expressing cells. In the present study, we developed an in vivo selection system for busulfan-resistant transgene (MGSTII)-containing chicken germ cells in adult testes as well as in vitro selection. The selection of transgenic germ cells in the adult stage of germline chimera is considered to be highly advantageous for the reproduction of donor-derived progeny. To test the reliability of this system, we produced a germline chimera containing a busulfan resistance gene, and subsequently assessed the efficiency of transgenic progeny production using busulfan treatment.
Results
Establishment of a MGSTII expressing chicken PGC line with in vitro resistance to busulfan.
To establish MGSTII-expressing chicken PGCs, we first constructed a piggyBac transposon vector designed for CMV-driven expression of enhanced green fluorescent protein (EGFP) and chickenized human MGSTII. Subsequently, cultured PGCs were transfected with piggyBac TK NeoR CMV GFP CMV MGSTII and transposase (CAGG-PBase) plasmids (Fig. 1A). Transfected PGCs (MGSTII-tg PGCs) were selected with G418 (300 μg/mL) culture medium for 1 month (Fig. 1B). MGSTII-tg PGCs exhibited robust GFP fluorescence and specifically expressed the MGSTII gene compared with WT PGCs (Fig. 1C). To analyze busulfan resistance in MGSTII-tg PGCs, we analyzed the time- and dose-dependent effects of busulfan treatment on cell viability. The effect of busulfan on WT and MGSTII-tg PGC proliferation was examined using a WST-1 assay after 48 h exposure to varying concentrations of busulfan. In cells treated with 1 μM, 2 μM, and 4 μM busulfan, the proliferation rate of MGSTII-tg PGCs was significantly higher than that of WT PGCs (Fig. 1D). In cultured PGCs, 1 μM and 2 μM busulfan exposure decreased cell numbers in WT PGCs relative to MGSII-tg PGCs, but 8 μM busulfan decreased the number of PGCs in both genotypes (Fig. 1E). These results suggested that MGSTII-tg PGCs had increased resistance to busulfan, and therefore that MGSTII overexpression conferred resistance to busulfan.

Establishment of the piggyBac CMV EGFP CMV MGSTII PGC cell line and validation of busulfan effect on MGSTII-expressing PGCs. (A) Schematic illustration of the piggyBac TK NeoR CMV EGFP CMV MGSTII vector with transposase and introduction into PGCs. Transgenic PGCs were selected with G418 for 30 days. (B) Establishment of the piggyBac TK NeoR CMV EGFP CMV MGSTII transgenic PGC cell line (MGSTII-tg PGCs) after selection. Scale bars, 200 μm. (C) Genomic DNA PCR analysis of MGSTII-tg PGCs using MGSTII- and GAPDH-specific primers. Wild-type (WT) PGCs treated with distilled water (−) were used as control. (D–E) Dose-dependent effect of busulfan on MGSTII-tg and WT PGCs. (D) WST1 assay of PGCs after 48 h of dose-dependent busulfan treatment (mean ± SD; n = 3). * P < 0.05, **P < 0.001. (E) Morphology of MGSTII-tg and WT PGCs in the presence of multiple busulfan dosages (0, 1, 2, and 8 μM). Vehicle control was treated with DMSO only. Scale bars, 50 μm.
Effect of busulfan on migrating PGCs
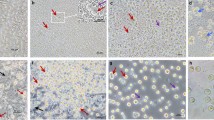
In subsequent studies, we examined the effect of busulfan on migrating PGCs in the blood stream and to the genital ridge. Avian PGCs circulate through embryonic blood vessels during HH 13–16 and migrate to the gonads by HH 28. Thus, to assess the effect of busulfan on embryonic PGCs, we injected 1 μM and 2 μM of busulfan into embryonic blood vessels at stage HH 13–16. At embryonic day 5.5 (HH 28), whole gonads were collected from control and busulfan-treated embryos, and DAZL immunostaining was conducted to identify germ cell distribution. In control gonads (animals injected with DMSO vehicle control), DAZL+ PGCs were dispersed in the entire gonad, but DAZL+ PGCs were completely eliminated in gonads of the 1 μM and 2 μM busulfan-treated groups (Fig. 2A).

Effect of busulfan in embryonic gonads after MGSTII-tg PGC transplantation. (A) In vivo effect of chicken PGCs under busulfan treatment. Endogenous PGCs were detected by DAZL immunostaining in HH28 embryonic gonads with or without busulfan treatment. Scale bars, 100 μm. (B) Migration assay of MGSTII-tg and GFP-tg PGCs treated with 1 μM busulfan. Approximately 1,000 GFP-tg or MGSTII-tg PGCs were injected into the dorsal aortas of chicken embryos at HH 13–16. Fluorescent cells were observed and counted in recipient embryonic gonads at HH 28. Scale bars, 100 μm. (C) Number of PGCs migrated to the gonads of HH 28 embryos injected (i.v.) with 1,000 PGCs at HH 13–16 (mean ± SD; n = 3 for each group). *P < 0.05, **P < 0.01, ***P < 0.001.
To assess busulfan resistance in embryonic stages, we further conducted an in vivo migration assay using MGSTII-tg PGCs exposed to busulfan. Approximately 1,000 GFP-tg PGCs17 or MGSTII-tg PGCs were injected into the bloodstream of recipient HH 13–16 embryos with or without 1 μM busulfan. In untreated PGCs, the number of PGCs was similar in whole embryonic gonads at HH 28 in recipient embryos injected with GFP-tg PGCs and embryos injected with MGSTII-tg PGCs (Fig. 2B). However, in the busulfan-treated embryos, the number of migrated PGCs was dramatically decreased in embryos injected with GFP-tg PGCs relative to embryos injected with MGSTII-tg PGCs (Fig. 2B). The mean number of migrated gonadal PGCs in the left gonads of embryos injected with 1 μM busulfan-treated MGSTII -tg PGCs (174 ± 24.45) was significantly higher than not only in embryos injected with 1 μM busulfan-treated GFP-tg PGCs (9 ± 7.54), but also in embryos injected with untreated GFP-tg PGCs (88 ± 20.59) and untreated MGSTII-tg PGCs (112.67 ± 32.47). Similarly, the mean number of migrated cells in the right gonads of embryos injected with busulfan-treated MGSTII-tg PGCs (161 ± 33.08) was also significantly higher than in other groups (82 ± 24.06 in untreated GFP-tg PGCs, 58 ± 24.02 in untreated MGSTII-tg PGCs, and 4.3 ± 3.21 in busulfan-treated GFP-tg PGCs) (Fig. 2C). These results suggested that MGSTII-tg PGCs were resistant to busulfan in vivo, as demonstrated by in vivo migration activity. We also examined the survivability and hatchability of recipients after transfer of PGCs with busulfan treatment into embryonic blood vessels (at HH 13–16) (Supplementary Table 1). The survivability (at E6) and hatchability of recipients were 50% lower in the busulfan only-treated group that in the untreated control group. The survivability (at E6) and hatchability of recipients were not lower in the busulfan-treated/untreated groups injected with transgenic PGCs (GFP-tg PGCs and MGSTII-tg PGCs) than in the busulfan only-treated group (Supplementary Table 1). These results suggest that busulfan does not directly affect the development or hatching of recipient embryos.
Production of MGSTII transgenic chickens and detection of genomic integration site
To produce hMGSTII-expressing transgenic chickens, cultured MGSTII-tg PGCs were injected into HH 13–16 Korean Ogye (KO) chicken embryos. A total of 39 embryos were injected with MGSTII-tg PGCs and 21 hatched (hatchability was 53.84%) (Supplementary Table 2). The donor cells (MGSTII-tg PGCs) were male PGCs from White Leghorn (WL) chickens; therefore, only male KO recipients were selected as putative germline chimeras by genomic DNA sexing PCR (data not shown). Eight progenies were determined to be male (0396, 0398, 0399, 0411, 0412, 0415, 0416, and 0418). After development for about 5 months, five founder males (0398, 0399, 0411, 0412, and 0416) reached sexual maturity. Among these, two germline chimeric chickens (0398 and 0412: hereafter, a “M” is placed in front of the animal number to designate founder germline chimeras e.g., “M0398” and “M0412”) were randomly selected and testcrossed with wild-type WL hens (Supplementary Table 2). The efficiencies of donor MGSTII-tg PGC-derived chicks from two germline chimeras (M0398 and M0412) were 79.74% and 75.61%, respectively (Table 1). Among the donor-derived progeny, transgenic chicks were validated through GFP expression, which was absent in non-transgenic chicks (Fig. 3A). Analyses of GFP expression and transgene genomic PCR were performed, identifying that approximately half of donor germ cell-derived chicks were transgenic (33 of 63 chicks, 52.38% from M0398, and 14 of 31 chicks, 45.16% from M0412) (Fig. 3B, Table 1). In G1 transgenic chickens, GFP expression was measured in one transgenic male (TG0276) and one transgenic female (TG0235), and robust GFP expression was detected until sexual maturation (Fig. 3C). In subsequent analyses, the genomic transgene integration sites were identified by genome walking analysis. The piggyBac TK NeoR CMV GFP CMV MGSTII transgene was integrated into the intergenic region of chromosome 14 in TG0276 and into the intergenic region of chromosome 4 in TG0285 (Fig. 3D). Both loci had conserved TTAA sequences, in which the piggyBac transposon could be inserted by transposase.

GFP expression in of MGSTII-tg chicks and identification of transgene insertion sites. (A) Donor MGSTII-tg PGCs-derived chicks. GFP-expressing chicks were distinguishable from non-transgenic hybrids using a fluorescent excitation lamp fitted with the appropriate filters. (B) Screening of MGSTII-tg chicks via genomic DNA PCR analysis. Genomic DNA PCR analysis of transgenic G1 chicks using MGSTII- and GAPDH-specific primers. MGSTII-tg PGCs and non-tg genomic DNA samples were used as positive and negative controls, respectively. (C) Detection of GFP expression in adult TG chickens (20 weeks) in both male (TG 0276) and female (TG 0285) animals using a fluorescent excitation lamp fitted with the appropriate filters. (D) Identification of the transgene integration site in transgenic chickens. The transgene was integrated into the intergenic region of chromosome 14 in TG 0276, and into the intergenic region of chromosome 4 in TG 0285. Black color nucleotides indicate genomic DNA sequences, yellow color nucleotides indicate transposon recognition sequences (TTAA), and red color nucleotides indicate the 5′ of piggyBac vector sequences.
Enhanced transgenic chicken production rate after busulfan treatment
After evaluating the donor-derived offspring and transgenic chicken production rates in untreated animals, the same germline chimeras (M0398 and M0412) and two additional germline chimeras (M0399 and M0411) received a single intraperitoneal injection of 40 mg/kg busulfan. Busulfan-treated germline chimeras were designated by placing a “B” in front of the animal number, for example B0398, B0399, B0411, and B0412. To confirm the distribution of GFP+ germ cells in testes, 10 µm paraffin sections from germline chimera WT and busulfan non-treated control testes were examined under a fluorescent microscope after immunostaining for GFP as a marker for transplanted transgenic germ cells (Fig. 4A–C). In WT testes, GFP+ cells were not observed (Fig. 4A), but in testes of germline chimera untreated with busulfan (M0416), GFP+ germ cells were sparsely dispersed adjacent to the basements of seminiferous tubules (Fig. 4B). Surprisingly, the distribution of GFP+ germ cells in testes of busulfan-treated germline chimera was significantly increased compared with non-treated controls and WT testes (Fig. 4C). To determine the increase in transgenic offspring production efficiency, busulfan-treated germline chimeras were testcrossed by insemination of female WL chickens. Unexpectedly, all busulfan-treated germline chimeras exhibited a decreased hatch rate (35.59–45.31%) compared with the non-treated control group (70.69–94.05%) even in the same germline chimeras used in the busulfan non-treated group (B0398, B0412) (Tables 1, 2, Fig. 4D). Despite the higher efficiency of donor-derived offspring production in the busulfan-treated group relative to control (94.68% vs. 78.33%, respectively), this relationship was not statistically significant (Tables 1, 2, Fig. 4E). However, the production rate of transgenic chickens was significantly enhanced in the busulfan-treated group (80.95%) relative to the non-treated group (45.16%) (Tables 1, 2, Fig. 4F). In particular, transgenic chicken production efficiency was significantly improved by busulfan treatment even in the same germline chimeric roosters. The transgenic chicken production efficiency was significantly improved by busulfan treatment, from 52.38 to 95.23% in M0398 and 45.16% to 80.95% in M0412.

Improved transgenic chicken production rate after busulfan injection. (A–C) Cross-sections of testes from adult germline chimeras, and detection of GFP-expressing donor germ cells in adult germline chimeric testes with or without busulfan treatment. (A) WT rooster testes were used as a negative control. GFP expression was not observed. (B) Testes of germline chimeric roosters without busulfan treatment. GFP+ cells were sparsely distributed adjacent to the basement membranes of seminiferous tubules. (C) Testes from germ chimeric roosters 2 weeks after busulfan treatment. GFP+ cells were present throughout the seminiferous tubules. DAPI was used to stain nuclei in each panel. Scale bars, 200 μm (upper panel) and 50 μm (lower and smaller panel). (D–F) Analysis of progeny production rates of germline chimera for 8 weeks. (D) Hatching rate of G1 offspring from germline chimeras with or without busulfan treatment. (E) Efficiencies of donor-derived offspring production with or without busulfan treatment. (F) Efficiencies of MGSTII-tg offspring (G1) production with or without busulfan treatment. Data are presented as means ± SD from each independent group (progeny production of each week) for 8 weeks. *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
The production of transgenic cells and organisms provides a platform to assess gene functions, improve human health, enhance production of useful materials, protect against environmental threats, and control disease in livestock1. In this regard, the conventional means for transgenic animal production, involving introduction of foreign genomic materials into fertile eggs, and newer genome editing technologies such as CRISPR/Cas9 have been successfully utilized. In mammalian systems, genome editing systems are highly efficient, with the desired genotype reaching almost 100%8,9,10. If only small gene deletions are required, further technology development might not be necessary.
However, production of transgenic animals through transgene overexpression and targeted gene insertion in germline-competent stem cells is still required in some contexts such as for gene homologous recombination and precise targeted gene insertion38. Thus far, cell-mediated systems do not efficiently and reliably produce transgenic animals. At the initial stage, the frequency of stable integration into the genomes of host cells is very low. Therefore, transgene cassettes that include resistance genes to dominant selection markers have been used to increase efficiency; for example, neomycin phosphotransferase II against the neomycin derivative G418, puromycin N-acetyltransferase against puromycin, and hygromycin B phosphotransferase against hygromycin B39. This strategy can induce homogenization of transgene-integrated cells and supply the appropriate cell population for transgenesis. Despite drug selection for transgenic cell lines, the efficiency of desired transgenic littermate production is not enhanced, as the process is irreversible after transfer to recipient embryos. To overcome this limitation, a method to increase the proportion of donor cells in vivo is required.
A recent study demonstrated that transgenic chickens expressing a fluorescent protein (mCherry or GFP) and avian leukosis virus (ALV)-resistant genome-edited chickens are effectively produced by transplanting PGCs into sterilized recipient testes40,41. Despite the relatively high production efficiency (more than 60% in some cases), this method requires intramagnal insemination, which may limit mass production of transgenic/genome-edited progenies due to technical difficulties. Another research group reported a germ cell sterilized model through targeted DDX4 gene disruption20, which enables production of donor-derived offspring with high efficiency using DDX4-knockout embryos as recipients42. More recently, another germ cell sterilized recipient model was generated by conditional knockout of the DAZL gene43. These methods can effectively increase the proportion of donor cells in the recipient and therefore be used to effectively conserve rare breeds and produce transgenic animals. However, germ cell sterilized model systems must be established separately to produce the proposed animal, which limits their wide application.
In the present study, we demonstrated that transgenic chicken production efficiency was increased in vivo by use of a busulfan resistance cassette, as busulfan is specifically toxic to endogenous germline cells in chimeric animals. Busulfan, an alkylating agent with antispermatogonial activity, is highly cytotoxic to germ cells specifically, including PGCs and SSCs in mouse, rat, and avian testes30,31,33. Busulfan treatment of chicken embryos also increased germline chimera production27,34. Busulfan induces germ cell damage and germ cell apoptosis mediated by loss of the c-kit/SCF pathway, a mechanism that is conserved in multiple germ line cell types, including PGCs and spermatogonia44. It was therefore expected that high-efficiency transgenic animal production would be possible simply by developing busulfan-resistant germ cells expressing the transgene of interest. Previous studies36 suggested that overexpression of human GST, MGSTII, confers resistance to busulfan in HEK293 cells. In this regard, we overexpressed hMGSTII in chicken PGCs using the piggyBac transposon, and confirmed significant resistance to 1 μM, 2 μM, and 4 μM busulfan relative to WT PGCs. Interestingly, busulfan is cytotoxic not only to PGCs in early embryos29,34, but also to circulating PGCs in blood vessels after exogenous PGC transplantation. Moreover, the number of PGCs migrated to the recipients’ gonads (both left and right) was significantly increased by treatment of MGSTII-tg PGCs with busulfan prior to transplantation, and was increased relative to both vehicle- and busulfan-treated GFP-tg PGCs. We also evaluated the harmful effect of busulfan treatment on developing embryos under several conditions. Although survivability was lower in all experimental groups than in the untreated control group, the hatching rate did not significantly differ between the busulfan-treated (busulfan only and transgenic PGCs with busulfan) and untreated (transgenic PGCs without busulfan) groups. Therefore, egg shell windowing and cell/busulfan injection are considered to cause stress, not busulfan itself. These results indicate that use of the MGSTII transgene to confer busulfan resistance improved the migration efficiency of transplanted PGCs, as busulfan decreases endogenous PGCs without eliciting other side effects on embryo development and hatching.
In our previous reports, germline transmission efficiency was 90.4–98.9%, and over half of the transgenic chicks (52.2%) were heterozygous17. In another study, the 2 × ERE-OVcEGF transgene was integrated into chicken PGCs using the piggyBac transposon, and the average rate of germline transmission was 92.2%, with 47.1% of transgenic animals being heterozygous24. However, these high efficiencies are not always consistent in chicken germline chimeras. In two types of chickens produced using a similar system, the efficiency of germline transmission and transgenesis was highly variable and inefficient19, and only one transgenic chick were obtained from 518 progeny chicks (0.193%)16. Further, this variance in germline transmission efficiency is exacerbated when combined with targeted genome editing technologies. In targeted knockout of chicken ovalbumin in chicken PGCs using TALEN, the germline transmission efficiency was 40.84%, and only 8.04% of progeny had disruption of genomic ovalbumin18. In a study in which the GFP gene was knocked in to the chicken sex chromosome (Z chromosome) using TALEN, 17 targeted genome-edited chicks were derived from 372 hatched chicks20. Use of the CRISPR/Cas9 system on chicken PGCs resulted in germline transmission efficiency of 3.1%, and 62.5% of donor germ cell-derived progenies were genome modified22. These phenomena are due in part to multicopy transgene integration in transgenic models, and non-homogenous selection of genome-edited germ cells during the drug screening process.
In the present study, the germline transmission efficiencies were 79.74% and 75.61% in two different germline chimera (M 0398 and M 0412), and almost half of the progeny exhibited GFP fluorescence and transgene integration (52.38% and 45.16%, respectively). After intraperitoneal injection of a single dose of busulfan emulsion (40 mg/kg) as in previous reports using chicken and quail33,45, germline transmission efficiency was increased to 94.68% on average. Further, the production efficiency of transgenic progeny after busulfan treatment improved to 80.95%. Among the chimeras tested, in the 0398 and 0412 germline chimera used in both the busulfan-treated groups and non-treated groups, the transgenic progeny production efficiency was 95.23% and 80.95%, respectively. However, the fertility of germline chimeras after treatment with busulfan was significantly decreased. In control germline chimeras, the fertility of germline chimeras after busulfan treatment was only 38.59%, which was significantly lower than that of chimeras without busulfan treatment (70.68%). One possible explanation is that SSCs have relatively high resistance to busulfan44, and from a haploid round spermatid stage are extremely sensitive to alkylating agents such as cyclophosphamide46. Therefore, diploid cells, including spermatogonia and spermatocytes, could be resistant to busulfan-induced cytotoxicity, while the transgene-free haploid germ cells such as spermatocytes, spermatids, and sperm of donor cells and endogenous germ cells were depleted. In those stages, even haploid germ cells from donor-derived cells are highly susceptible to busulfan. As a result, the entire population of germ cells decreases, and haploid germ cells containing the MGSTII transgene are relatively increased, such that fertilization rate is decreased, but production efficiency of transgenic progeny is increased.
In transgenic animal production, the use of genetically modified stem cells with germline competency, including pluripotent stem cells and germ cells such as PGCs and SSCs, is required not only for transgene overexpression in target cells, but also for targeted gene insertion and homologous recombination. Thus far, strategies to increase production efficiency of transgenic animals have been limited to drug selection during homogenous cell cultivation in vitro. The in vivo selection system developed in the present study using a busulfan resistance gene combined with busulfan treatment demonstrated that donor-derived transgenic production efficiency can be increased even in adult recipients. To confirm the enhancement of in vitro and in vivo donor cell proportions demonstrated in this study, additional experiments using several gene cassettes, including MGSTII, and further validation in other model animals are needed. This strategy can be applied in multiple species to improve the production efficiency of transgenic and genome-edited animals.
Materials and methods
Experimental animals and animal care
The care and experimental use of chickens was approved by the Institute of Laboratory Animal Resources, Seoul National University. Chickens were maintained according to a standard management program at the University Animal Farm, Seoul National University, Korea. All procedures, including chicken maintenance, reproduction, and sample collection, were governed by standard operating protocols according to a standard management program at the University Animal Farm, Seoul National University and the Animal Genetic Engineering Laboratory at Seoul National University.
Construction of human MGSTII expression vector
Codons of the human microsomal glutathione-S-transferase II (MGSTII) gene were optimised for expression in the hen using the Gallus gallus codon database (https://www.kazusa.or.jp/codon). A codon-optimized human MGSTII gene were integrated into piggyBac TK NeoR CMV GFP FRT backbone vector from our previous study19. Briefly, codon-optimized MGSTII CDS inserted into backbone vector by using HindIII, NotI restriction enzymes. After that, CMV MGSTII cassette was cloned by PCR including XhoI restriction site at the 5′ end and 3′ end. This cloned cassette was integrated into piggyBac TK NeoR CMV GFP FRT backbone by using XhoI restriction enzyme and produce final vector piggyBac TK NeoR CMV GFP CMV MGSTII.
Transfection and G418-selection of PGCs
The methods of cultivation, transfection and G418-selection of WL male PGC cells were followed by our previous report17. Briefly, male PGCs were cultured on mitotically inactivated mouse fibroblast cells (MEFs) in knockout Dulbecco's Modified Eagle's Medium (KO-DMEM) (Invitrogen, Life Technologies, Carlsbad, CA, USA) supplemented with 20% (v/v) fetal bovine serum (Invitrogen, Life Technologies), 2% (v/v) chicken serum (Sigma-Aldrich, St. Louis, MO, USA), 1 × nucleoside mix (EMD Millipore, Temecula, CA, USA), 2 mM L-glutamine, 1 × nonessential amino acid mix, β-mercaptoethanol, 10 mM sodium pyruvate, 1 × antibiotic antimycotic mix (Invitrogen, Life Technologies), and human basic fibroblast growth factor (10 ng/mL; Koma Biotech, Seoul, Korea). PGCs were cultured at 37℃ in an atmosphere of 5% (v/v) CO2 and 60–70% relative humidity. The piggyBac TK NeoR CMV GFP CMV MGSTII expression vector and transposase (CAGG-PBase, pCyL43) were cotransfected into the PGC line using Lipofectamine 2000 (Invitrogen, Life Technologies). One day after transfection, G418 (300 mg/mL) was added to culture medium to enable selection of transfected PGCs for 1 month.
In vitro assay to assess the effect of busulfan resistance on MGSTII-expressing PGCs
WT and MGSTII-tg PGCs were incubated at 50,000 cells per well in 96-well tissue culture plates with varying concentrations of busulfan (1, 2, 4, 6, and 8 μM, Sigma-Aldrich) dissolved in dimethyl sulfoxide (DMSO). To assess the busulfan resistance of MGSTII-tg PGCs, cells were incubated in the specified concentrations of busulfan for 48 h. After incubation, cell viability was assessed using the WST-1 method (Roche Diagnostic, Basel, Switzerland). Briefly, WST-1 was added (15 μL/well) and cells were incubated for 4 h. The absorbance (A450-A650) of formazan dye produced by metabolically active cells was detected using an Epoch microplate reader (BioTek, Winooski, VT, USA).
Transplantation of MGSTII-tg PGCs into recipient embryos
To produce germline chimeric chickens through injection of MGSTII-tg PGCs into recipient embryos, we made a small hole at the pointed end of each recipient KO egg at Hamburger and Hamilton (HH) stages HH13–16, and microinjected a 2 μL aliquot containing at least 3,000 MGSTII-tg PGCs into the dorsal aortas of recipient embryos. Egg holes were sealed with paraffin film, and eggs were incubated with the pointed end down prior to further screening and eventual hatching. To assess the effect of busulfan on MGSTII-tg PGCs, approximately 1,000 GFP-tg PGCs, as generated in our previous report17, or MGSTII-tg PGCs were injected with 1 μM busulfan solution at HH13–16. Three days after transplantation, the gonads from a sample of the recipient embryos were collected at HH 28, and the presence of GFP-expressing PGCs in the gonads were imaged by fluorescence microscopy (Nikon) and manually counted.
Test-cross analysis and validation of transgenic chicken production efficiency
After sexual maturation, putative germline chimeras were testcrossed by mating with WL female chickens to generate transgenic chickens derived from transplanted donor PGCs. Transgenic (TG) chickens were identified using a fluorescent excitation lamp with detection filters (BLS Ltd., Budapest, Hungary), and were confirmed by genomic DNA PCR. To evaluate the donor-derived production efficiency and transgenic chick production efficiency, adult germline chimeras were injected with 40 mg/kg busulfan. Briefly, 40 mg busulfan dissolved in 1 mL N,N-dimethyl formamide (Merck, Darmstadt, Germany) was injected intraperitoneally into germline chimeras. Two weeks after busulfan injection, busulfan-treated germline chimeras were testcrossed by mating with wildtype WL female chickens. At least 20 progeny animals from both groups of germline chimeras (busulfan-treated or non-treated) were analyzed for fertilization rate and transgenic chick production efficiency by measuring hatching rates, fluorescence expression, and genotypes for 8 weeks.
PCR analysis and identification of the transgene integration site
To detect the presence of the integrated transgene and distinguish between the wild-type (WT) and MGSTII-tg loci, transgene specific primer sets (MGST F: 5′-CCA CCA TGG CAG GCA ACA GC-3′ and MGST R: 5′-CCG CTC AGA ATT GCC GCC TC-3′) were used. Genomic DNA PCR from WT and MGSTII-tg was also performed using GAPDH primers (F: 5′-GGT GGT GCT AAG CGT GTT AT-3′; R: 5′-ACC TCT GCC ATC TCT CCA CA-3′) as a control. Each PCR was performed in a total volume of 20 μL containing 100 ng genomic DNA from WT and MGSTII-tg, 10 × PCR buffer (BioFACT, Daejeon, Korea), 10 mM dNTPs, 5 pmol of each primer, and 0.5 U Taq polymerase (BioFACT) in the following thermocycling conditions: 5 min at 94 °C, followed by 35 cycles of 30 s at 94 °C, 30 s at 60 °C, 30 s at 72 °C, and, finally, 10 min at 72 °C.
The transgene (piggyBac CMV GFP CMV MGSTII TK NeoR) insertion site was identified using the Genome Walker Kit (Takara, Japan) according to the manufacturer’s protocol and our previous report24. Gene-specific primers were designed from the known DNA sequence of the piggyBac transgene to move upstream of the gene in genomic DNA. Genomic DNA was digested with four different restriction enzymes (EcoRV, DraI, PvuII, and SspI) ligated with adaptors, and PCR was performed using the polymerase mix. PCR products were excised from agarose gel, purified using a Power Gel Extraction Kit (Promega, Madison, WI, USA), and subsequently cloned into the pGEM-T Easy Vector (Promega). Cloned PCR products were sequenced using an ABI Prism 3730 XL DNA Analyzer (Applied Biosystems, Foster City, CA, USA). Sequences of the 5′- flanking regions were analyzed using the Basic Local Alignment Search Tool (BLAST) Assembled Genome database (http://blast.ncbi.nlm.nih.gov/BLAST.cgi) and the UCSC Genome Bioinformatics browser (http://www.genome.ucsc.edu) to identify transgene integration sites in the transgenic chickens’ genomes.
Immunohistochemistry
The procedures of testis section and immunostaining were followed by our previous report47. Adult testes of WT chickens and busulfan-treated or untreated germline chimeric chickens were paraffin-embedded and sectioned (thickness, 10 μm). After deparaffinization, sections were washed three times with 1 × phosphate-buffered saline (PBS) and blocked with a blocking buffer (5% goat serum and 1% bovine serum albumin in PBS) for 1 h at room temperature. Sections were then incubated at 4 °C overnight with a rabbit anti-GFP primary antibody (Invitrogen, Carlsbad, CA, USA) (1:200 dilutions in blocking buffer). After washing three times with PBS, sections were incubated with fluorescence-conjugated secondary antibodies (Alexa Fluor 594 or 488, Invitrogen) for 1 h at room temperature. After washing three times with PBS, sections were mounted with Prolong Gold antifade reagent with DAPI and imaged using a confocal fluorescence microscope (Carl Zeiss Inc, Oberkocken, Germany).
Statistical analyses
To analyze dose-dependent effects of busulfan on MGSTII PGCs and WT PGCs, a two-way ANOVA was used to determine statistical significance. Significant differences between busulfan-treated and busulfan non-treated groups were examined using a one-way ANOVA. P < 0.05 was considered indicative of statistical significance. ***p < 0.001, **p < 0.01, *p < 0.05.
Ethics statement
All experimental procedures and care of chickens was approved by the Institute of Laboratory Animal Resources, Seoul National University, and all methods were carried out in accordance with ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines and approved by the Institutional Animal Care and Use Committee (IACUC, SNU‐190,401‐1–1) of Seoul National University, Korea.
Approval for animal experiments
All experimental procedures and care of chickens was approved by the Institute of Laboratory Animal Resources, Seoul National University (SNU‐190,401‐1–1), and all methods were carried out in accordance with guidelines and regulations of the Institutional Animal Care and Use Committee of Seoul National University (IACUC, SNU-200519–2), Korea. All procedures, including chicken maintenance, reproduction, and sample collection, were governed by standard operating protocols according to a standard management program at the University Animal Farm, Seoul National University and the Animal Genetic Engineering Laboratory at Seoul National University.
References
Wheeler, M. B., Walters, E. M. & Clark, S. G. Transgenic animals in biomedicine and agriculture: outlook for the future. Anim. Reprod. Sci. 79, 265–289. https://doi.org/10.1016/s0378-4320(03)00168-4 (2003).
Ittner, L. M. & Gotz, J. Pronuclear injection for the production of transgenic mice. Nat. Protoc. 2, 1206–1215. https://doi.org/10.1038/nprot.2007.145 (2007).
Gordon, J. W. & Ruddle, F. H. Gene transfer into mouse embryos: production of transgenic mice by pronuclear injection. Methods Enzymol. 101, 411–433. https://doi.org/10.1016/0076-6879(83)01031-9 (1983).
Gordon, J. W., Scangos, G. A., Plotkin, D. J., Barbosa, J. A. & Ruddle, F. H. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc. Natl. Acad. Sci. U. S. A. 77, 7380–7384. https://doi.org/10.1073/pnas.77.12.7380 (1980).
Mashimo, T. Gene targeting technologies in rats: zinc finger nucleases, transcription activator-like effector nucleases, and clustered regularly interspaced short palindromic repeats. Dev. Growth Differ. 56, 46–52. https://doi.org/10.1111/dgd.12110 (2014).
Liu, H. et al. TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell 14, 323–328. https://doi.org/10.1016/j.stem.2014.01.018 (2014).
Niu, Y. et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156, 836–843. https://doi.org/10.1016/j.cell.2014.01.027 (2014).
Sung, Y. H. et al. Highly efficient gene knockout in mice and zebrafish with RNA-guided endonucleases. Genome Res. 24, 125–131. https://doi.org/10.1101/gr.163394.113 (2014).
Wang, H. et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918. https://doi.org/10.1016/j.cell.2013.04.025 (2013).
Zhou, J. et al. One-step generation of different immunodeficient mice with multiple gene modifications by CRISPR/Cas9 mediated genome engineering. Int. J. Biochem. Cell Biol. 46, 49–55. https://doi.org/10.1016/j.biocel.2013.10.010 (2014).
Suter, D. M. et al. Rapid generation of stable transgenic embryonic stem cell lines using modular lentivectors. Stem Cells 24, 615–623. https://doi.org/10.1634/stemcells.2005-0226 (2006).
Robertson, E., Bradley, A., Kuehn, M. & Evans, M. Germ-line transmission of genes introduced into cultured pluripotential cells by retroviral vector. Nature 323, 445–448. https://doi.org/10.1038/323445a0 (1986).
Auerbach, W. et al. Establishment and chimera analysis of 129/SvEv- and C57BL/6-derived mouse embryonic stem cell lines. Biotechniques 29, 1024–1032. https://doi.org/10.2144/00295st04 (2000).
Yamamoto, S. et al. Derivation of rat embryonic stem cells and generation of protease-activated receptor-2 knockout rats. Transgenic Res. 21, 743–755. https://doi.org/10.1007/s11248-011-9564-0 (2012).
Han, J. Y. & Park, Y. H. Primordial germ cell-mediated transgenesis and genome editing in birds. J. Anim. Sci. Biotechnol. 9, 19. https://doi.org/10.1186/s40104-018-0234-4 (2018).
Macdonald, J. et al. Efficient genetic modification and germ-line transmission of primordial germ cells using piggyBac and Tol2 transposons. Proc. Natl. Acad. Sci. U. S. A. 109, E1466-1472. https://doi.org/10.1073/pnas.1118715109 (2012).
Park, T. S. & Han, J. Y. piggyBac transposition into primordial germ cells is an efficient tool for transgenesis in chickens. Proc. Natl. Acad. Sci. U. S. A. 109, 9337–9341. https://doi.org/10.1073/pnas.1203823109 (2012).
Park, T. S., Lee, H. J., Kim, K. H., Kim, J. S. & Han, J. Y. Targeted gene knockout in chickens mediated by TALENs. Proc. Natl. Acad. Sci. U. S. A. 111, 12716–12721. https://doi.org/10.1073/pnas.1410555111 (2014).
Lee, H. J. et al. Site-specific recombination in the chicken genome using Flipase recombinase-mediated cassette exchange. FASEB J. 30, 555–563. https://doi.org/10.1096/fj.15-274712 (2016).
Taylor, L. et al. Efficient TALEN-mediated gene targeting of chicken primordial germ cells. Development 144, 928–934. https://doi.org/10.1242/dev.145367 (2017).
Oishi, I., Yoshii, K., Miyahara, D. & Tagami, T. Efficient production of human interferon beta in the white of eggs from ovalbumin gene-targeted hens. Sci. Rep. 8, 10203. https://doi.org/10.1038/s41598-018-28438-2 (2018).
Lee, H. J. et al. Targeted gene insertion into Z chromosome of chicken primordial germ cells for avian sexing model development. FASEB J. 33, 8519–8529. https://doi.org/10.1096/fj.201802671R (2019).
van de Lavoir, M. C. et al. Germline transmission of genetically modified primordial germ cells. Nature 441, 766–769. https://doi.org/10.1038/nature04831 (2006).
Park, T. S. et al. Deposition of bioactive human epidermal growth factor in the egg white of transgenic hens using an oviduct-specific minisynthetic promoter. FASEB J. 29, 2386–2396. https://doi.org/10.1096/fj.14-264739 (2015).
Park, K. J. et al. Gamma-irradiation depletes endogenous germ cells and increases donor cell distribution in chimeric chickens. Vitro Cell Dev. Biol. Anim. 46, 828–833. https://doi.org/10.1007/s11626-010-9361-8 (2010).
Trefil, P. et al. Restoration of spermatogenesis and male fertility by transplantation of dispersed testicular cells in the chicken. Biol. Reprod. 75, 575–581. https://doi.org/10.1095/biolreprod.105.050278 (2006).
Nakamura, Y. et al. Germline replacement by transfer of primordial germ cells into partially sterilized embryos in the chicken. Biol. Reprod. 83, 130–137. https://doi.org/10.1095/biolreprod.110.083923 (2010).
Song, Y., D’Costa, S., Pardue, S. L. & Petitte, J. N. Production of germline chimeric chickens following the administration of a busulfan emulsion. Mol. Reprod. Dev. 70, 438–444. https://doi.org/10.1002/mrd.20218 (2005).
Lee, H. et al. Compensatory proliferation of endogenous chicken primordial germ cells after elimination by busulfan treatment. Stem Cell Res. Ther. 4, 136. https://doi.org/10.1186/scrt347 (2013).
Vasiliausha, S. R. et al. Seminiferous epithelium damage after short period of busulphan treatment in adult rats and vitamin B12 efficacy in the recovery of spermatogonial germ cells. Int. J. Exp. Pathol. 97, 317–328. https://doi.org/10.1111/iep.12195 (2016).
Moisan, A. E., Foster, R. A., Betteridge, K. J. & Hahnel, A. C. Dose-response of RAG2-/-/gammac-/- mice to busulfan in preparation for spermatogonial transplantation. Reproduction 126, 205–216 (2003).
Jones, P. & Jackson, H. Estimation of the duration of spermatogenesis in japanese quail, Coturnix coturnix japonica, using antispermatogonial chemicals. J. Reprod. Fertil. 31, 319–322 (1972).
Tagirov, M. & Golovan, S. The effect of busulfan treatment on endogenous spermatogonial stem cells in immature roosters. Poult. Sci. 91, 1680–1685. https://doi.org/10.3382/ps.2011-02014 (2012).
Nakamura, Y. et al. Increased proportion of donor primordial germ cells in chimeric gonads by sterilisation of recipient embryos using busulfan sustained-release emulsion in chickens. Reprod. Fertil. Dev. 20, 900–907. https://doi.org/10.1071/rd08138 (2008).
Zohni, K., Zhang, X., Tan, S. L., Chan, P. & Nagano, M. C. The efficiency of male fertility restoration is dependent on the recovery kinetics of spermatogonial stem cells after cytotoxic treatment with busulfan in mice. Hum. Reprod. 27, 44–53. https://doi.org/10.1093/humrep/der357 (2012).
Harkey, M. A., Czerwinski, M., Slattery, J. & Kiem, H. P. Overexpression of glutathione-S-transferase, MGSTII, confers resistance to busulfan and melphalan. Cancer Investig. 23, 19–25 (2005).
Hayes, J. D., Flanagan, J. U. & Jowsey, I. R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 45, 51–88. https://doi.org/10.1146/annurev.pharmtox.45.120403.095857 (2005).
Gerlai, R. Gene targeting using homologous recombination in embryonic stem cells: the future for behavior genetics?. Front. Genet. 7, 43. https://doi.org/10.3389/fgene.2016.00043 (2016).
Nakatake, Y. et al. Kinetics of drug selection systems in mouse embryonic stem cells. BMC Biotechnol. 13, 64. https://doi.org/10.1186/1472-6750-13-64 (2013).
Koslova, A. et al. Precise CRISPR/Cas9 editing of the NHE1 gene renders chickens resistant to the J subgroup of avian leukosis virus. Proc. Natl. Acad. Sci. U. S. A. 117, 2108–2112. https://doi.org/10.1073/pnas.1913827117 (2020).
Trefil, P. et al. Male fertility restored by transplanting primordial germ cells into testes: a new way towards efficient transgenesis in chicken. Sci. Rep. 7, 14246. https://doi.org/10.1038/s41598-017-14475-w (2017).
Woodcock, M. E. et al. Reviving rare chicken breeds using genetically engineered sterility in surrogate host birds. Proc. Natl. Acad. Sci. U. S. A. 116, 20930–20937. https://doi.org/10.1073/pnas.1906316116 (2019).
Ballantyne, M. et al. Single generation allele introgression into pure chicken breeds using Sire Dam Surrogate (SDS) mating. bioRxiv https://doi.org/10.1101/2020.09.03.281204 (2020).
Choi, Y. J. et al. Murine male germ cell apoptosis induced by busulfan treatment correlates with loss of c-kit-expression in a Fas/FasL- and p53-independent manner. FEBS Lett. 575, 41–51. https://doi.org/10.1016/j.febslet.2004.08.034 (2004).
Kim, Y. M. et al. Production of germline chimeric quails following spermatogonial cell transplantation in busulfan-treated testis. Asian J. Androl. 20, 414–416. https://doi.org/10.4103/aja.aja_79_17 (2018).
Trasler, J. M., Hales, B. F. & Robaire, B. Paternal cyclophosphamide treatment of rats causes fetal loss and malformations without affecting male fertility. Nature 316, 144–146. https://doi.org/10.1038/316144a0 (1985).
Jung, K. M. et al. Identification and characterization of primordial germ cells in a vocal learning Neoaves species, the zebra finch. FASEB J. 33, 13825–13836. https://doi.org/10.1096/fj.201900760RR (2019).
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. NRF-2019R1I1A1A01061770) and by the Korea government (MSIT) (No. NRF-2015R1A3A2033826) and by the BK21 FOUR Program of the Department of Agricultural Biotechnology, Seoul National University, Seoul, Korea.
Author information
Authors and Affiliations
Contributions
J.Y.H. and Y.M.K. designed the research. Y.M.K., K.J.P., J.S.P. and K.M.J. carried out and analyzed the experiments. Y.M.K. and K.J.P. wrote the manuscript and interpreted and critically reviewed the data. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, Y.M., Park, K.J., Park, J.S. et al. In vivo enrichment of busulfan-resistant germ cells for efficient production of transgenic avian models. Sci Rep 11, 9127 (2021). https://doi.org/10.1038/s41598-021-88706-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-88706-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
