
Abstract
This study compared the pharmacokinetic and glucodynamic profiles of biosimilar SAR341402 insulin aspart to Japan-approved insulin aspart (NovoRapid) in healthy Japanese males. In this single-center, randomized, double-blind, single-dose, two-period, crossover study, subjects received 0.3 U/kg of SAR341402 or NovoRapid before undergoing a 10 h euglycemic clamp procedure. Plasma insulin aspart concentrations and blood glucose levels were measured, and glucose infusion rates (GIRs) were assessed. Primary endpoints were maximum plasma insulin aspart concentration (INS-Cmax), area under the plasma insulin concentration–time curve to the last quantifiable concentration (INS-AUClast), area under the GIR–time curve during the clamp (GIR-AUC0–10 h), and maximum GIR (GIRmax). Forty subjects were randomized with 39 completing both treatment periods. Pharmacokinetic exposure showed a mean ratio between products of 1.00 (90% confidence interval [CI] 0.94–1.05) for INS-Cmax and 1.02 (90% CI 1.00–1.04) for INS-AUClast. Glucodynamic activity showed a mean ratio between products of 1.00 (95% CI 0.93–1.06) for GIR-AUC0–10 h and 1.01 (95% CI 0.95–1.08) for GIRmax. The 90% CIs for pairwise treatment ratios were within the predefined equivalence range of 0.80–1.25. Both treatments were well tolerated. We concluded that similar pharmacokinetic exposure and glucodynamic potency were shown for SAR341402 and NovoRapid in healthy Japanese males.
Similar content being viewed by others

Introduction
SAR341402 (SAR-Asp; Sanofi, Paris, France) is a rapid-acting insulin aspart product1, developed in accordance with relevant United States (US), European (EU), and Japanese guidelines for development of biosimilar insulin products2,3,4,5,6. Insulin aspart is the active ingredient of an insulin analog product available in Japan and EU as NovoRapid, and as NovoLog in the US. SAR-Asp has an amino acid sequence and structure identical to NovoLog/NovoRapid (NN-Asp; Novo Nordisk, Bagsværd, Denmark) and is formulated at a concentration of 100 U/mL. NN-Asp has been approved for use in adults, adolescents, and children with diabetes in many countries, including Japan, for ~ 20 years7,8.
Similar pharmacokinetic exposure and pharmacodynamic activity were shown for SAR-Asp versus both US-reference and EU-reference NN-Asp in a euglycemic clamp study in subjects with type 1 diabetes (T1D)9. Subsequently, similar efficacy, safety and immunogenicity of SAR-Asp and NN-Asp were reported in a multinational, randomized phase 3 study among patients with T1D and type 2 diabetes (T2D) using insulin glargine 100 U/mL (Lantus) as the basal insulin10,11. SAR-Asp and NN-Asp were also well-tolerated in insulin pump users with T1D treated for 4-weeks12.
For approval of any biosimilar insulin in Japan, local guidelines6 state that it is necessary to show comparability in pharmacokinetic exposure and pharmacodynamic activity versus the locally available reference product. The present study was designed to show similar pharmacokinetic and glucodynamic profiles for SAR-Asp versus Japan-reference NovoRapid (hereafter called NN-Asp-Jp) in healthy Japanese subjects.
Subject and method
Subjects
Subjects were enrolled at Hakata Clinic and included Japanese males aged 20–55 years (both inclusive) with a body mass index of 18.0–28.0 kg/m2, certified as healthy by a comprehensive clinical assessment that included his past medical history, normal vital signs, normal 12-lead electrocardiogram (ECG) parameters and laboratory parameters within the normal ranges.
Study design
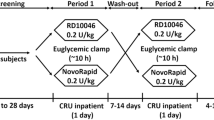
This was a randomized, single-center, double-blind, single-dose, 2-treatment, 2-period, crossover study undertaken in 2018–2019 (trial registration: www.clinicaltrials.jp, identifier: JapicCTI-205380, registered 20/07/2020). Subjects received each treatment once. The trial was approved by an ethical review board (Hakata Clinic, Fukuoka, Japan) and conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonization guidelines for Good Clinical Practice. Written informed consent was provided by all subjects before study entry.
Following a screening visit 3–28 days before the first period, subjects enrolled into the study were admitted to the study clinic on the day before dosing. On the day of treatment, they were randomized (computer-generated by sponsor) to one of the two treatment sequences (see Supplementary Fig. S1). Subjects received a single subcutaneous dose of 0.3 U/kg SAR-Asp or NN-Asp-Jp in the first treatment period in randomized order followed by the other drug in the second treatment period. A wash-out period of 7–18 days separated each treatment period, with an end-of-study visit performed 4–8 days after the last dose.
Treatments
SAR-Asp (100 U/mL) solution for injection was manufactured by Sanofi (Frankfurt, Germany), with NN-Asp-Jp provided as the commercially available formulation. To maintain double-blinding (with respect of the Investigator, subject and Sponsor) and consistency of dosing methodology, an independent pharmacist at the study site was responsible, with an assistant, for preparing the treatments for each subject.
Bioanalytical methods
Venous blood samples for pharmacokinetic analysis were collected before dosing of study drug, and then every 15 to 60 min during the 10 h clamp period. Samples were centrifuged within 20 min of collection. Plasma was collected and kept frozen (− 60 to − 80 °C) until analysis. Plasma concentrations of SAR-Asp and NN-Asp-Jp were analyzed by using a validated liquid chromatography-tandem mass spectrometry (LC–MS/MS) assay at Syneos Health (Quebec, Canada). Plasma concentrations within the validated concentration range (100–8000 pg/mL) were used to calculated pharmacokinetic parameters. Inter-assay precision (% coefficient of variation [CV]) and inter-assay accuracy (%bias) during validation were ≤ 15%.
Pharmacodynamic evaluation using euglycemic clamp
Each dosing visit included a 10 h automated euglycemic clamp procedure (STG-55, Artificial Endocrine Pancreas; Nikkiso Co, Ltd, Tokyo, Japan)13. The premise of performing the clamp procedure is that the blood glucose lowering effect of an administered insulin is antagonized by a variable infusion of glucose so that blood glucose is maintained (or clamped) at a target level2,14. The metabolic profile of the investigated insulin is characterized by the glucose infusion rate (GIR) needed to keep blood glucose as close as possible at its predefined target level during the glucose clamp15.
Following an overnight fast of at least 10 h, subjects were connected to the clamp device and their baseline blood glucose was determined (via glucose oxidase sensors that measure whole-blood glucose levels) before dosing of the study drug. For each subject, this was calculated as the mean of four glucose measurements at 30, 20, 10 and 1 min before administration of the study drugs. The clamp procedure was not performed in those subjects having a baseline glucose level less than 70 mg/dL (3.92 mmol/L). After dosing, onset of insulin action in each subject was when the blood glucose dropped below the target level of the clamp procedure, which was when it was 5 mg/dL (0.28 mmol/L) less than their fasting baseline value2.
Blood glucose levels were measured in 1-min intervals, and using a predefined algorithm the clamp device automatically administered a variable infusion of 10% glucose in response to changes in glucose to maintain each subject at their target glucose level. The GIR, being the amount of external glucose needed to keep a subject’s blood glucose concentration at its target level, was continuously measured, and recorded by the STG-55 device. GIR profiles reflected the metabolic effects of SAR-Asp and NN-Asp-Jp over time.
Safety assessment
The safety and tolerability of single doses of SAR-Asp and NN-Asp-Jp were assessed by 12-lead ECG, vital signs, routine laboratory parameters, physical examination, and reporting of adverse events (AEs). AEs were classified using MedDRA (Medical Dictionary for Regulatory Activities) 21.1. The safety population included all randomized patients who received at least one dose of study insulin, analyzed according to the treatment received. The treatment-emergent AE period included the time from the first dose of study drug up to 72 h after the last dose in each treatment period.
Pharmacokinetic and pharmacodynamic parameters
The pharmacokinetic analysis dataset included subjects who completed at least one treatment period, had measurable insulin aspart concentrations and no major or critical deviations. Parameter estimates for SAR-Asp and NN-Asp-Jp were calculated by using standard noncompartmental methods with Phoenix WinNonlin 8.1 (Certara, Princeton, NJ). Area under the plasma insulin aspart concentration–time curve was calculated by the trapezoidal method from 0 to the time of the last concentration above the limit of quantification (INS-AUClast) and extrapolated to infinity (INS-AUCinf). Maximum plasma insulin aspart concentration observed (INS-Cmax) and INS-AUClast were the primary pharmacokinetic endpoints of the study. INS-AUCinf was a secondary endpoint.
The GIR over time was the primary pharmacodynamic parameter measured during the clamp procedure. The area under the body weight-standardized GIR time curve [GIR-AUC] measured insulin mediated glucose uptake into tissues. Subjects completing at least one clamp procedure with no major or critical deviations were included in the pharmacodynamic analysis dataset. Individual GIR values in each treatment group were standardized for body weight and subjected to a smoothing procedure using a locally weighted scatterplot smoothing (LOESS) function (SAS, PROC LOESS, factor 0.06). A smoothing factor of 6% was used based on the expected morphology of the GIR-profiles. Individual blood glucose levels were also subjected to a smoothing procedure. Fitted data were used to calculate the pharmacodynamic parameters, including the primary parameters of maximum smoothed body weight standardized GIR (GIRmax) and GIR-AUC over 10 h (GIR-AUC0–10 h), and the secondary parameter of time to reach GIRmax (GIR-tmax).
Parameters used to assess clamp quality included the mean and individual %CV of blood glucose measurements during euglycemia, and the absolute difference of individual mean blood glucose measurements from the clamp target level, as described previously9.
Statistical analyses
The study aimed to show similarity (equivalence) in pharmacokinetic exposure and glucodynamic activity of a single dose (0.3 U/kg) of SAR-Asp to NN-Asp-Jp under fasting conditions. To achieve this, 26 and 32 evaluable subjects, respectively, were required. These calculations were based on estimates of within-subject variability from a prior SAR-Asp study in subjects with T1D9, and those reported in applicable studies performed in healthy subjects16,17. A within-subject standard deviation (SD) of 0.175 and 0.180 was assumed for the natural log-transformed pharmacokinetic (INS-Cmax and INS-AUClast) and pharmacodynamic (GIRmax and GIR-AUC0–10 h) parameters, respectively, for a true treatment ratio between the two formulations of 0.93 and 1.07, respectively. The planned sample sizes provided at least 90% power to show equivalence with a type 1 error of 5% and 2.5% for the pharmacokinetic and pharmacodynamic parameters, respectively. To allow for dropouts, the study planned to recruit at least 36 individuals (18 per sequence).
Log-transformed pharmacokinetic and pharmacodynamic parameter estimates for SAR-Asp and NN-Asp-Jp were analyzed using a linear mixed-effects model with period, sequence, and treatment as fixed effects and subject as a random effect. For each parameter, the model-based difference in treatment means along with the confidence limits (90% for pharmacokinetic parameters, 95% for pharmacodynamic parameters) was back-transformed to provide estimates for the ratio of geometric means (gMean) between treatments (SAR-Asp/NN-Asp-Jp) and the corresponding confidence limits. Similarity for the pharmacokinetic parameters (INS-AUCinf and INS-Cmax) and bioequipotency for the pharmacodynamic parameters (GIR-AUC0–10 h and GIRmax) was concluded if the confidence limits (90% and 95% confidence intervals [CIs], respectively) of the treatment ratios were entirely within the 0.80–1.25 equivalence interval, in agreement with regulatory guidance2,18. Data was analyzed using SAS v9.4 (SAS Institute, Cary, NC).
Results
Forty Japanese male subjects were randomized and treated, with thirty-nine completing both treatment periods. One subject withdrew from the study because of an AE (road traffic accident) during the washout period before dosing in Period 2. The treatment sequence of this subject was NN-Asp-Jp/SAR-Asp and therefore the pharmacokinetic and pharmacodynamic populations for SAR-Asp were missing. Clamp data during period 2 from five subjects were excluded because of operational errors of the devices (two during SAR-Asp treatment and three during NN-Asp-Jp treatment). In view of these operational errors, an additional four subjects were included in the study. Pharmacodynamic parameters for SAR-Asp and NN-Asp-Jp were available for 36 and 38 subjects, respectively. Baseline characteristics of the included subjects are given in Table 1.
Pharmacokinetics
Single 0.3 U/kg doses of SAR-Asp and NN-Asp-Jp resulted in mean insulin concentration-versus-time profiles that were virtually superimposable (Fig. 1A). The extent of exposure, indicated by gMean INS-AUClast and INS-Cmax values, was similar between the groups (Table 2), with the gMean ratios close to 1 and the corresponding 90% CIs for each parameter contained within the predefined equivalence interval (0.80–1.25). This confirmed equivalent exposure of the two treatments. The secondary endpoint AUCinf showed similar results to AUClast. Low to moderate between-subject variability in the pharmacokinetic parameters was observed, with CVs between 26 and 32%.

Linear arithmetic mean insulin aspart plasma time profiles (A), mean smoothed LOESS fits of body weight standardized GIR versus time profiles (GIR values less than 0 at baseline are results of the LOESS smoothing) (B). The horizontal line in (A) is the lower level of quantification (100 pg/mL). GIR glucose infusion rate, LOESS locally weighted scatterplot smoothing.
Pharmacodynamics
In the pharmacodynamic analysis, the mean smoothed bodyweight normalized GIR profiles during the euglycemic clamp procedure (from time 0 to 10 h postdose) were nearly superimposable for SAR-Asp and NN-Asp-Jp (Fig. 1B). The overall pharmacodynamic effects of both treatments were similar, displaying a short time-action profile.
The extent of glucose-lowering, indicated by gMean GIR-AUC0–10 h and GIRmax values, was similar for SAR-Asp and NN-Asp-Jp (Table 2), with the gMean ratios close to 1 and the corresponding 95% CIs for each parameter entirely within the equivalence interval (0.80–1.25). This confirmed equipotency of the two treatments. Between-subject variability for GIR-AUC0–10 h and GIRmax was low, as shown by CVs between 21 and 24%. The median GIR-tmax for SAR-Asp (2.82 h) was similar to that for NN-Asp-Jp (2.35 h).
Performance of the clamp
Several parameters were used to assess clamp quality during euglycemia (Table 3). Individual mean blood glucose levels were similar for both treatments (mean values of 79.51 and 79.89 mg/dL for SAR-Asp and NN-Asp-Jp, respectively). Individual variability of blood glucose measurements was low with median CV% values of 7.05% and 5.90% for SAR-Asp and NN-Asp-Jp, respectively. Similarly, absolute differences between individual mean blood glucose measurements and the blood glucose target level were low (mean of 2.31 mg/dL for SAR-Asp and 1.74 mg/dL for NN-Asp-Jp).
Safety and tolerability
Single doses of both insulin aspart products were well-tolerated. There were no serious AEs, AEs of special interest or treatment-emergent AEs reported during the study. One subject (sequence of NN-Asp-Jp/SAR-Asp) reported AEs of back pain and neck pain following a road traffic accident during the washout (post treatment) period following Period 1 and withdrew his consent for further participation before dosing in Period 2. A few predefined potentially clinically significant abnormalities in laboratory tests and ECG parameters were reported but with no noticeable differences between SAR-Asp and NN-Asp-Jp. The majority of the abnormalities in laboratory values and ECG parameters were present at baseline prior to administration of SAR-Asp and NN-Asp-Jp. The investigator assessed that none of abnormalities were clinically significant.
Discussion
In this crossover study in healthy Japanese males, single doses of SAR-Asp insulin aspart solution showed similar pharmacokinetic exposure and glucodynamic activity to the commercially available NN-Asp-Jp formulation, using a euglycemia clamp device to measure insulin action. Between-subject variability estimates for both treatments were low, and both were well tolerated.
The design of this crossover clamp study, involving administration of single-doses of each treatment, is consistent with regulatory guidance for the evaluation of biosimilar insulins2. A similar design was used in a trial that compared insulin exposure and pharmacodynamic activity for SAR-Asp with US-reference NN-Asp and EU-reference NN-Asp9. We assessed insulin exposure from the plasma insulin concentration–time profiles and insulin activity as glucose utilization during the euglycemic clamp. The crossover design allowed each subject to receive both treatments, so that a comparison between the two treatments could be made on the same subject. This allowed within-subject comparisons of pharmacokinetic and glucodynamic parameters between the two products to minimize potential within- and between-subject variability.
The study was conducted in normal-weight healthy subjects as this provides a homogeneous population that is sensitive to insulin, thereby enabling detection of any potential treatment-related differences2,18. Healthy subjects are also recommended in bioequivalence studies to reduce pharmacokinetic and/or pharmacodynamic variability as they avoid potential confounding effects such as concomitant disease and medications2,18. Only male subjects were included as it was uncertain if the known insulin sensitivity in females during the menstrual cycle might affect the study results2. One concern with conducting insulin bioequivalence studies in healthy subjects is that the presence of endogenous insulin is not easily distinguished from exogenous insulin administration with available assays2, thereby interfering with the pharmacokinetic and dynamic measurements. In this respect, the bolus of the two rapid-acting insulin treatments should have adequately suppressed endogenous insulin for the duration of the clamp, meaning that no C-peptide correction was required2. In addition, endogenous insulin was suppressed by clamping blood glucose levels below the subject’s fasting glucose with plasma insulin aspart concentrations of both treatments measured using a validated and sensitive LC–MS/MS assay.
The insulin dose selected for this study under fasting conditions (0.3 U/kg) provided strong effects in the euglycemic clamp shown by a substantial increase in glucose demand reflected by the sizable GIR increase over the 10 h clamp duration and has been used in other similar rapid-acting insulin clamp studies2,9,19. The short duration of action and rapid clearance of insulin aspart meant that a clamp duration of 10 h was sufficient to adequately account for individual variations in insulin elimination and the duration of pharmacodynamic activity. It also minimized the time during which subjects were required to remain fasted and undergo the various clamp procedures20. The washout period of 7 to 18 days between the two dosing periods ensured that insulin concentrations were below the lower limit of quantification before the second treatment period.
The euglycemic clamp procedure in this study directly assessed the glucodynamic properties of SAR-Asp and NN-Asp-Jp. Clamp studies remain widely used to evaluate insulin action with the technique considered to be the reference standard for evaluating insulin sensitivity in human subjects14,20. They are also recommended for use in pharmacokinetic/dynamic studies that aim to show biosimilarity between two insulins2. The quality of the clamp performance is important for the interpretation of data. Here, the individual variability (CV%) of blood glucose per clamp was similar between treatments, with median CV values ranging from 5.9 to 7.1%. This was indicative of stable blood glucose concentrations during the clamp procedure with blood glucose levels during the clamp maintained close to their target level. These findings indicate that the GIR during the clamp was appropriate to measure the hypoglycemic effect of the two insulins.
Strengths of this study include the crossover design that allows for a within-subject comparison of the pharmacokinetic and pharmacodynamic response for the two insulins using the same subcutaneous doses, thereby enabling subjects to act as their own control. The glucose clamp technique and blinding of the investigator- and subject also avoided investigator-related bias. The requirement for standardized and well-controlled conditions (e.g., a long fasting period, administration of fixed insulin doses) is a recognized limitation of the euglycemic glucose clamp technique. However, the evaluation of the two treatments using a glucose clamp in phase 1 trials provides optimal sensitivity for detecting potential differences between the insulin treatments9,14. Establishing similar efficacy (i.e., change in HbA1c), safety and immunogenicity in real-life conditions is subsequently evaluated in larger phase 3 trials14.
Finally, rising insulin costs remain a concern for people with diabetes, their families and health care providers. The development of new biosimilar insulin products over the last few years, including rapid-acting insulins like reported here, has the potential to reduce drug treatment costs and thereby facilitate greater access of insulin treatment for people with diabetes. The reporting of such biosimilar trial data will increase the credibility of these valuable products and thereby provide benefit for physicians and their patients.
Conclusion
This study showed that SAR-Asp had similar pharmacokinetic exposure and glucodynamic activity compared with Japanese-approved insulin aspart formulation (NN-Asp-Jp).
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
European Medicines Agency. Insulin aspart Sanofi. Summary of product characteristics, 4 May 2021 (accessed 10 June 2021); https://www.ema.europa.eu/en/documents/product-information/insulin-aspart-sanofi-epar-product-information_en.pdf.
European Medicines Agency. Guideline on non-clinical and clinical development of similar biological medicinal products containing recombinant human insulin and insulin analogues, 26 February 2015 (accessed 10 June 2021); https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-non-clinical-clinical-development-similar-biological-medicinal-products-containing_en-0.pdf.
US Food and Drug Administration Center for Drug Evaluation and Research (CDER). Guidance for industry. Diabetes mellitus: Developing drugs and therapeutic biologics for treatment and prevention. February 2008:1–30 (accessed 10 June 2021); https://wayback.archive-it.org/7993/20191211232220https://www.fda.gov/media/71289/download.
US Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry: Questions and answers on biosimilar development and the BPCI Act (Revision 1), December 2018:1–19 (accessed 10 June 2021); https://www.fda.gov/media/119258/download.
US Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for Industry. New and revised draft Q&As on biosimilar development and the BPCI Act (revision 2) (draft guidance), December 2018:1–14 (accessed 10 June 2021); https://www.fda.gov/media/119278/download.
Japan Ministry of Health Labour and Welfare (MHLW). Guidelines for the quality, safety and efficacy assurance of follow-on biologics. Notification No. 0304007. March 2009 (accessed 10 June 2021); http://www.pmda.go.jp/files/000153851.pdf.
Liebl, A. et al. Ten years of experience with biphasic insulin aspart 30: From drug development to the latest clinical findings. Drugs 72, 1495–1520. https://doi.org/10.2165/11635490-000000000-00000 (2012).
Liebl, A., Mohan, V., Yang, W., Strojek, K. & Linjawi, S. 15 years of experience with biphasic insulin aspart 30 in type 2 diabetes. Drugs R. D. 18, 27–39. https://doi.org/10.1007/s40268-018-0228-x (2018).
Kapitza, C., Nosek, L., Schmider, W., Teichert, L. & Nowotny, I. Single-dose euglycemic clamp study demonstrating pharmacokinetic and pharmacodynamic similarity between SAR341402 insulin aspart and US- and EU-approved versions of insulin aspart in subjects with type 1 diabetes. Diabetes Technol. Ther. 22, 278–284. https://doi.org/10.1089/dia.2019.0351 (2020).
Garg, S. K. et al. Efficacy and safety of insulin aspart biosimilar SAR341402 versus originator insulin aspart in people with diabetes treated for 26 weeks with multiple daily injections in combination with insulin glargine: A randomized open-label trial (GEMELLI 1). Diabetes Technol. Ther. 22, 85–95. https://doi.org/10.1089/dia.2019.0382 (2020).
Garg, S. K. et al. Safety, immunogenicity, and glycemic control of insulin aspart biosimilar SAR341402 versus originator insulin aspart in people with diabetes also using insulin glargine: 12-month results from the GEMELLI 1 trial. Diabetes Technol. Ther. 22, 516–526. https://doi.org/10.1089/dia.2020.0008 (2020).
Thrasher, J. et al. Safety and tolerability of insulin aspart biosimilar SAR341402 versus originator insulin aspart (NovoLog) when used in insulin pumps in adults with type 1 diabetes: A randomized, open-label clinical trial. Diabetes Technol. Ther. 22, 666–673. https://doi.org/10.1089/dia.2019.0446 (2020).
Tsukamoto, Y. et al. Evaluation of a novel artificial pancreas: closed loop glycemic control system with continuous blood glucose monitoring. Artif. Organs 37, E67–E73. https://doi.org/10.1111/aor.12068 (2013).
Heise, T. et al. Euglycaemic glucose clamp: What it can and cannot do, and how to do it. Diabetes Obes. Metab. 18, 962–972. https://doi.org/10.1111/dom.12703 (2016).
Benesch, C., Heise, T., Klein, O., Heinemann, L. & Arnolds, S. How to assess the quality of glucose clamps? Evaluation of clamps performed with ClampArt, a novel automated clamp device. J. Diabetes Sci. Technol. 9, 792–800. https://doi.org/10.1177/1932296815576957 (2015).
Heinemann, L., Weyer, C., Rauhaus, M., Heinrichs, S. & Heise, T. Variability of the metabolic effect of soluble insulin and the rapid-acting insulin analog insulin aspart. Diabetes Care 21, 1910–1914. https://doi.org/10.2337/diacare.21.11.1910 (1998).
Engwerda, E. E. C., Tack, C. J. & de Galan, B. E. Pharmacokinetic and pharmacodynamic variability of insulin when administered by jet injection. J. Diabetes Sci. Technol. 11, 947–952. https://doi.org/10.1177/1932296817699638 (2017).
US Food and Drug Administration Center for Drug Evaluation and Research (CDER). Guidance for industry. Clinical pharmacology data to support a demonstration of biosimilarity to a reference product. December 2016:1–15 (accessed 10 June 2021); https://www.fda.gov/media/88622/download.
Kapitza, C. et al. Similar pharmacokinetics and pharmacodynamics of rapid-acting insulin lispro products SAR342434 and US- and EU-approved Humalog in subjects with type 1 diabetes. Diabetes Obes. Metab. 19, 622–627. https://doi.org/10.1111/dom.12856 (2017).
Linnebjerg, H. et al. Comparison of the pharmacokinetics and pharmacodynamics of LY2963016 insulin glargine and EU- and US-approved versions of Lantus insulin glargine in healthy subjects: Three randomized euglycemic clamp studies. Diabetes Care 38, 2226–2233. https://doi.org/10.2337/dc14-2623 (2015).
Acknowledgements
Sanofi K.K. (Tokyo, Japan) was the sponsor of the study and was responsible for the design, data collection and performing all statistical analyses. Oberon Ltd (London, UK) provided editorial support, funded by Sanofi.
Funding
Sanofi K.K., Tokyo, Japan.
Author information
Authors and Affiliations
Contributions
W.S., H.T., M.K., H.M., I.N.: Conception and design of the study. M.S., T.Y.: Subject recruitment, data acquisition and clinical management. H.T.: Statistical analysis. W.S., H.T., M.K., H.M., I.N.: Data analysis. All authors contributed to interpretation of the data and manuscript preparation. All authors are fully responsible for the content and approved the final version.
Corresponding author
Ethics declarations
Competing interests
M.S. has no conflict of interest. His current affiliation is Kashiihara Hospital, and this manuscript does not represent opinions of his current affiliation. T.Y. declares no conflict of interest. W.S., H.T. and M.K. are employees of Sanofi. I.N. is a former employee of Sanofi and a current consultant to Sanofi. H.M. is a former employee of Sanofi and a current employee of Novartis. H.M. declares no conflict of interest as this manuscript does not represent opinions of his current affiliation.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shiramoto, M., Yoshihara, T., Schmider, W. et al. Pharmacokinetic and pharmacodynamic similarity between SAR341402 insulin aspart and Japan-approved NovoRapid in healthy Japanese subjects. Sci Rep 11, 22931 (2021). https://doi.org/10.1038/s41598-021-02410-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-02410-z
This article is cited by
-
Survey of Data Package and Sample Size of Comparative Clinical Studies for Biosimilar Developments from PMDA Assessments
Pharmaceutical Medicine (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
