
Abstract
The Queensland fruit fly, Bactrocera tryoni, is a major pest of Australian horticulture which has expanded its range in association with the spread of horticulture over the last ~ 150 years. Its distribution in northern Australia overlaps that of another fruit fly pest to which some authors accord full species status, Bactrocera aquilonis. We have used reduced representation genome-wide sequencing to genotype 359 individuals taken from 35 populations from across the current range of the two taxa, plus a further 73 individuals from six of those populations collected 15–22 years earlier. We find significant population differentiation along an east–west transect across northern Australia which likely reflects limited but bidirectional gene flow between the two taxa. The southward expansion of B. tryoni has led to relatively little genetic differentiation, and most of it is associated with a move into previously marginal inland habitats. Two disjunct populations elsewhere in Australia and three on Melanesian islands are each clearly differentiated from all others, with data strongly supporting establishment from relatively few founders and significant isolation subsequently. Resequencing of historical samples from one of the disjunct Australian populations shows that its genetic profile has changed little over a 15-year period, while the Melanesian data suggest a succession of ‘island hopping’ events with progressive reductions in genetic diversity. We discuss our results in relation to the control of B. tryoni and as a model for understanding the genetics of invasion and hybridisation processes.
Similar content being viewed by others

Introduction
The Queensland fruit fly (Bactrocera tryoni Froggatt, 1897; 'Qfly') is both a major pest of horticulture in Australia and some Pacific islands and a useful model for understanding genetic processes associated with range expansion and invasion1. Qfly was originally confined to the relatively wet tropical and sub-tropical east coast of Queensland (QLD) and the north eastern coast of New South Wales (NSW), with populations in the northern-most regions of the Northern Territory (NT) (nNT) and northern Western Australia (nWA) recognised by some authors as a distinct species, the Northern Territory fruit fly (NTfly, B. aquilonis)2,3,4. Over the last ~ 150 years, however, B. tryoni has expanded southward in association with the development of horticultural industries5. No infestations of Qfly were reported in the Sydney area (latitude ~ 33°5′ South) before the twentieth century6, but by the 1960s Qfly were found in Gippsland in Victoria (latitude ~ 37°5′ South)7. Later expansions included incursions further inland in QLD, NT and southern Australia4, with opportunistic invasions into New Caledonia, French Polynesia, Pitcairn Island and Cook Island in the Pacific1. Given increasing trade and horticultural activities in Australia and the region, climate modelling suggests a high likelihood of further expansion, under both current and future climate scenarios8,9.
Phenological work on Qfly in its native range suggests that host availability, as well as climate, are principal determinants of seasonal abundance, with rapid population increases in spring and summer and substantial decreases in autumn and winter10. This seasonal host availability effect is consistent with the range expansion which has occurred post-European settlement, and the associated introduction of new host crops. Many of the new crops depend on irrigation, and Yonow and Sutherst8 found the presence of irrigation to be a strong predictor of Qfly presence in northern, eastern and southern Australia. Several studies have reported strong plastic responses to low temperature in southerly populations11,12, as well as heavy dependence on overwintering refugia13,14, but there is little evidence for inherited geographic variation in cold resistance15. There is heritable ecotypic variation in heat and desiccation resistance, but these two traits are not strongly correlated with each other, or with latitude, suggesting complex patterns of geographic differentiation15.
Broadly consistent with all the distributional and ecological data, early population genetic studies with microsatellite markers indicated significant genetic differences across the species range16,17. In coastal eastern Australia there was an essentially panmictic population in the central eastern coast of Queensland (Rockhampton-Brisbane) with some differentiation to the north and south. Moreover there was also a progressive divergence westward along the north coast into the northern parts of the Northern Territory and northern Western Australia (nNT/nWA). An apparently distinct and potentially isolated population in central Australia was also identified4,17. As noted, several authors have classified the nNT/nWA populations as B. aquilonis, which is mainly distinguished from B. tryoni by a slightly paler thorax and abdomen, and more pointed aculeus on the ovipositor2,3,18,19,20. However, Drew and Lambert19 noted that the differences in colouration ‘are so slight as to be of questionable value for species-level discrimination’, and Wang et al.21 reported a Nei’s genetic distance of just 0.014 between the two taxa based on microsatellites. Cameron et al.17 argued that the nNT/nWA populations are simply progressively extreme samples of an essentially continuous clinal pattern of genetic differences from the east coast populations. There is as yet no genetic data on the island populations in the South Pacific but a specific succession of ‘island hopping’ invasions has been proposed based on the dates of the first records on those islands; Qfly was first reported in New Caledonia and the Loyalty Islands around 1969, in French Polynesia a year later, and subsequently in Cook Islands (albeit it was later eradicated from the latter)22.
The present study explores Qfly/NTfly population structure from across the known geographic range using over two thousand single nucleotide polymorphisms (SNPs) generated from genome-wide reduced representation resequencing. We examine both the spatial and temporal dimensions of the invasion process within Australia by sampling expansions southwards and inland and sampling and resampling from the centre and margins of the earlier range over a 15–22-year interval. Additionally, we characterise extant Pacific island populations to test the ‘island hopping’ hypothesis and investigate the genetics of disjunct as opposed to continuous spatial range expansions. Finally, we discuss the relevance of our results to the ecotypic variation, future distribution and management of the Qfly.
Materials and methods
Insect sampling
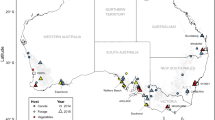
We sampled a total of 368 individuals from 35 localities between 2015 and 2018, and for six locations we also had a total of 83 samples from 1994 to 2003 courtesy of the earlier microsatellite-based studies4, 16. Some flies were collected as adults from lure traps (BioTrap, Victoria, Australia) and then preserved in 100% ethanol. Others were collected as larvae from infested fruits, reared through to adults as part of the previous studies15 and then frozen at − 80 °C. Between four and 20 individuals were analysed per collection. Six B. neohumeralis (a closely related sister species in the tryoni complex) individuals which were used as an out-group in one analysis were collected from infested fruits from Cape Tribulation (16°08′ South), Brisbane (27°41′ South) and Mareeba (17°00′ South) and processed as per the Qfly and NTfly samples above. All flies were identified morphologically according to the species descriptions in Drew20, except that, like many of the previous authors, we could not consistently distinguish nNT/nWA flies from eastern states Qfly (although concurring that some of the former were somewhat paler). Figure 1 and Table 1 summarise all the populations analysed. (The maps in Fig. 1 and subsequent figures were prepare using ggmap23 and ggplot224 in R version 3.6.1 (https://www.r-project.org/)25, with final edits in Inkscape 0.92.4, https://inkscape.org/).

Geographic distribution of the Qfly and NTfly and our sampling sites. Distribution of the Australian populations within the Qfly native, expanded range and disjunct populations, our 35 collections sites and the former Fruit Fly Exclusion Zone (FFEZ). AS Alice Springs (Northern Territory, NT), AR Ardlethan (New South Wales, NSW), BB Batemans Bay (NSW), BV Bega Valley (NSW), BO Borroloola (NT), BW Bowen (Queensland, QLD), BR Brisbane (QLD), BM Broome (Western Australia, WA), CR Cairns (QLD), CA Canberra (Australian Capital Territory, ACT), CT Cape Tribulation (QLD), CY Cape York (QLD), CL Cloncurry (QLD), CO Coen (QLD), CK Cooktown (QLD), DW Darwin (NT), GR Griffith (NSW), HU Hughenden (QLD), KA Katherine (NT), KU Kununurra (WA), LI Loyalty Island (Pacific Islands, PI), MP Mapoon (QLD), MA Mareeba (QLD), MK Mataranka (NT), MI Mt Isa (QLD), NA Narrabri (NSW), NC New Caledonia (PI), RO Rockhampton (QLD), SH Shepparton (Victoria, VIC), SY Sydney (NSW), TH Tahiti (PI), TC Torrens Creek (QLD), TO Townsville (QLD), UC Utchee Creek (QLD), WP Weipa (QLD). Asterisk denote localities sampled in both 2015–2018 and 1994–2003.
DNA extraction, sequencing and SNP filtering
Individual flies were transferred from ethanol or − 80 °C storage into 96-well plates at − 20 °C and sent to Diversity Arrays Technology Pty Ltd (Canberra, Australia) for DNA extraction as per Kilian et al.26. PstI and SphI restriction enzyme digestion, ligation of < 200 bp fragments to a bar coding adapter, PCR amplification, and Illumina HiSeq2500 sequencing were completed following the DArTseq protocol of Georges et al.27. SNPs were called from the resulting sequence reads using the DArT analytical pipeline as described by Georges et al.27 and the resulting dataset was read into R, together with associated metadata, using the helper functions of the dartR R package28. Filtering of SNPs involved excluding those which were (a) not scored consistently in two replicate runs of the protocol (see George et al.27 for details), (b) had < 10 × or > 250 × read depth or c) a call rate < 90% (i.e. SNPs not found in ≥ 10% of the individuals). Filtering of individuals then involved excluding those with a call rate < 90% (i.e. individuals with ≥ 10% of the SNPs missing). Finally, we only retained one randomly chosen SNP from any sequenced tag containing multiple SNPs. After filtering, the data from nine individuals in the N + E + D dataset (see below) were excluded, as were 23 individuals from the temporal dataset (the nine above plus four more from the 2015–2018 samples and ten from the 1994–2013 samples).
Data structure
The analyses below were structured around four sets of samples:
-
1.
Native range (N) populations: the 20 2015–2018 collections in the Native Qfly/NTfly range.
-
2.
Expansion (E) populations: samples from 10 populations in the essentially contiguous expansion southward and inland.
-
3.
Disjunct (D) populations: samples from Broome, Alice Springs, and the Melanesian islands, and
-
4.
Temporal data: samples from six locations, collected 15–22 years earlier (1994–2003) for the previous microsatellite studies, plus the 2015–2018 collections from the same locations.
Depending on the question, the analyses below were conducted on the three spatial datasets (N, N + E, N + E + D) or the temporal dataset.
Population structure
The first of two tests for population structure we carried out on all datasets was a Discriminant Analysis of Principal Components (DAPC)29, which was implemented in the R package “adegenet”30 and used the localities sampled as priors for populations. The optimal number of Principal Components (PCs) to be retained was selected using alpha score optimisation with the optim.a.score function in the “adegenet” package, retaining 83, 77, 51, and 12 PCs from the N, N + E, N + E + D and temporal datasets, respectively (Fig. S1). The Bayesian information criterion (BIC) was used to identify the best number of discriminant functions to represent the data. We retained two discriminant functions for the N and N + E, and five and three discriminant functions for the N + E + D and temporal datasets, respectively (see Fig. S1).
The second test for population structure used sparse, non-negative matrix factorisation (sNMF31) to estimate individual fly admixture coefficients. This involves an algorithm which is similar to the Bayesian clustering program structure32 but uses non-negative matrix factorisation and least-square optimisation to facilitate working with large datasets. The R package LEA31 implements this algorithm in the snmf function. The SNP data were converted first into a Structure-like input format with the gl2faststructure function from the dartR package and then into the required format for the snmf function with the helper function struct2geno from the LEA package. The snmf algorithm was run with K values for all the datasets, with 100 repetitions per K value and other options set to default values in all cases. The value of K that best explained the results was selected using the cross-entropy criterion as detailed by the LEA package authors31 (Fig. S2).
Isolation by distance
We conducted two analyses to test for any isolation by distance in the N + E dataset. First we ran a Mantel test of the linearised genetic distances between populations (Nei’s D/1 − Nei’s D, calculated using the R package StAMPP33) against the geographical distance (Euclidean distance, calculated using the distance function in the R package vegan34). Secondly, we conducted a redundancy analysis (RDA) of allele frequencies against polynomial functions of latitude and longitude (maximum power two, calculated with poly and forward selected with ordistep from the vegan package34) in this dataset as per Meirmans35. The overall model and specific functions within it were tested for significance using the anova.cca function from the vegan package34 with 1,000 permutations for each test. We then multiplied the percentage of variance accounted for by the latitude and longitude of each population (i.e. the constrained variance of the RDA) by the overall Nei’s D for all populations to obtain the percentage of the total genetic variation that is explained by the spatial variables35,36.
Phylogenetics
We investigated the origins of the contiguous expansion (E) and disjunct invasion (D) populations by inferring a population tree from the N + E + D dataset using TreeMix37. TreeMix was preferred over traditional phylogenetic approaches because of our primary interest in tracing the origin of the range expansion and disjunct (E and D) populations. We created an input file of allele frequencies for every contemporary sample according to the TreeMix manual. We then built a maximum likelihood tree from the allele frequencies using the default 500 SNP block setting, rooting the trees with B. neohumeralis and visualising them with the R script provided with the software38.
Fixed difference and other genetic parameters
We tested for fixed differences between populations in the N + E + D dataset using the gl.collapse.recursive function in the dartR package28. This method sums the fixed differences between each pair of populations and combines the members of each pair which do not have a significant number of fixed differences, where the number of differences expected due to sampling error is estimated from a thousand bootstrapping runs with the gl.collapse.pval function. The process is repeated until no further non-significant pairs are found28. The resulting set of collapsed populations can be thought of as diagnostic operational taxonomic units (OTUs) between which there is strong support for an absence of recent gene flow.
We also estimated genetic diversity summary statistics (allele richness, Shannon diversity, and heterozygosity) for all populations, using the formulae of Sherwin et al.39 implemented in the gl.diversity function in the dartR package28. Additionally we calculated Fst between all populations in the N + E + D dataset with the gl.fst.pop function from the dartR package and tested their significance using 1,000 bootstrap replications.
Temporal variation
The temporal dataset contained six pairs of samples from previous Qfly population genetic studies4,16,17 and contemporary populations collected from the same localities (Table 1). We first tested for overall differences due to the six localities and two sampling times in the genetic distances between the 12 samples (calculated with the helper function dist in R) using the poppr.amova function in the poppr package and simulations with 1,000 permutations generated with the randtest function40. We then screened for changes within each population pair over the 15–22 years by calculating Fst between the two temporal samples from each locality with the gl.fst.pop function from the dartR package and testing their significance using 1,000 bootstrap replications.
Results
After quality filtering, the data comprised 2,785 SNPs for 359 individuals for the 2015–2018 samples for the spatial analyses and 2,344 SNPs (~ 58% of them also in the 2,785 above) from 138 individuals for the temporal analyses (i.e. the six populations scored on both the 2015–2018 and 1994–2003 datasets).
Population structure in the native and expanded range
We used a combination of clustering (DAPC) and admixture coefficients (sNMF) analyses to test for differentiation among the 20 populations we sampled from the Qfly/NTfly native range (i.e., dataset N). DA1 in the DAPC analysis accounted for 31.5% of the variance and separated most of the nNT/nWA populations from the QLD populations, with Darwin (DA) and Borroloola (BO) as intermediates. DA2, accounting for 16% of the variance, separated Darwin populations from the others (Fig. S3). The cross-entropy algorithm in sNMF yielded the lowest cross-entropy for a K of two, with less support for other values of K (3–10; Fig. S2). Similar to the DAPC results, sNMF differentiated the NTfly and Qfly populations, but it also showed considerable individual variation within them (Fig. 2). Some Darwin and Borroloola individuals showed 50% or more QLD ancestry, whereas Kununurra (KU) individuals shared little ancestry with the east coast populations. Notably Kununurra is furthest from the east coast among these populations while Borroloola is closest, and Darwin is a major sea- and airport city, with significant trade and travel links to the east coast. Among the QLD samples, the more northerly coastal populations (Cape York, Mapoon, Coen, Weipa, Cook Town and Cairns, i.e. CY, MP, CO, WP, CT, CR) included individuals showing significant nNT/nWA ancestry, whereas individuals in the most southerly population, Brisbane (BR), had the lowest level of admixture. The two inland QLD populations, Hughenden (HU) and Torrens Creek (TC), are relatively isolated geographically and, while relatively northerly, they also showed relatively little nNT/nWA ancestry.

Analysis of Qfly and NTfly native range populations. The ancestry coefficients for 20 populations from the Qfly and NTfly native range are presented. (A) Estimates of admixture proportions inferred with sNMF for the Qfly and NTfly native dataset (N) with the best supported number of ancestral populations (K = 2). (B) Geographical distribution of the Qfly and NTfly within its native range. Pie charts represent the admixture proportion of every individual for each population estimated with sNMF. Population abbreviations are as per Fig. 1.
The sNMF analysis including the southerly and north-western QLD populations (i.e., the N + E dataset) found a K value of two yielded the lowest cross-entropy (Fig. S2) and showed the southern populations had mainly QLD and little or no NTfly ancestry (Fig. 3a). However the DAPC analysis also showed that the three most inland southerly populations, Griffith, Ardlethan and Shepparton (GR, AR and SH respectively) were somewhat further separated from the nNT/nWA group than the rest (Fig. 3b). The DAPC analysis also showed the two most inland northern populations, Mt. Isa (MI) and Cloncurry (CL), had mostly QLD ancestry but with greater admixture with nNT/nWA than many of the populations to their east (Fig. 3).

Analysis of the Qfly expanded range populations. (A) Estimates of admixture proportions inferred with sNMF for the Qfly and NTfly native and Qfly expanded range dataset (N + E) with the best supported number of ancestral populations (K = 2). (B) DAPC of individuals within the Qfly and NTfly native and Qfly expanded dataset. Colours are representative of the admixture proportions of every individual estimated with sNMF. Population abbreviations are as per Fig. 1.
Consistent with a genetic structure suggestive of isolation by distance, a Mantel test of genetic vs geographic distance between all pairs of populations in the N + E dataset yielded a significant positive correlation with geographic distance (Mantel’s r = 0.37, P = 0.001). However, the redundancy analysis (RDA; Fig. S4) showed this only explained 7.6% of the population differences, equivalent to a value of Nei’s D of 0.01 (F7, 283 = 3.31, P = 0.001). Most of the spatial effect was related to longitude (RDA1 = 61%, F1, 283 = 14.17, P < 0.001), which is consistent with the distinction between the QLD and nNT/nWA ancestry groups. However, the low overall explanatory value of distance indicates there is also significant gene flow unrelated to distance and probably facilitated by horticultural trade among the native range and contiguous expansion populations.
Origins of the disjunct populations
DAPC and sNMF analyses including all 35 contemporary (i.e. N + E + D) samples identified the five geographically disjunct populations as genetically distinct (Fig. 4). The three Pacific Island populations were separated on both DA1 and DA2, with New Caledonia (NC) and Loyalty Island (LI) most similar, and with Tahiti (TH) as their next nearest neighbour, although Tahiti was even more divergent from all mainland populations. The two disjunct Australian populations, Broome (BM) and Alice Springs (AS), were also well separated from the other Australian samples in the DAPC plot, but they were also well separated from each other. The sNMF analysis identified the lowest cross-entropy for a K value of five (Fig. S2), corresponding to ancestry groupings of QLD and nNT/nWA as identified above, as well as the Pacific Islands (as a cluster), Alice Springs and Broome.

Analysis of the disjunct populations. (A) Estimates of admixture proportions inferred with sNMF for the native, expanded, and disjunct dataset (N + E + D) with the best supported number of ancestral populations (K = 6). (B) DAPC of individuals within the native, expanded, and disjunct dataset (N + E + D). Colours are representative of the admixture proportions of every individual estimated with sNMF. Population abbreviations are as per Fig. 1. Also see Fig. S5 for an IQ-tree phylogeny of all individuals in the expansion and disjunct datasets.
A TreeMix maximum likelihood tree of all 35 contemporary samples, using B. neohumeralis as the outgroup (Fig. 5), had little resolution (i.e. low branch length/drift parameters) among the native range and expansion populations (nor did an IQ-tree maximum likelihood tree of all N + E + D individuals; Fig. S5). TreeMix placed the northern nNT/nWA populations together with one another but separated from the QLD and expansion populations by somewhat higher drift parameters. The five disjunct populations were all separated by relatively high drift parameters from all the other populations above. Broome apparently derived from its geographically most proximal population, the nNT/nWA group, although the source of the Alice Springs population remains unclear (Fig. 5; Fig. S5). Finally, the three Pacific Island populations were closest relatives of each other and joined by a long branch to populations from the Australian east coast, with Tahiti having drifted further than the other two.

TreeMix analysis for the origin of the contiguous expansion and disjunct populations. The maximum-likelihood tree inferred with TreeMix depicts phylogenetic relationships of the 35 populations in the N + E + D dataset with B. neohumeralis as an outgroup. Colours are representative of the admixture proportions of every individual estimated with sNMF, as in Fig. 4. Population abbreviations are as per Fig. 1.
The fixed difference analysis, testing for populations with high differentiation in the contemporary samples (N + E + D), found no populations or groups of populations were distinguished by a statistically significant number of fixed differences (OUT = 1, P > 0.05, Table S1). Shannon diversity statistics and heterozygosity values were also relatively low, the latter particularly for the disjunct populations (Table S2). Fst values were likewise low across all pairwise comparisons (mean Fst value of 0.09), albeit slightly higher for comparisons involving disjunct populations (Table S3). These results confirm the relatively high levels of genetic similarity across the native range and expansion populations, and further suggest that the disjunct populations are recent invasions from small founding populations.
Temporal variation
The six populations from which we had both contemporary and 1994–2003 collections included three nNT/nWA and two QLD ancestry groups, plus the disjunct Alice Springs population. DAPC and sNMF analyses of the 138 individuals whose sequences survived the filtering in these six population pairs (Fig. 6) showed little change in population structure and differentiation over the last 15–22 years. An AMOVA indicated that 91% of the variance was explained by geographic differentiation (\(\phi =\) 0.08, P < 0.0001) but only about 1% by temporal changes, although the latter was still statistically significant (\(\phi =\) 0.01, P < 0.001). Such temporal changes as were found were largely in Darwin (Fst = 0.017, P < 0.001) and Cairns (Fst = 0.014, P < 0.001); in the latter the ancestry group principally associated with Alice Springs was more common in the older samples. More notably, both the broad nNT/nWA vs QLD and Alice Springs vs the rest distinctions were seen in both sampling periods. While the fixed difference analyses and Shannon diversity statistics suggest relatively high levels of genetic similarity among contiguous populations and relatively recent origins of the disjunct populations, the temporal data thus indicate that the nNT/nWA vs QLD distinction and the uniqueness of the Alice Springs population have changed little in the last 15–22 years.

Analysis of temporal variation in six Qfly populations. (A) Estimates of admixture proportions inferred with sNMF for the historical and contemporary populations groups with the best supported number of ancestral populations (K = 3). (B) DAPC of individuals from the historical and contemporary population groups. Colours are representative of the admixture proportions of every individual estimated with sNMF. Population abbreviations are as per Fig. 1.
Discussion
There is little genetic differentiation across the native and expanded Qfly range down the east coast of Australia, but larger differences are evident along an east/west transect across the north of Australia. These results concur with the pattern of differences obtained in earlier allozyme19 and microsatellite studies17. As noted, some authors have suggested the existence of two distinct taxa in northern Australia, B. tryoni in the east and B. aquilonis in the northern parts of the Northern Territory and northwest Western Australia. Cameron et al.17 suggested their microsatellite data could reflect recent gene flow between those taxa, aided by increased production and trade of horticultural commodities across northern Australia18. The fact that our Darwin samples show more similarity to the east coast populations than do other northern NT populations supports the notion that trade contributed to the introgression. The absence of significant numbers of fixed differences even between the most widely separated populations along the transect suggests that the taxonomic distinction might not have amounted to species status, at least in recent times. However, research on ecotypic variation15 did not include any minimally introgressed B. aquilonis (the most B. aquilonis-like collection in that study being Darwin), so it remains possible that the populations further west than those in our analysis are ecologically distinct from B. tryoni. There are several examples in the literature whereby insect species have retained ecotypic distinction in the face of quite high levels of genetic introgression41,42. The east/west transect covered here crosses a number of biogeographical barriers which have led to differentiation between closely related species of amphibians43, birds44,45,46, other insects47,48, mammals49 and plants50 across different geological periods51,52.
Populations from the southerly Qfly expansion down the east coast show little genetic differentiation from the north coast populations, except for progressively less evidence of nNT/nWA genotypes. This fits with an expectation53, 54 that these southerly coastal populations would have derived from the adjacent native Qfly range rather than the more distant nNT/nWA.
Although the differences are not large, the three more interior southern expansion populations, Griffith, Ardlethan and Shepparton, are all somewhat displaced from their coastal counterparts and more distinct from the nNT/nWA genotypes in the DAPC plot. These populations are in regions previously classified as ecologically marginal for the Qfly8, and are the closest in our sample to a region previously recognised as a Fruit Fly Exclusion Zone (FFEZ)55 (Fig. 1). The FFEZ covered the Murrumbidgee Irrigation Area of NSW and adjacent Riverland areas of South Australia, Victoria and NSW, but all except the South Australian Riverland lost its FFEZ designation in July 2013 because of the growing Qfly populations immediately adjacent to it, and increasingly frequent Qfly outbreaks within it55. Microsatellite studies showed that those incursions were generally from the adjacent populations53,54,56. Given our evidence that these adjacent populations show some differentiation from Qflies elsewhere, the intensive Sterile Insect Technique (SIT) control programmes now under way in what was the FFEZ57,58 may benefit from using the adjacent populations as source material for their mass release strains.
Our data for the three disjunct Melanesian populations support an island-hopping model in which flies migrated from the east coast of Australia into New Caledonia and Loyalty Island (~ 1,600 and 1,800 km east, respectively), which in turn became the source of migrants to the more distant Tahiti (~ 6,400 km further east)22. Thus our data show a progressive reduction of genetic diversity (Tables S2, S3) and increasing differentiation from their Australian ancestors (Fig. 4) during this process.
A similar loss of genetic diversity and clear differentiation is seen in one of the disjunct Australian populations, Broome, in the far northwest of Australia, but in this case the most likely source was the nNT/nWA population (~ 1,130 km east and separated by the Kimberley range). Our data did not identify the source of the other disjunct Australian population, Alice Springs, in central Australia. Notwithstanding its isolation it is not deficient in genetic diversity. This is evident in our temporal analysis, which shows the genetic composition of the Alice Springs population has changed very little in the 15 years between samples, suggesting its founding population was large enough to avoid bottleneck issues and there has been little immigration since.
Given that all five of the disjunct populations show a level of genetic differentiation (i.e., high drift parameter values) indicative of low gene flow with other populations, they could all be good candidates for local eradication programs based on SIT.
We can compare our evidence for genetic differentiation across the Qfly/NTfly range with the data of Popa-Báez et al.15 for ecotypic differentiation in relation to abiotic stress resistance. They only studied 12 populations, mostly from the east coast of Australia but also including Darwin, Alice Springs and Griffith. Darwin and Alice Springs were found to be distinguished by relatively high heat resistance, and Griffith by relatively high desiccation resistance, all of which is consistent with the genetic differentiation from the east coast populations evident in the present study. Among their ten east coast populations, however, Cape Tribulation (CT, their most northerly population) and Sydney (SY) were found to have relatively high desiccation resistance, and Sydney relatively high heat resistance as well, which would not have been predicted from our data. Furthermore, a separate sample of the Sydney population was collected and tested two years later for desiccation resistance and again found to have relatively high resistance, suggesting it was a stable, inherited feature of the population15. While our spatial analysis is based on over 2,700 SNPs taken randomly from across the genome, it can clearly miss phenotypically important genetic differences whose distribution across the species range departs from those seen from the overall trends evident in the summary statistics from our data.
It is interesting to compare the results presented in this study and Popa-Báez et al.15 with those of Qin et al.59 on B. dorsalis, another widely distributed tephritid pest. They screened for differences in four taxa previously recognised as different species from a wide range of locations in Africa, Asia and Hawaii, using microsatellite and morphometric (wing length) analysis. Their microsatellite work revealed differences between the African and Hawaiian samples on one hand and the east Asian populations on the other, with only minor differences in wing length among their samples. In contrast, the stronger evidence of genetic differentiation and ecotypic variation evident in the current study and Popa-Báez et al.15 might in part reflect a greater power of our DArT data and the multiple stress response traits measured by Popa-Báez et al.15, but it could also indicate a stronger tendency for local differentiation in Qfly.
Data availability
The SNPs data and scripts used in this study are available in the Figshare Population genomics of the Queensland fruit fly repository (https://doi.org/10.6084/m9.figshare.11390043.v1) and GitHub repository (https://github.com/Angel-Popa/Qfly_pop-gen), respectively.
References
Clarke, A. R., Powell, K. S., Weldon, C. W. & Taylor, P. W. The ecology of Bactrocera tryoni (Diptera: Tephritidae): what do we know to assist pest management? Ann. Appl. Biol. 158, 26–54 (2011).
Sultana, S., Baumgartner, J. B., Dominiak, B. C., Royer, J. E. & Beaumont, L. J. Impacts of climate change on high priority fruit fly species in Australia. PLoS ONE 15, e0213820 (2020).
May, A. W. S. New species and records of Dacinae (Diptera: Trypetidae) from northern Australia. Aust. J. Entomol. 4, 58–66 (1965).
Osborne, R. et al. Australian distribution of 17 species of fruit flies (Diptera: Tephritidae) caught in cue lure traps in February 1994. Aust. J. Entomol. 36, 45–50 (1997).
Dominiak, B. C. & Daniels, D. Review of the past and present distribution of Mediterranean fruit fly (Ceratitis capitata Wiedemann) and Queensland fruit fly (Bactrocera tryoni Froggatt) in Australia. Aust. J. Entomol. 51, 104–115 (2012).
Froggatt, W. W. Report on parasitic and injurious insects 1907–8. Part III. In Fruit Flies: A General Account of the Flies Belonging to the Family Trypetidae (New South Wales Department of Agriculture, 1909).
May, A. W. S. An investigation of fruit flies (Trypetidae: Diptera) in Queensland. 1. Introduction, species, pest status and distribution. Qld. J. Agric. Sci. 20, 1–82 (1963).
Yonow, T. & Sutherst, R. W. The geographical distribution of the Queensland fruit fly, Bactrocera (Dacus) tryoni, in relation to climate. Aust. J. Agric. Res 49, 935–953 (1998).
Sultana, S., Baumgartner, J. B., Dominiak, B. C., Royer, J. E. & Beaumont, L. J. Potential impacts of climate change on habitat suitability for the Queensland fruit fly. Sci. Rep. 7, 13025 (2017).
Muthuthantri, W. S. N. Population Phenology of the Tropical Fruit Fly, Bactrocera tryoni (Froggatt) (Diptera: Tephritidae), in Queensland, Australia. M.Sc. thesis, Queensland University of Technology (2008).
Meats, A. Rapid acclimation to low temperature in the Queensland fruit fly, Dacus tryoni. J. Insect Physiol. 19, 1903–1911 (1976).
Meats, A. Critical periods for developmental acclimation to cold in the Queensland fruit fly, Dacus tryoni. J. Insect Physiol. 29, 943–946 (1983).
Fletcher, B. S. The ecology of a natural population of the Queensland fruit fly, Dacus tryoni. IV The immigration and emigration of adults. Aust. J. Zool. 21, 541–565 (1973).
Clarke, A. R., Merkel, K., Hulthen, A. D. & Schwarzmueller, F. Bactrocera tryoni (Froggatt) (Diptera: Tephritidae) overwintering: an overview. Austral Entomol. 58, 1–6 (2018).
Popa-Báez, A. D. et al. Climate stress resistance in male Queensland fruit fly varies among populations of diverse geographic origins and changes during domestication. BMC Genetics (in press).
Yu, H. et al. Microsatellite analysis of the Queensland fruit fly Bactrocera tryoni (Diptera: Tephritidae) indicates spatial structuring: implications for population control. Bull. Entomol. Res. 91, 139–147 (2001).
Cameron, E. C., Sved, J. A. & Gilchrist, A. S. Pest fruit fly (Diptera: Tephritidae) in northwestern Australia: one species or two?. Bull. Entomol. Res. 100, 197–206 (2010).
Allwood, A. J. & Angeles, T. S. Host records of fruit flies (family Tephritidae) in the Northern Territory. Qld. J. Agric. Anim. Sci. 36, 105–113 (1979).
Drew, R. A. I. & Lambert, D. M. On the specific status of Dacus (Bactrocera) aquilonis and D. (Bactrocera) tryoni (Diptera: Tephritidae). Ann. Entomol. Soc. Am. 79, 870–878 (1986).
Drew, R. A. I. The tropical fruit flies (Diptera: Tephritidae: Dacinae) of the Australasian and Oceanian regions. Mem. Qld. Museum 26, 1–521 (1989).
Wang, Y., Yu, H., Raphael, K. & Gilchrist, A. S. Genetic delineation of sibling species of the pest fruit fly Bactrocera (Diptera: Tephritidae) using microsatellites. Bull. Entomol. Res. 93, 351–360 (2003).
Vargas, R. I., Piñero, J. C. & Leblanc, L. An overview of pest species of Bactrocera fruit flies (Diptera: Tephritidae) and the integration of biopesticides with other biological approaches for their management with a focus on the Pacific region. Insects 6, 297–318 (2015).
Kahle, D. & Wickham, H. ggmap: spatial visualization with ggplot2. R J. 5, 144–161 (2013).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer, New York, 2016).
R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.r-project.org/ (2018).
Kilian, A. et al. Diversity arrays technology: A generic genome profiling technology on open platforms. In Data Production and Analysis in Population Genomics (eds Pompanon, F. & Bonin, A.) 67–89 (Humana Press, Totowa, 2012).
Georges, A. et al. Genomewide SNP markers breathe new life into phylogeography and species delimitation for the problematic short-necked turtles (Chelidae: Emydura) of eastern Australia. Mol. Ecol. 27, 5195–5213 (2018).
Gruber, B., Georges, A., Berry, O. & Unmack, P. dartR: Importing and analysing SNP and silicodart data generated by genome-wide restriction fragment analysis. R package version 1.3.3 https://cran.r-project.org/web/packages/dartR/index.html (2019).
Jombart, T. et al. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet. 11, 94 (2010).
Jombart, T. adegenet: an R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Frichot, E. & François, O. LEA: an R package for landscape and ecological association studies. Methods Ecol. Evol. 6, 925–929 (2015).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Pembleton, L. W., Cogan, N. O. I. & Forster, J. W. StAMPP: an R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 13, 946–952 (2013).
Oksanen, J. et al. vegan: community ecology package. R package version 2.5–3 https://cran.r-project.org/package=vegan (2019).
Meirmans, P. G. Seven common mistakes in population genetics and how to avoid them. Mol. Ecol. 24, 3223–3231 (2015).
McVean, G. A genealogical interpretation of principal components analysis. PLoS Genet. 5, e1000686 (2009).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012).
Pickrell, J. K. & Pritchard, J. K. TreeMix. 22/11/16 https://bitbucket.org/nygcresearch/treemix/wiki/Home (2016).
Sherwin, W. B., Chao, A., Jost, L. & Smouse, P. E. Information theory broadens the spectrum of molecular ecology and evolution. Trends Ecol. Evol. 32, 948–963 (2017).
Excoffier, L., Smouse, P. E. & Quattro, J. M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitocondrial DNA restriction data. Genetics 131, 479–491 (1992).
Anderson, C. J. et al. Hybridization and gene flow in the mega-pest lineage of moth, Helicoverpa. Proc. Natl. Acad. Sci. 115, 5034–5039 (2018).
Thurman, T. J., Szejner-Sigal, A. & McMillan, W. O. Movement of a Heliconius hybrid zone over 30 years: a Bayesian Approach. J. Evol. Biol. 32(9), 974–983 (2019).
Catullo, R. A., Lanfear, R., Doughty, P. & Keogh, J. S. The biogeographical boundaries of northern Australia: evidence from ecological niche models and a multi-locus phylogeny of Uperoleia toadlets (Anura: Myobatrachidae). J. Biogeogr. 41, 659–672 (2014).
Peñalba, J. V., Joseph, L. & Moritz, C. Current geography masks dynamic history of gene flow during speciation in northern Australian birds. Mol. Ecol. 28, 630–643 (2019).
Ford, J. Hybrid zones in Australian birds. Emu-Austral Ornithol. 87, 158–178 (1987).
Cracraft, J. Origin and evolution of continental biotas: Speciation and hitorical congruence within the australian avifauna. Evolution 40, 977–996 (1986).
Braby, M. F. Biogeography of butterflies in the Australian monsoon tropics. Aust. J. Zool. 56, 41 (2008).
Cook, L. G. & Gullan, P. J. The gall-inducing habit has evolved multiple times among the eriococcid scale insects (Sternorrhyncha: Coccoidea: Eriococcidae). Biol. J. Linn. Soc. 83, 441–452 (2004).
Eldridge, M. D. B., Potter, S., Johnson, C. N. & Ritchie, E. G. Differing impact of a major biogeographic barrier on genetic structure in two large kangaroos from the monsoon tropics of Northern Australia. Ecol. Evol. 4, 554–567 (2014).
Chappill, J. A., Wilkins, C. F. & Crisp, M. D. Taxonomic revision of Jacksonia (Leguminosae: Mirbelieae). Aust. Syst. Bot. 20, 473 (2007).
Edwards, R. D., Crisp, M. D., Cook, D. H. & Cook, L. G. Congruent biogeographical disjunctions at a continent-wide scale: Quantifying and clarifying the role of biogeographic barriers in the Australian tropics. PLoS ONE 12, e0174812 (2017).
Bowman, D. M. J. S. et al. Biogeography of the Australian monsoon tropics. J. Biogeogr. 37, 201–216 (2010).
Gilchrist, A. S. & Meats, A. W. The genetic structure of populations of an invading pest fruit fly, Bactrocera tryoni, at the species climatic range limit. Heredity. 105, 165–172 (2010).
Sved, J. A., Yu, H., Dominiak, B. & Gilchrist, A. S. Inferring modes of colonization for pest species using heterozygosity comparisons and a shared-allele test. Genetics 163, 823–831 (2003).
Dominiak, B. C. & Mapson, R. Revised distribution of Bactrocera tryoni in Eastern Australia and effect on possible incursions of Mediterranean fruit fly: development of Australia’s Eastern trading block. J. Econ. Entomol. 110, 2459–2465 (2017).
Gilchrist, A. S., Dominiak, B., Gillespie, P. S. & Sved, J. A. Variation in population structure across the ecological range of the Queensland fruit fly, Bactrocera tryoni. Aust. J. Zool. 54, 87–95 (2006).
Horticulture Innovation Australia. Establishment of the Qfly SITplus facility in southern Australia (HG14035). https://www.horticulture.com.au/growers/help-your-business-grow/research-reports-publications-fact-sheets-and-more/hg14035/ (2018).
Horticulture Innovation Australia. SITplus: Port Augusta Qfly SIT factory pilot operation (FF15000). https://www.horticulture.com.au/growers/help-your-business-grow/research-reports-publications-fact-sheets-and-more/ff15000/ (2019).
Qin, Y. J. et al. Population structure of a global agricultural invasive pest, Bactrocera dorsalis (Diptera: Tephritidae). Evol. Appl. 11, 1990–2003 (2018).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2014).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q. & Vinh, L. S. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2017).
Yu, G., Smith, D. K., Zhu, H., Guan, Y. & Lam, T. T. Y. ggtree: an R Package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 8, 28–36 (2017).
Acknowledgements
Many samples used in this work were collected as part of an earlier study15. For the additional collections used here we thank: Ian Lacey, Bridget Henshaw and Bill Woods (Department of Agriculture and Food Western Australia) for helping us collect samples in Broome and Kununurra; Rodney Freedman and Brian Thistleton (Northern Territory Department of Primary Industries and Resources) for helping us collect samples in Darwin, Katherine and Mataranka; Peter Bidgood and Brian Thistleton for helping us collect samples in Alice Springs; Cindy Morgan (Roper Gulf Regional Council) for helping us locate samples in Borroloola; David Britton and Isarena Schneider (Northern Australia Quarantine Strategy), and Jane Royer (Queensland Department of Agriculture and Fisheries) for collecting samples in the Cape York Peninsula and northern Queensland; Dr Christian Mille and Sylvie Cazeres (Institut Agronomique néo-Calédonien, New Caledonia) for collecting samples in New Caledonia and Loyalty Island; and Dr Julie Grandgirard (Direction de l’Agriculture, Tahiti) for collecting samples in Tahiti. This research was conducted as part of the SITplus collaborative fruit fly program, specifically a component of the project Raising Q-fly Sterile Insect Technique to World Standard (HG14033) funded by the Hort Frontiers Fruit Fly Fund (part of the Hort Frontiers strategic partnership initiative developed by Hort Innovation), with co-investment from Macquarie University and contributions from the Australian Government. We also acknowledge CSIRO for its generous support of this project.
Author information
Authors and Affiliations
Contributions
A.D.P.B.: study design, data collection, analysis and interpretation, co-wrote the manuscript; R.C.: study design, analysis and interpretation, co-wrote the manuscript; S.F.L.: study design, data collection, analysis and interpretation, co-wrote the manuscript; H.L.Y.: data collection and analysis; R.G.M.: data collection; M.F.: data collection; J.A.S.: data collection; E.C.C.: data collection; O.R.E.: study design and interpretation; P.W.T.: study design and interpretation; J.G.O.: study design, data analysis and interpretation, co-wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Popa-Báez, ÁD., Catullo, R., Lee, S.F. et al. Genome-wide patterns of differentiation over space and time in the Queensland fruit fly. Sci Rep 10, 10788 (2020). https://doi.org/10.1038/s41598-020-67397-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-67397-5
This article is cited by
-
Genomic signals of local adaptation across climatically heterogenous habitats in an invasive tropical fruit fly (Bactrocera tryoni)
Heredity (2024)
-
The blowfly Chrysomya latifrons inhabits fragmented rainforests, but shows no population structure
Oecologia (2023)
-
Using PVA and captive breeding to balance trade-offs in the rescue of the island dibbler onto a new island ark
Scientific Reports (2022)
-
Tracing the origins of recent Queensland fruit fly incursions into South Australia, Tasmania and New Zealand
Biological Invasions (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
