
Abstract
Prion diseases comprise a fatal neuropathy caused by the conversion of prion protein from a cellular (PrPC) to a pathological (PrPSc) isoform. Previously, we obtained an RNA aptamer, r(GGAGGAGGAGGA) (R12), that folds into a unique G-quadruplex. The R12 homodimer binds to a PrPC molecule, inhibiting PrPC-to-PrPSc conversion. Here, we developed a new RNA aptamer, r(GGAGGAGGAGGAGGAGGAGGAGGA) (R24), where two R12s are tandemly connected. The 50% inhibitory concentration for the formation of PrPSc (IC50) of R24 in scrapie-infected cell lines was ca. 100 nM, i.e., much lower than that of R12 by two orders. Except for some antibodies, R24 exhibited the lowest recorded IC50 and the highest anti-prion activity. We also developed a related aptamer, r(GGAGGAGGAGGA-A-GGAGGAGGAGGA) (R12-A-R12), IC50 being ca. 500 nM. The structure of a single R12-A-R12 molecule determined by NMR resembled that of the R12 homodimer. The quadruplex structure of either R24 or R12-A-R12 is unimolecular, and therefore the structure could be stably formed when they are administered to a prion-infected cell culture. This may be the reason they can exert high anti-prion activity.
Similar content being viewed by others


Introduction
Prion diseases are fatal neurodegenerative disorders including Creutzfeldt–Jakob disease in humans, bovine spongiform encephalopathy in cattle, scrapie in sheep and goats, and other diseases1,2,3,4,5,6. Many studies revealed that the conformational transition of prion protein (PrP) from a normal cellular form (PrPC) into an abnormal form (PrPSc) is a key event in the pathogenesis of prion diseases. Although the mechanism of transition from PrPC to PrPSc and the structure of PrPSc remain unknown, the structure of PrPC was used for development of drugs against prion diseases. It was reported that some antibodies7, nanobody8, aptamers9,10 and chemical compounds11,12,13,14,15,16,17 against PrPC successfully reduced the amount of PrPSc in cells persistently infected with the transmissible spongiform encephalopathy (TSE) agent.
Previously we reported that an anti-PrPC aptamer, r(G1G2A3G4G5A6G7G8A9G10G11A12) (R12), tightly binds to PrPC and reduces the PrPSc level in mouse neuronal cells persistently infected with the human TSE agent18,19,20. It was revealed that R12 binds to two portions of bovine PrP (bPrP), P1 (residues 25–35 of bPrP) and P16 (residues 108–119 of bPrP), in the N-terminal intrinsically disordered region of PrP19. We also determined the structure of R12 in a complex with the binding peptide P16 (GQWNKPSKPKTN) to elucidate the mechanism by which R12 exhibits high affinity20,21. It was revealed that R12 folds into a unique quadruplex structure composed of a G:G:G:G tetrad plane and a G(:A):G:G(:A):G hexad plane, and that two R12 molecules bind in a tail-to-tail manner to form a homodimer. It was found that the R12 homodimer simultaneously binds to two portions of one PrPC molecule (Fig. 1A), resulting in tight binding and stabilization of PrPC20,21. Here, we developed a new RNA aptamer, r(G1G2A3G4G5A6G7G8A9G10G11A12G13G14A15G16G17A18G19G20A21G22G23A24) (R24), containing two tandem R12 sequences. R24 was supposed to unimolecularly form a similar structure made of two R12 molecules. Therefore, under the conditions applied for the assay with prion-infected cells, it was expected that the structure of R24 is stably formed and that R24 exerts anti-prion activity to block the pathological conformational conversion of PrP. In fact, R24 showed much higher anti-prion activity than R12. Furthermore, another related aptamer, r(G1G2A3G4G5A6G7G8A9G10G11A12-A13-G14G15A16G17G18A19G20G21A22G23G24A25) (R12-A-R12), was also supposed to unimolecularly form a similar structure composed of two R12 molecules. It was revealed that R12-A-R12 also exhibited much higher anti-prion activity than R12, as expected. Then, we determined the structure of R12-A-R12 by NMR and deduced the mode of interaction of R12-A-R12 with PrP. These studies confirmed the origin of the high anti-prion activity of R12-A-R12 and R24, and provided further insight. The information on the sequences of potent aptamers and the structural bases of their anti-prion activity revealed in this study may be used to develop aptamer-based drugs against prion diseases.

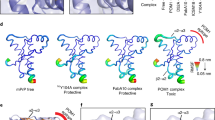
Schematic models of RNA aptamers. (A) R12 homodimer in complex with PrPC 20. The structure of the C-terminal region of bPrP was drawn by using the coordinates of accession number 1DX0 of the Protein Data Bank42. (B,C) Two candidate architectures of R24. It is assumed that the A12 residue links two quadruplexes in a back to front (B) or a back to back manner. (C) The two-fold symmetrical axis that correlates two quadruplexes at the top and bottom is shown for each structure, respectively.
Results and Discussion
Structure-based development of RNA aptamers having higher anti-prion ability to block the pathological conformational conversion of PrP than the R12 homodimer
We attempted to develop an RNA aptamer that exhibits higher anti-prion ability to block the pathological conformational conversion of PrP than R12 on the basis of the structure of R12. The unique quadruplex structure of a R12 monomer and its dimeric architecture are essential for tight binding to PrPC20. When R12 is dissolved in the cell culture for the assay, however, the R12 homodimer architecture may be destabilized, resulting in a decrease in the activity. Therefore, we tandemly connected two R12 monomers and obtained R24. R24 is expected to unimolecularly form a structure that resembles the structure formed by two R12 molecules. Two candidate structures were assumed for the overall architecture of R24 (Fig. 1B,C). In both cases, the positions of G1 - G11 of R24 are the same as those in the top R12 molecule of the R12 homodimer (Fig. 1A). In the Fig. 1B model, the positions of G13 - A24 of R24 are the same as those of G1 - A12 in the bottom R12 molecule of the R12 homodimer (Fig. 1A), as a result of the linkage of the two quadruplex units in a back side to front side manner via the A12 residue. On the other hand, in the Fig. 1C model, the A12 residue links the two quadruplexes in a back side to back side manner. The two-fold symmetrical axis that correlates two quadruplex units is shown in Fig. 1B,C, respectively. It should be noted that the latter architecture is the same as that found for d(GGA)8, whose structure was determined previously22. The real structure of R24 will be discussed later.
The anti-prion activity of R24 was examined using mouse neuronal cells (GT1-7) persistently infected with the human TSE agent (Fukuoka-1 strain), which was designated as GT + FK12,15,17,23. Either R24, an anti-prion drug, GN8 (positive control), or a solution containing 100 mM KCl and 10 mM K-phosphate (pH 6.2) (negative control) was added to the cell culture containing GT + FK12. R24 was found to significantly reduce the PrPSc level (Fig. 2A). 5 μM R24 added to the cell culture almost completely abolished the formation of PrPSc (Fig. 2A). Actually, the amount of PrPSc with 5 μM R24 was approximately 1% of that in the control. The 50% inhibitory concentration (IC50) for the formation of PrPSc of R24 was 100 nM (Supplementary Fig. S1A and Table 1). For R12, it was administered to the cell culture to a final concentration of 10 μM. Then, it turned out that the PrPSc level was reduced to 49 ± 17% of the control, that is to say, nearly 50% of the control. This suggests that the IC50 value of R12 is ca. 10 μM (Table 1). Thus, it is indicated that R24 possesses much higher, ca. 100-fold higher, anti-prion activity than R12. Furthermore, except for some antibodies7, R24 exhibited the lowest recorded IC50 determined by the prion-infected cell-based assay among all anti-prion materials involving nanobody8, RNA/DNA aptamers9,10, and chemical compounds11,12,13,14,15,16,17.

Anti-prion activity of R24 and R12-A-R12. Western blotting of PrP in GT + FK cells after the addition of various concentrations of either R24 (A) or R12-A-R12. (B) Lane 1 is a negative control, in which a buffer solution containing 100 mM KCl and 10 mM K-phosphate (pH 6.2) was added to the cell culture. Lane 2 is a positive control, in which 1.4 μM GN8, an anti-prion drug, was added. The experiments were carried out once for R24 and three times for R12-A-R12, respectively.
The dissociation constant of the R12-bPrP complex was examined by filter binding assay18,20 and determined to be 18 nM21. The dissociation constant of the R24-bPrP complex was examined in the same way and determined to be 61 nM. The magnitude of the dissociation constant turns out to be the same order of 10−8 M for both R12 and R24.
We attempted structural analysis of R24 to confirm whether its structure resembles that of the R12 homodimer, as we expected. The imino proton signals of R24 were observed at 10.8–12 ppm (Fig. 3B), indicating the formation of a quadruplex. However, the quality of the NMR spectrum was not good enough for detailed structural analysis. To overcome this, we carried out screening to find an RNA that shows well-resolved imino proton signals among a series of RNAs with modified R24 sequences (Supplementary Table S1 and Fig. S2). As a result, an RNA sequence, R12-A-R12, was found to give a high-quality NMR spectrum (Fig. 3C). We then carried out an anti-prion assay using R12-A-R12, which demonstrated that R12-A-R12 can also reduce the PrPSc level significantly (Fig. 2B), with an IC50 value of ca. 500 nM (Supplementary Fig. S1B and Table 1). The IC50 of R12-A-R12 is of the same order of magnitude as that of R24, 10−7 M. Thus, it turned out that the potency of R12-A-R12 is also high. Therefore, we decided to carry out the structural analysis of R12-A-R12 as a representative RNA aptamer that exhibits much higher anti-prion activity than R12. It might also be a case that since the sequence of R12-A-R12 is almost the same as that of R24, the overall architecture of R12-A-R12 is expected to be similar, if not the same, as that of R24.

Imino proton spectra of anti-PrPC aptamers: (A) r(GGAGGAGGAGGA) (R12), (B) r(GGAGGAGGAGGAGGAGGAGGAGGA) (R24) and (C) r(GGAGGAGGAGGA-A-GGAGGAGGAGGA) (R12-A-R12). Spectra were recorded in H2O solvent at 15 °C. The assignments are indicated by residue number.
The structure determination of R12-A-R12 by NMR
To confirm that R12-A-R12 folds into a unimolecular structure that resembles the structure of the R12 homodimer, we performed NMR analysis of R12-A-R12. First of all, the line width for the structure of R12-A-R12 is almost the same as that for the homodimer structure of R12 (Fig. 3A,C). This indicates that the molecular weight of the structure of R12-A-R12 is almost the same as that of the homodimer structure of R12 comprising 24 residues in total. As R12-A-R12 comprises 25 residues, it was revealed that the structure of R12-A-R12 is a unimolecular one and that R12-A-R12 does not form a dimer. The resonance assignment was conducted in the same way as reported for R1219,20. The nonexchangeable protons were assigned by NOESY, TOCSY, and DQF-COSY experiments. Figure 4A shows sequential NOESY connectivities for fingerprint base proton/H1′. Imino protons were assigned by means of a JRHMBC experiment that gave NH (imino proton)-C5 and H8-C5 correlation peaks for each guanine base (G)24. The characteristic interresidue NOEs between GNH (NH of G)/GNH2 (amino protons of G) and GH8 (H8 of G), and those between GNH and GNH established the formation of four G:G:G:G tetrad planes for G2:G5:G8:G11, G15:G18:G21:G24, G1:G4:G7:G10 and G14:G17:G20:G23 (Fig. 4B,C). Since the latter two tetrad planes show NOEs between GNH/GNH2 and AH8 (H8 of adenine base (A)), and GNH2 and ANH2 (amino protons of A), the formation of G1(:A3):G4:G7(:A9):G10 and G14(:A16):G17:G20(:A22):G23 hexad planes was confirmed.

Assignments and identification of tetrad and hexad planes, together with the mode of hexad-hexad stacking, of R12-A-R12. (A) The fingerprint base proton/H1′ region of the NOESY spectrum in D2O solvent recorded with a mixing time of 400 ms at 15 °C. (B,C) Imino-imino (B) and imino-H8 (C) cross peaks of the NOESY spectrum in H2O solvent recorded with a mixing time of 400 ms at 5 °C. The cross peaks between the tetrad and hexad planes are underlined and those between two hexad planes are double-underlined. (D) The elucidated overall architecture of R12-A-R12.
The interresidue AH2 (H2 of A)-AH1′ (H1′ of A) and AH2-AH8 NOEs between A3 and A22, and A9 and A16 suggest that the G1(:A3):G4:G7(:A9):G10 hexad plane stacks on the G14(:A16):G17:G20(:A22):G23 hexad plane, as shown in Fig. 4D. Additionally, this mode of stacking of two hexad planes is supported by identification of NOEs between G1 and G23, G4 and G20, G7 and G17, and G10 and G14.
The structure of R12-A-R12 was calculated on the basis of NMR restraints obtained as described previously19,22 (Supplementary Table S2) by using the XPLOR-NIH program, as for R1219,20. Ten of the obtained lowest energy structures were further refined by NMR-based molecular dynamics (MD) simulations, which utilize NMR restraints with explicit water molecules by using the AMBER 16 program. The statistics for the ten refined structures are listed in Supplementary Table S2. The refined structure derived from the structure with the lowest XPLOR-NIH energy was selected as a representative structure. The structure of R12-A-R12 is shown in Fig. 5. R12-A-R12 comprises two tetrad and two hexad planes, whose arrangement is consistent with that shown in Fig. 4D. A6, A12, A13, A19 and A25 are not involved in either a tetrad or hexad plane. The two parallel quadruplexes within R12-A-R12 are packed in a tail-to-tail manner through stacking between two hexad planes (Figs. 4D and 5). Figure 5C–E show the stacking between planes. It should be noted that the bottom quadruplex of the R12 homodimer structure is rotated by 180° in the arrangement of the R12-A-R12 structure. Overall, it was revealed that one R12-A-R12 molecule forms a structure that resembles the structure of the R12 homodimer.

The structure of R12-A-R12. G2, G5, G8, and G11 (yellow), G1, A3, G4, A6, G7, A9, and G10 (red), G14, A16, G17, A19, G20, A22, and G23 (blue), G15, G18, G21, and G24 (green) are color coded. (A) A stereo view of the superimposed 10 refined structures. (B) The representative structure. (C) Stacking configuration of the G2:G5:G8:G11 tetrad and the G1(:A3):G4:G7(:A9):G10 hexad. (D) Stacking configuration of the G15:G18:G21:G24 tetrad and the G14(:A16):G17:G20(:A22):G23 hexad. (E) Stacking configuration of two hexads.
The deduced modes of interaction of R12-A-R12 as well as R24 with PrPC
Previously, we determined the structure of the R12 homodimer in a complex with two molecules of the binding peptide of PrP, P16, by NMR20,21. Here, we constructed a model of a single R12-A-R12 molecule in a complex with two P16 peptides. The structure of R12-A-R12 determined in this study was superimposed on the previously determined homodimer structure of R12 in a complex with two P16 peptides and then the coordinates of the R12 homodimer were removed (Fig. 6A).

Models of either R12-A-R12 or R24 in a complex with PrPC. (A) The model structure of R12-A-R12 in a complex with two P16 (residues 108–119 of bPrP: GQWNKPSKPKTN) peptides. The structure was constructed by the replacement of the atomic coordinates of two R12 molecules in the R12-P16 complex (PDB ID: 2RU7) by those of one R12-A-R12 molecule. The red tube indicates the chain connecting the C5′ atoms of R12-A-R12. The magenta and purple tubes indicate the chain connecting the Cα atoms of each P16 monomer, respectively. (B) Close-up views of the interactions between R12-A-R12 and the upper P16 in the model. The G5 and K5, A9 and K10, G11 and K8, and G8 and W3 pairs are indicated by pink, green, orange and cyan, respectively. (C) Close-up views of the interactions between R12-A-R12 and the lower P16 in the model. The G24 and K5, A16 and K10, G18 and K8, and G15 and W3 pairs are indicated by pink, green, orange and cyan, respectively. (D,E) A schematic model of either R12-A-R12 (D) or R24 (E) in a complex with PrPC.
In the case of the R12 homodimer in a complex with two P16 peptides, we have recently pointed out that the effects of hydration are crucial in the binding and as a consequence the charge and shape complementarities within the binding interface play essential roles in stabilizing the complex21. Three Lys residues, K5, K8 and K10, of the P16 peptide came in contact with the phosphate groups of the G5, G11 and A9 residues of R12, respectively, reflecting the charge complementarity20,21. The stacking of a Trp residue, W3, of the P16 peptide and the G8 residue of R12 occurred as paradigmatic part of the shape complementarity20,21. These complementarities are basically conserved in the model of R12-A-R12 in a complex with two P16 peptides as well. First, the model is characterized by the contact of K5:G5, K8:G11 and K10:A9 and the stacking of W3:G8, though the K5:G5 distance is greater (Fig. 6B). Secondly, the model shows the contact of K5:G24, K8:G18 and K10:A16 and the stacking of W3:G15, though the K5:G24 distance is greater (Fig. 6C). These results indicate that the binding mode of a single R12-A-R12 molecule and two P16 peptides is physically the same as that of the R12 homodimer and two P16 peptides.
It was shown that the R12 homodimer binds to P16 and another binding peptide of PrP, P1, with almost the same affinity20. Therefore, for the R12 homodimer, we concluded that it tightly binds to PrPC in the mode shown in Fig. 1A, resulting in the inhibition of the PrPC-to-PrPSc conversion20. It is reasonable to suppose that a single R12-A-R12 molecule tightly binds to PrPC in the mode shown in Fig. 6D, resulting in the inhibition of the PrPC-to-PrPSc conversion. The same mode is also likely for R24 (Fig. 6E). It should be added that the structure of R24 in Fig. 6E corresponds to one of the two candidate architectures discussed above, Fig. 1C.
Conclusion
In this study, we rationally designed new potent anti-PrPC aptamers, R24 and R12-A-R12, on the basis of the structure of R12 and proved their high anti-prion abilities to block the pathological conformational conversion of PrP by means of a cell-based assay.
In order to elucidate the origin of high anti-prion activity, we determined the structure of R12-A-R12 by NMR. It was revealed clearly that a single R12-A-R12 molecule forms a unique quadruplex structure that resembles that of the R12 homodimer, as intended. Then, by use of the determined structure of R12-A-R12, a model of R12-A-R12 in a complex with two molecules of binding peptide P16 of PrP was constructed on the basis of the previously determined structure of the R12 homodimer in a complex with two P16 peptides. The constructed model indicated that R12-A-R12 can interact with and trap two P16 peptides, as the R12 homodimer does. Finally, it was deduced that the mode of the specific interactions of R12-A-R12 with PrPC is similar to that of the R12 homodimer with PrPC.
Initially, we speculated that the unimolecular structure is stable even at the low RNA concentration, 10 μM, applied for the cell-based anti-prion assay, while the homodimer structure is not so stable at the low RNA concentration due to dissociation of the homodimer and that this could be a reason why RNA that can unimolecularly form the unique quadruplex structure exhibits higher anti-prion activity. However, the dissociation constant of R12-bPrP is 18 nM21. As the homodimer structure of R12 is essential for tight binding of PrP20, this result indicates that a certain amount of the homodimer structure still remains at the RNA concentration of 18 nM, which is much lower than 10 μM applied for the cell-based assay. Thus, the dissociation of the homodimer at the concentration applied for the cell-based assay cannot be a determinant to lower the anti-prion activity of R12.
When the R12 homodimer was administered to a prion-infected cell culture, the homodimer architecture of R12 may be destabilized and transiently destroyed by components in the culture. The transient destruction of the homodimer architecture may also cause the exposure of a fragile single-stranded R12 monomer to nucleases present in the cell culture and thereby degradation of the R12 molecule. The transient destruction and/or resultant degradation could cause the decrease in the anti-prion activity of R12. In contrast, the unimolecularly formed structure of R12-A-R12 is supposed to be more resistant to the transient destruction of the structure and/or resultant degradation in a cell culture, because two quadruplexes of R12-A-R12 are connected by covalent bonds. This is suggested to be the origin of the high anti-prion activity of R12-A-R12.
R24 exhibited even higher anti-prion activity. Except for some antibodies7, R24 exhibited the lowest recorded IC50 determined by the prion-infected cell-based assay among all anti-prion materials involving nanobody8, RNA/DNA aptamers9,10, and chemical compounds11,12,13,14,15,16,17. Thus, R24 is a promising anti-prion material. As the sequence of R24 is almost the same as that of R12-A-R12, it is likely that the structure and mode of the interaction with PrPC of R24 are similar to those of R12-A-R12. The broadness of the imino proton spectrum of R24 hindered the structural determination of R24. This broadness may reflect the intermolecular aggregation of R24 with the high RNA concentration used for NMR analysis. In the case of a cell-based assay in which the RNA concentration is low, this possible aggregation is not likely to occur, which would be a reason why R24 exhibited high anti-prion activity like R12-A-R12. It was also noted that the dissociation constant of the R24-PrP complex is the same order of that of the R12-PrP complex. Resistance of the unimolecularly formed structure of R24 to the transient destruction and/or resultant degradation under cell culture may be why anti-prion activity of R24 is much higher than that of R12.
Although several anti-prion materials involving aptamers9,10,25,26, compounds11,12,13,15,16,17, and Fab7 have been reported, little is known about the structure and interaction at high resolution. The crystal structure was reported for a complex between a compound, promazine, and the C-terminal region of PrPC27. Crystal structures have also been reported for PrPC in a complex with either an antibody28,29 or nanobody8. In all three cases, the C-terminal region of PrP was bound by each anti-prion material. The NMR structure has been reported only for the octapeptide portion of the N-terminal region of PrPC in a complex with a polymer compound, pentosane polysulfate, the coordinates of pentosane polysulfate not being provided14. The structures and interactions of R1220,21 and R12-A-R12 are the only information reported for anti-prion aptamers at high resolution. This is also the only information reported for an anti-prion material that binds to the N-terminal region of PrPC other than the octapeptide region. Therefore, the information obtained in this study is generally valuable for the development of potent anti-prion materials.
Alzheimer’s disease (AD) is one of the most common neurodegenerative disorders and the link between amyloid β (Aβ) and AD has been suggested30,31. It was reported that PrPC that is anchored to the cell surface acts as a receptor of the Aβ oligomer32. The interaction of Aβ oligomers with PrPC reportedly causes the neurotoxicity of Aβ oligomers such as inhibition of long-term potentiation32. Thus, it is thought that compounds that can disrupt the formation of the PrPC-Aβ oligomer complex may prevent AD. Recently, we reported that R12 can disrupt the PrPC-Aβ complex through tight binding of R12 to PrPC33. This result suggested the therapeutical potential of R12 for treating AD pathology. As R24 and R12-A-R12 tightly bind to PrPC and exhibit higher anti-prion activity than R12 in the cell-based assay, they may also disrupt the PrPC-Aβ complex, even more efficiently than R12. Therefore, R24 and R12-A-R12 may have the therapeutical potential for treating not only prion but also AD pathology.
The development of drugs against prion diseases is waited right now. Nucleic acid-based drugs are widely recognized as next-generation ones. We demonstrated the way of rational developing of nucleic acids that exhibit much higher activity. Rational interpretation of the achieved much higher anti-prion activity is also provided on the basis of the structure determination. The substantial advance of the achievement of the low IC50 value of 100 nM opens the realistic possibility of the application of these RNAs for therapeutic use.
Methods
Sample preparation
R24 [r(GGAGGAGGAGGAGGAGGAGGAGGA)], R12-A-R12 [r(GGAGGAGGAGGA-A-GGAGGAGGAGGA)], and other RNAs with modified R24 sequences, that were synthesized, purified by high-performance liquid chromatography and desalted, were purchased from Japan Bio Services Co., Ltd. Oligonucleotides were dissolved in a solution comprising 10 mM KCl, 10 mM K-phosphate (pH 6.2), and 1.0 mM DSS, which was used as an internal chemical shift reference. The concentration of R12-A-R12 was ca. 1.0 mM for structural determination, and those of oligonucleotides were 50–100 μM for screening. Each sample was heated at 95 °C for 5 min, followed by quick cooling on ice.
Evaluation of anti-prion activity
The anti-prion activity of either R24 or R12-A-R12 was examined using mouse neuronal cells (GT1–7) persistently infected with the human TSE agent (Fukuoka-1 strain), designated as GT + FK12,15,17,23, as reported previously20. The source is cells described in the report by Milhavet et al.23 Either R24 or R12-A-R12 was dissolved in a buffer solution containing 10 mM K-phosphate (pH 6.2) and 100 mM KCl. The samples were heated at 95 °C for 5 min, quickly cooled on ice and then stored at 4 °C before use. Approximately 1.5 × 105 cells were plated in each well of a six-well plate, and drug treatment was started 15 h later. Either R24 or R12-A-R12 was administered to the cell culture to a final concentration of 0.5 nM, 5 nM, 50 nM, 500 nM or 5000 nM, respectively, and additionally to 300 nM and 1000 nM for R12-A-R12. The buffer and GN8 solutions were used as negative and positive controls, respectively. During incubation, it was confirmed by the observation and cell counting with microscope that the cells were alive and still attached to the bottom of the well after treatment with the RNA. After 72 h incubation, cells were lysed in 150 μl of 1×Triton X-100-deoxycholate lysis buffer containing 150 mM NaCl, 0.5% of Triton X-100, 0.5% of sodium deoxycholate and 50 mM Tris–HCl (pH 7.5), and then the supernatant was collected. Each sample concentration was adjusted to 2 mg of protein per milliliter. Samples were digested with 20 μg/ml of proteinase K for 30 min at 37 °C. Western blotting for PrPSc was performed as described previously20. For detection of PrPSc, PrP M-20 antibody (Santa Cruz Biotechnology) was used as the primary antibody. The signals were visualized with Super-Signal (Pierce Biotechnology) and scanned using a LAS-1000 UV mini analyzer (Fuji Film). The experiments were carried out once for R24 and three times for R12-A-R12, respectively. The curves that allowed obtaining the IC50 values of R24 and R12-A-R12 are shown in Supplementary Fig. S1A,B, respectively. For R12, it was administered to the cell culture to a final concentration of 10 μM. After 72 h incubation, the PrPSc level was obtained in the same way. This experiment was carried out four times.
Determination of the dissociation constant with filter binding assay
5′-end γ-32P-labeled R24 was mixed with varying concentrations of bPrP to give a total volume of 25 μl in a solution comprising 20 mM Tris-HCl (pH 7.5) and 10 mM KCl. After 20 min incubation, each mixture was passed through a nitrocellulose filter and washed with 500 μl of the solution. The amount of bound R24 was measured with BAS 2500 (Fuji Film), and binding activities were calculated as the percentage of input R24 in the bPrP-R24 complex retained on the filter. We determined the dissociation constant (Kd) using GraphPad PRISM and non-linear regression curve fitting, and a one site-binding hyperbola equation (R24 binding (%) = Bmax × [bPrP]/(Kd + [bPrP]), where Bmax is the maximum bound at the saturating bPrP concentration)18,20.
NMR spectroscopy
One-dimensional 1H NMR spectroscopy, nuclear Overhauser effect spectroscopy (NOESY), total correlation spectroscopy (TOCSY), double-quantum filtered correlation spectroscopy (DQF-COSY), 1H-13C heteronuclear single quantum coherence (HSQC), and heteronuclear multiple bond coherence with jump and return solvent suppression (JRHMBC) were performed at 5, 15 and 25 °C with Bruker AVANCE III HD 600 MHz and AVANCE III HD 950 MHz spectrometers equipped with a cryoprobe. NMR data were processed and analyzed with XWIN-NMR/TopSpin (Bruker), NMRPipe34 and Sparky35.
Structure calculation
The distance restraints between protons were estimated from NOESY spectra with mixing times of 100 ms and 400 ms, as described previously19,22. Dihedral angle restraints for the δ, and endocyclic ν0, ν1, ν2, ν3 and ν4 torsion angles were derived from 3JH1′-H2′ and 3JH3′-H4′ couplings from DQF-COSY spectra as described previously19,22. Structural calculations were performed using these restraints including hydrogen-bonding restraints for G:G and G:A base pairs, and planarity restraints for the tetrad and hexad with a simulated annealing protocol supplied with XPLOR-NIH v. 2.2636,37, as described previously19,20. NMR restraints and structural statistics are shown in Supplementary Table S2. Ten structures that were selected from 100 calculations on the basis of the criterion of the smallest residual energy were further refined by MD simulation as described below.
Structure refinement
The structure of R12-A-R12 obtained by XPLOR-NIH was further refined by all-atom MD simulations with explicit water molecules. Ten structures derived on structural calculation with XPLOR-NIH were employed as the initial structures for the MD simulation. An MD simulation with NMR restraints on the basis of the NPT ensemble at 298 K under periodic boundary conditions was performed using the AMBER16 program38. The force-field parameters adopted for RNA and water molecules were ff99OL339,40 and TIP4P-FB41, respectively. The time step was set at 2.0 fs and a 50-ns simulation was carried out. Detailed protocols are described in Supplementary Methods.
Building of a structural model of R12-A-R12 in a complex with P16 peptides
A refined structure that was derived through a MD simulation from the lowest XPLOR-NIH energy was superimposed on the structure of the R12 homodimer in a complex with two P16 peptides (PDB ID: 2RU7) using the Match Maker module of UCSF Chimera. At this time, the numbers of the residues constituting the tetrad and hexad of the bottom R12 molecule of the homodimer were changed to correspond to those of the respective residues at the same positions in the R12-A-R12 molecule. Then, the atomic coordinates of R12 homodimer were removed.
Data deposition
The atomic coordinates for the ensemble of the ten refined structures of R12-A-R12 were deposited in the Protein Data Bank (PDB), and are available under accession code 6K84.
References
Prusiner, S. B. Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 (1982).
Prusiner, S. B. Molecular biology and transgenetics of prion diseases. Crit. Rev. Biochem. Mol. Biol. 26, 397–438 (1991).
Pan, K. M. et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 90, 10962–10966 (1993).
Prusiner, S. B., Scott, M. R., DeArmond, S. J. & Cohen, F. E. Prion protein biology. Cell 93, 337–348 (1998).
Prusiner, S. B. Prions. Proc. Natl. Acad. Sci. USA 95, 13363–13383 (1998).
Weissmann, C. The state of the prion. Nat. Rev. Microbiol. 2, 861–871 (2004).
Peretz, D. et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 412, 739–743 (2001).
Abskharon, R. N. et al. Probing the N-terminal β-sheet conversion in the crystal structure of the human prion protein bound to a nanobody. J. Am. Chem. Soc. 136, 937–944 (2014).
Proske, D. et al. Prion-protein-specific aptamer reduces PrPSc formation. Chembiochem 3, 717–725 (2002).
Rhie, A. et al. Characterization of 2′-fluoro-RNA aptamers that bind preferentially to disease-associated conformations of prion protein and inhibit conversion. J. Biol. Chem. 278, 39697–39705 (2003).
Vogtherr, M. et al. Antimalarial drug quinacrine binds to C-terminal helix of cellular prion protein. J. Med. Chem. 46, 3563–3564 (2003).
Kuwata, K. et al. Hot spots in prion protein for pathogenic conversion. Proc. Natl Acad. Sci. USA 104, 11921–11926 (2007).
Cronier, S., Beringue, V., Bellon, A., Peyrin, J. M. & Laude, H. Prion strain- and species-dependent effects of antiprion molecules in primary neuronal cultures. J. Virol. 81, 13794–13800 (2007).
Taubner, L. M., Bienkiewicz, E. A., Copie, V. & Caughey, B. Structure of the flexible amino-terminal domain of prion protein bound to a sulfated glycan. J. Mol. Biol. 395, 475–490 (2010).
Kimura, T., Hosokawa-Muto, J., Kamatari, Y. O. & Kuwata, K. Synthesis of GN8 derivatives and evaluation of their antiprion activity in TSE-infected cells. Bioorg. Med. Chem. Lett. 21, 1502–1507 (2011).
Risse, E. et al. Identification of a Compound That Disrupts Binding of Amyloid-β to the Prion Protein Using a Novel Fluorescence-based Assay. J. Biol. Chem. 290, 17020–17028 (2015).
Yamaguchi, K. et al. A designer molecular chaperone against transmissible spongiform encephalopathy slows disease progression in mice and macaques. Nat. Biomed. Eng. 3, 206–219 (2019).
Murakami, K., Nishikawa, F., Noda, K., Yokoyama, T. & Nishikawa, S. Anti-bovine prion protein RNA aptamer containing tandem GGA repeat interacts both with recombinant bovine prion protein and its β isoform with high affinity. Prion 2, 73–80 (2008).
Mashima, T., Matsugami, A., Nishikawa, F., Nishikawa, S. & Katahira, M. Unique quadruplex structure and interaction of an RNA aptamer against bovine prion protein. Nucleic Acids Res. 37, 6249–6258 (2009).
Mashima, T. et al. Anti-prion activity of an RNA aptamer and its structural basis. Nucleic Acids Res. 41, 1355–1362 (2013).
Hayashi, T. et al. Binding of an RNA aptamer and a partial peptide of a prion protein: Crucial importance of water entropy in molecular recognition. Nucleic Acids Res. 42, 6861–6875 (2014).
Matsugami, A., Okuizumi, T., Uesugi, S. & Katahira, M. Intramolecular higher order packing of parallel quadruplexes comprising a G:G:G:G tetrad and a G(:A):G(:A):G(:A):G heptad of GGA triplet repeat DNA. J. Biol. Chem. 278, 28147–28153 (2003).
Milhavet, O. et al. Prion infection impairs the cellular response to oxidative stress. Proc. Natl. Acad. Sci. USA 97, 13937–13942 (2000).
Phan, A. T. Long-range imino proton-13C J-couplings and the through-bond correlation of imino and non-exchangeable protons in unlabeled DNA. J. Biomol. NMR 16, 175–178 (2000).
Weiss, S. et al. RNA aptamers specifically interact with the prion protein PrP. J. Virol. 71, 8790–8791 (1997).
Bibby, D. F. et al. Application of a novel in vitro selection technique to isolate and characterise high affinity DNA aptamers binding mammalian prion proteins. J. Virol. Methods 151, 107–115 (2008).
Baral, P. K. et al. Structural basis of prion inhibition by phenothiazine compounds. Structure 22, 291–303 (2014).
Eghiaian, F. et al. Insight into the PrPC → PrPSc conversion from the structures of antibody-bound ovine prion scrapie-susceptibility variants. Proc. Natl. Acad. Sci. USA 101, 10254–10259 (2004).
Baral, P. K., Swayampakula, M., Aguzzi, A. & James, M. N. G. Structural characterization of POM6 Fab and mouse prion protein complex identifies key regions for prions conformational conversion. FEBS J. 285, 1701–1714 (2018).
Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002).
Selkoe, D. J. Alzheimer’s disease is a synaptic failure. Science 298, 789–791 (2002).
Laurén, J., Gimbel, D. A., Nygaard, H. B., Gilbert, J. W. & Strittmatter, S. M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 457, 1128–1132 (2009).
Iida, M. et al. The anti-prion RNA aptamer R12 disrupts the Alzheimer’s disease-related complex between prion and amyloid β. FEBS J. 286, 2355–2365 (2019).
Delaglio, F. et al. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995).
Goddard, T. D. & Kneller, D. G. SPARKY 3, University of California, San Francisco (2006).
Brünger, A. T. X-PLOR Version 3.1: A System for X-ray Crystallography and NMR., Yale University Press, New Haven (1992).
Schwieters, C. D., Kuszewski, J. J., Tjandra, N. & Clore, G. M. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 (2003).
Case, D.A. et al. AMBER 16. University of California, San Francisco (2016).
Pérez, A. et al. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys. J. 92, 3817–3829 (2007).
Zgarbová, M. et al. Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory. Comput. 7, 2886–2902 (2011).
Wang, L.-P., Martinez, T. J. & Pande, V. S. Building Force Fields - An Automatic, Systematic, and Reproducible Approach. J. Phys. Chem. Lett. 5, 1885–1891 (2014).
López Garcia, F., Zahn, R., Riek, R. & Wüthrich, K. NMR structure of the bovine prion protein. Proc. Natl. Acad. Sci. USA 97, 8334–8339 (2000).
Acknowledgements
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan [18H04550 and 18K19397 to M.K.; 17K07307 and 17H05878 to T.N.; and 16K07269 to T.M.]. J.H.L was supported by the Samsung Science and Technology Foundation [SSRF-BA1701-10]. This work was performed in part using the NMR spectrometers with the ultra-high magnetic fields under the Cooperative Research Program of Institute for Protein Research, Osaka University, NMRCR-18-05.
Author information
Authors and Affiliations
Contributions
T.M., T.N., F.N., S.N. and M. Katahira conceived the idea. T.M. performed the sample preparation, NMR experiments and structure determination. T.M. and L.J.-H. analyzed the NMR data. Y.O.K. and K.K. performed the assaying of anti-prion activity. T.H. and M. Kinoshita performed the structure refinement by MD simulation. T.M., T.N. and M. Katahira wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mashima, T., Lee, JH., Kamatari, Y.O. et al. Development and structural determination of an anti-PrPC aptamer that blocks pathological conformational conversion of prion protein. Sci Rep 10, 4934 (2020). https://doi.org/10.1038/s41598-020-61966-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-61966-4
This article is cited by
-
Therapeutic development of polymers for prion disease
Cell and Tissue Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
