
Abstract
Changes in the oxygen fugacity (fO2) of the Earth’s mantle have been proposed to control the spatial and temporal distribution of arc-related ore deposits, and possibly reflect the evolution of the atmosphere over billions of years. Thermodynamic calculations and natural evidence indicate that fluids released from subducting slabs can oxidise the mantle, but whether their oxidation potential varied in space and time remains controversial. Here, we use garnet peridotites from western Norway to show that there is a linear decrease in maximum fO2 with increasing depth in the mantle wedge. We ascribe this relation to changes in the speciation of sulfur released in slab fluids, with sulfate, controlling maximum oxidation, preferentially released at shallow depths. Even though the amount of sulfate in the Precambrian oceans, and thus in subducted lithologies, is thought to have been dramatically lower than during the Phanerozoic, garnet peridotites metasomatised during these two periods have a comparable fO2 range. This opens to the possibility that an oxidised mantle with fO2 similar to modern-day values has existed since the Proterozoic and possibly earlier. Consequently, early magmas derived from partial melting of metasomatised mantle may have had suitable fO2 to generate porphyry Cu-Au and iron-oxide Cu-Au deposits.
Similar content being viewed by others

Introduction
There has been a long running debate as to whether arc magmas inherit their elevated oxidation states from the mantle or acquire them during migration through the crust1,2. Since the onset of plate tectonics, subduction has introduced surficial material into the Earth’s mantle, some of which had the potential to modify its oxidation state3,4. It has been shown that fluids released through dehydration of subducting slabs may promote metasomatic oxidation of the overlying mantle wedge, by introducing oxidised carbon and sulfur5,6,7. Altered oceanic crust provides the greatest input of sulfur in subduction zones6, and the abundance of oxidised sulfur released from it is thought to vary as function of pressure and temperature8, potentially leading to changes in the oxidation potential of slab fluids with depth. The amount of oxidised species in subducted lithologies may have also changed during the Earth’s history. Due to the paucity of dissolved sulfate in seawater prior to the second great oxidation event7,9, the redox budget of early subduction zones may have been lower than during the Phanerozoic7. To investigate changes in mantle wedge fO2 as a function of depth we examined thirty-six new samples that we collected from four well-studied peridotite localities in the Western Gneiss Region (WGR) of Norway: Ugelvik, Bardane, Svartberget and Almklovdalen (Fig. 1a). These peridotites were initially modified by high degrees of partial melting in the Archean10,11, and record variable intensities of subsequent subduction-related metasomatic alteration at different times and depths (see below). Because these peridotites were mechanically emplaced into subducting continental crust they derive from the near-bottom end of the mantle wedge12. Based on previously published fO2 data for the Ugelvik and Bardane peridotites13,14, we selected five new samples to obtain the widest range of depths possible.

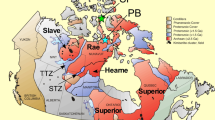
Samples localities. (a) Schematic map of the peridotite localities in the WGR of Norway considered in this study, modified from ref.17. (b) Location of other garnet peridotites considered from the literature. Diamonds represent kimberlite localities whereas yellow spots are orogenic garnet peridotites.
Geological Background
The Ugelvik peridotites have extremely depleted trace element compositions, implying that they escaped alteration by slab fluids11. From this locality we studied a garnet pyroxenite (UGL03; Fig. 2a). The proportion of the majorite component in garnet in this sample indicates equilibration at ~7.3 GPa (Methods). Metasomatism by slab fluids at ~500 Ma (ref.15) has been identified in the Bardane peridotite on the base of : i) strong enrichment in light-rare earth elements (LREE) and large-ion lithophile elements (LILE), such as Ba, Sr, Rb, U and Th; ii) elevated 87Sr/86Sr and low 143Nd/144Nd (ref.15,16); and iii) sulfur isotope signature compatible with fluid produced by dehydration of subducting slabs17. These fluids produced different assemblages; at ~3 GPa (M3-1 and M3-2 stages) and at ~6 GPa (M3-3 stage14). From Bardane we studied sample BDN02 (Fig. 2b), a phlogopite- and carbonate-bearing garnet pyroxenite recording the M3-1 event. The Svartberget peridotite experienced alteration by felsic slab-derived material at ~397 Ma that produced decimeter to meter wide veins with garnetite cores and pyroxenite margins strongly enriched in LILE and LREE, recording pressure of ~5.5 GPa (ref.18). SVT06 samples the transition between vein core and margin (Fig. 2c). The Almklovdalen peridotite was metasomatised by basaltic melt at ~1.0 Ga (ref.19) and the melt is preserved as pyroxenite veins formed at ~3.5 GPa (ref.20). The derivation of this metasomatic agent from a subducted slab is suggested by the sulfur isotopic composition of sulfides hosted in metasomatised domains17. From this locality we considered GSD08 (Fig. 2d), sampling the transition between garnet pyroxenite and metasomatised harzburgite, and GSD07, a garnet pyroxenite (Fig. 2e).

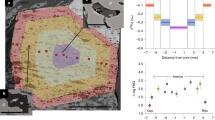
Examples of orogenic peridotite samples from the WGR of Norway considered in this study. On the left is reported the scanned thin section, and on the right the compositional maps of key areas where the XANES spectra were collected (white spots). The numbers associated with the white spots identify the spectra in in Table 1 and Supplementary Table S1 (a) UGL03; (b) BDN02; (c) SVT06; (d) GSD08; (e) GSD07.
Results and Discussion
The fO2 of selected samples was calculated by applying garnet oxybarometry21, using the Fe3+/Fetot of garnets measured by synchrotron Fe-edge X-ray absorption near edge structure spectroscopy (XANES; Methods). Results are reported in Table 1 and graphically in Fig. 3, together with literature data for the Bardane and Ugelvik peridotites13,14 and other garnet peridotites metasomatised by subduction at similar depths during the Phanerozoic22. In Fig. 3 for comparison we have also reported the fO2 of cratonic mantle xenoliths brought to the surface by kimberlite magmas from the Siberian, Kaapvaal and Slave cratons23,24,25,26,27,28,29,30 (Fig. 1b). Because the calculated fO2 of a garnet peridotite with fixed composition decreases with increasing pressure31, depth-related changes in oxidation state due to metasomatism can be obscured. One way to overcome this issue is to normalise the results to the fO2 of an unmetasomatised mantle end-member.

fO2-depth relation in orogenic peridotites and kimberlite xenoliths. Blue diamonds represent mantle metasomatised by Phanerozoic subductions, new data from this work is integrated with values from ref.13, the Ulten amphibole-bearing garnet peridotite, the magnesite-phlogopite garnet peridotite from Sulu22, and a garnet-websterite from Bardane14. The latter, unlike our samples, records the M3-3 metasomatic event in veins crosscutting the earlier assemblages. The spots represent the fO2 of peridotite xenoliths recovered from kimberlite magmas23,24,25,26,27,28,29,30. Blue spots are xenoliths with sinusoidal trace elements patterns in garnet, indicating metasomatism by fluids48, whereas red spots are xenoliths with garnet showing normal patterns, suggesting alteration by melts48. Grey spots are xenoliths without trace elements constrains. For the sake of comparison the fO2 of all samples was recalculated using the oxybarometric calibration of ref.21 and normalised to the modelled Archean pristine mantle fO2 (see Methods).
The fO2 of unmetasomatised mantle
It has been shown that the pre-metasomatic end-member of the Norwegian orogenic peridotites is a depleted harzburgite/dunite lithology generated by high-degrees of partial melting in the Archean10,11,32. A similar depleted end-member has also been suggested to represent the pristine unmetasomatised mantle that underlies Archean cratons worldwide33. By comparison, in areas where the continental crust is younger, the underlying mantle has largely been refertilised during major tectonic events, in some cases involving multiple subduction cycles33. These mantle end-members have been modelled by ref.33 by combining data from the orogenic peridotites of Norway and kimberlite xenoliths worldwide. They propose an Archean pristine mantle (APM) composition, represented by depleted dunite/harzburgite (Arc_9), and refertilised end-member compositions for Proterozoic (Pr_4) and Phanerozoic (Tc_1) mantle. Because of the large dataset of natural samples used in the definition of these end-members33 and their validation on geophysical basis33,34, we chose these as starting compositions for further fO2 modelling (Methods) with the aim of identifying the fO2 of an unmetasomatised mantle end-member. The Archean pristine mantle composition (Arc_9) has the lowest fO2 of all the considered end-members; ~95% of the considered samples, including both orogenic peridotites and kimberlite-hosted peridotite xenoliths, show higher fO2 (Fig. 4). Most of our samples, and the other orogenic peridotites for which there are published fO2 data, have been affected by some degree of metasomatic alteration13,14,22. We have also found that, on the basis of trace element concentrations in garnet, all kimberlite xenoliths for which there are relevant data23,24,28,29 have been affected by some degree of metasomatic alteration (Fig. 3). This can explain their higher fO2 compared to the modelled Arc_9 composition (Fig. 4). Only the samples from the Ugelvik peridotite, UGL01 and UGL03, have lower fO2 than Arc_9. This peridotite have experienced high degrees of partial melting in the Archean11 and the trace element concentrations in garnet show distinct steep REE patterns, which confirm its extremely depleted nature and indicate that it has not interacted with metasomatic fluids since the Archean11. Thus, the samples from the Ugelvik peridotite may represent the best Archean pristine mantle end-member for our metasomatised samples. Pr_4 and Tc_1 have higher fO2, averaging ~2 log units above Arc_9 (Fig. 4). Most of the considered samples (83%) have fO2 between the Arc_9 and Pr_4/Tc_1 fO2 values, in line with the different extents of metasomatic alteration reported for these samples14,28,35. Only the 17% of the samples show higher fO2 than Pr_4/Tc_1, and these may represent mantle domains altered by particularly oxidising agents. To constrain this calculation, we also modelled fO2-depth trajectories starting from the average bulk composition of harzburgite and lherzolite xenoliths from ref.36, which had Fe2O3 concentrations directly measured by Mössbauer spectroscopy36. With this approach the averaged harzburgite (5 samples), which is representative of depleted mantle, and has 0.09 wt% more Fe2O3 than Arc_9, plots ~0.3 log units above the Arc_9 curve, which is less than the uncertainty in the fO2 calculation21. The average lherzolite composition (14 samples), representative of fertile mantle, has Fe2O3 content of 0.20 wt%, giving fO2 values between Tc_1 and Pr_4 at most pressures. This confirms that Tc_1 and Pr_4 are representative of fertile mantle, whereas Arc_9 best represents the pre-metasomatic Archean pristine mantle (APM) protolith for the Norwegian peridotites and for the cratonic mantle samples.

fO2-depth trends for different mantle end-members. Grey diamonds and spots represent respectively orogenic garnet peridotites and garnet peridotite xenoliths hosted in kimberlites reported in Fig. 3.
A redox profile through subduction zone mantle
In order to graphically visualise changes in oxidation state of mantle samples as a function of depth we normalised the fO2 of samples reported in Fig. 3 to the fO2 of the modelled APM. It can be seen that all metasomatised samples have higher fO2 than APM and define a trend of decreasing fO2 with increasing depth, down to ~200 km, below which fO2 values are in the range of unmetasomatised mantle or lower. This suggests that with increasing depth the oxidative potential of subduction decreases, and becomes minimal below ~200 km, effectively creating a zonation in mantle wedge fO2. The main oxidants in slab fluids are carbon and sulfur7, and although melts might preferentially mobilise Fe3+ (ref.37) its abundant addition to the mantle wedge has been excluded5,38,39. Reduction of oxidised carbon (e.g., CO2) and sulfur (e.g., SO42− or S3−) during mantle metasomatism can increase mantle fO2 via redox reactions3,6,13. Carbonate minerals, diamond and graphite are present in samples from western Norway metasomatised at depths between ~100 and ~200 km13,14,22, indicating that carbon was mobilised in fluids across a wide range of depths. CO2 reduction can increase fO2 up to carbonate stability, defined by the EMOD/G line (enstatite-magnesite-olivine-diamond/graphite, Fig. 3; ref.40). Further oxidation requires reduction of oxidised sulfur. In subducting oceanic crust the dominant sulfate mineral is anhydrite and it is dissolved at relatively shallow depths in fluids released across the blueschist-eclogite transition8. Instead, at greater depths sulfur is thought to be released, through conversion of pyrite to pyrrhotite, in the form of H2S (ref.41) or S3− (ref.42).
Samples metasomatised at depths ≤160 km have fO2 above the EMOD/G buffer (Fig. 3), requiring the introduction of oxidised sulfur. In contrast, mantle metasomatised at deeper levels has fO2 below the EMOD/G buffer, suggesting that only shallower fluids introduced a sufficient amount of oxidised sulfur to exceed the EMOD/G buffer. Barite (BaSO4) is the dominant sulfur-bearing mineral in some samples metasomatised at depths ≥160 km, such as those from the Bardane (BDN03; Ref.13) and Svartberget (SVT06) peridotite, which intuitively suggests that sulfate was carried in the fluid. However, it has been previously suggested13 that because Ba is immobile in presence of SO42− (ref.43), it is more likely that S3− was the dominant sulfur species in those metasomatic fluids. Oxidation by reduction of S6+ in sulfate to S2− to form sulfide allows conversion of 8 moles of Fe2+ to Fe3+ for each mole of sulfate reduced, whereas 3 moles of S3− need to be reduced to oxidise 5 moles of ferrous iron. Therefore, sulfate is a stronger oxidant than S3− and the observed decreasing fO2 with increasing depth is reconcilable with preferential release of S6+ at shallower depths, and progressive increase in S3−/S6+ ratio as depth increases. Shallowly released fluids would also progressively remove carbonate by dissolution44 leaving deeper fluids comparatively depleted in CO2.
The other key influence on the variation in mantle oxidation with depth is the amount of oxidising agent added to a given mantle domain. There is a progressive decrease in the amount of fluid released from subducting slabs as hydrous phases are progressively diminished after a peak in fluid production across the blueschist-eclogite transition8, becoming minimal below ~200 km45. So, it is anticipated that there should be less fluid-driven metasomatism at deeper levels, although in the Norwegian samples metasomatised from ~100 to ~200 km depth we did not find considerable differences in the amount of H2O-bearing phases, or degree of alteration. In addition, fluid/melt with a given redox budget produces stronger, more localised oxidation when focused through narrow zones than when it interacts pervasively with larger mantle domains6,7. This variation in volume of fluid and extent of fluid-focusing would produce scatter in the fO2 of metasomatised mantle, potentially explaining the broad range in fO2 of the orogenic peridotite samples at any given depth (Fig. 3).
We suggest that the preferential release of different oxidants as a function of depth, in combination with a progressive decrease in the amount of fluid released from the slab, are responsible for the observed zonation in mantle wedge fO2. These factors provide an explanation for the decreasing fO2 of arc magmas with increasing distance from the trench observed in different arcs worldwide46,47, and related spatial variations in ore deposit types, where Cu-Au porphyries that require higher fO2 are distributed closer to the trench8.
The oxidation potential of subduction through time
In Fig. 3, we have reported also the fO2 of garnet peridotite xenoliths brought to the surface by kimberlite magmas from the Siberian, Kaapvaal, and Slave Cratons23,24,25,26,27,28,29,30 (Fig. 1b). These are grouped on the basis of the trace element composition of garnet48: blue spots represent samples metasomatised by fluids, whereas red spots are samples metasomatised by melts. It can be seen that metasomatism in the cratonic mantle produced an increase in fO2 comparable to that promoted by subduction fluids during the Phanerozoic. Many of these samples have fO2 above the EMOD/G line, therefore requiring reduction of oxidised sulfur (Fig. 3). Information regarding sulfate stability at mantle conditions is limited; however, experiments have shown that at 1.5 GPa sulfate becomes stable in basaltic melt at fO2 ≥ FMQ + 0.5 (ref.49), and that the stability of sulfate in aqueous fluids at similar pressures requires even higher fO2 (ref.50). Because the fO2 of deeper mantle is consistently lower than FMQ (ref.31), oxidised sulfur in cratonic mantle is unlikely to be derived from unaltered asthenosphere; instead, the most plausible source is subducted material. The cratonic mantle sampled by kimberlites formed as residue after high degrees of partial melting largely in the Archean and records different events of subsequent refertilisation33. Trace element signatures and isotopic composition of metasomatic silicates in kimberlite peridotite xenoliths51 and diamonds52 indicate that subduction fluids played a major role in the refertilisation of the cratonic mantle. The ages of subduction events that modified these mantle domains can be constrained by considering the ages of eclogite xenoliths recovered from kimberlites. These are thought to represent oceanic crust that was subducted and dehydrated in the sub-continental lithospheric mantle53,54. Dating of eclogite xenoliths across the Siberian, Kaapvaal and Slave cratons yielded Mesoarchean to Paleoproterozoic ages55,56,57, and because the amalgamation of these cratons largely ended in the Proterozoic58, the possibility that the cratonic mantle has been significantly affected by younger subduction events is unlikely. Therefore, the oxidised sulfur necessary for the oxidation of the cratonic mantle to values above the EMOD/G buffer was likely supplied by ancient, pre-Phanerozoic, subduction events.
This observation suggests that the oxidative potential of subduction may have not changed over billions of years and that similar proportions of oxidised sulfur to that subducted in the Phanerozoic were introduced into the mantle during the Proterozoic and possibly Archean. This is in apparent conflict with the suggestion that dominantly anoxic conditions and minimal sulfate existed in the deep oceans9, and thus in subducted lithologies, in the Proterozoic. Alternatively, it may be that sulfate may not need to be present to produce oxidised slab fluids containing sulfate species; sulfate could be produced during slab dewatering by sulfide oxidation via reduction of other redox-sensitive elements. During subduction of serpentinised lithospheric mantle, the Fe3+/∑Fe of serpentine minerals decreases, and it has been argued that this reduction of ferric iron promotes sulfide oxidation, but only in the most intensely serpentinised lithosphere, generating sulfate-bearing fluids that migrate into the mantle wedge38,59. Importantly, the genesis of oxidised fluids through this process is independent of the oxygenation of the oceans or the atmosphere, because serpentinisation is driven by H2O dissociation60, and thus would have occurred during the Archean and Proterozoic. However, the altered oceanic crust is thought to be by far the most important fluid contributor to the sub-arc mantle during subduction6,45, whereas the lithospheric mantle and sediment veneer are only minor contributors. Perhaps the simplest explanation is that sulfate was present in sufficient abundance in Proterozoic oceans to drive oxidation of the oceanic crust initiating a global redox cycle. The data reported in Fig. 2 suggest that Proterozoic magmas derived from partial melting of metasomatised mantle likely had suitable fO2 for the genesis of porphyry Cu-Au deposits37, and other ore deposits associated with oxidised magmas such as iron-oxide copper gold (IOCG) deposits, and some magmatic sulfide deposits61. The predominantly Phanerozoic distribution of porphyry Cu-Au deposits may thus not be related to major changes in mantle wedge fO2 (ref.7), but rather to preservational bias because they form in the upper levels of mountain belts7,62. IOCG deposits are relatively abundant in the Proterozoic and may be associated with partial melting of oxidised metasomatised mantle.
Methods
Synchrotron XANES spectroscopy and MAIA X-ray fluorescence imaging
Fe K-edge XANES spectroscopy has been used to determine the Fe3+/Fetot ratio in garnets. The experiment was conducted at the Australian Synchrotron X-ray Fluorescence Microscopy (XFM) beamline63 using a single element Vortex detector. Multiple XANES spectra were acquired for each garnet to constrain intra-grain variations in Fe3+/Fetot. The spots where the spectra were acquired are reported in Fig. 1 and Supplementary Fig. 2. The raw data were normalised using the mback software64. The Fe3+/Fetot ratio in garnet was calculated using the technique described by ref.65. This is based on the establishment of an empirical calibration curve, which relates the ratio of the intensity at 7138.4 and 71.61.7 eV in the XANES spectra of a series of standard garnets to their Fe3+/Fetot values determined by Mössbauer spectroscopy. The Fe K-edge XANES spectra of garnets in the studied samples were acquired in the same analytical session of those in the samples from Bardane (BDN03) and Ugelvik (UGL01) previously published in ref.13, thus the same standards (Supplementary Table S1) and calibration curve (Supplementary Fig. S1) of ref.13 have been used for the calculation of the Fe3+/Fetot ratios in this study. Results from the XANES experiment for the unknowns are reported in Supplementary Table S1. The precision of the XANES analyses from standard deviation is 0.0037 and the error for the Mössbauer analysis of the standards is 0.01. Elemental maps were acquired at the Australian Synchrotron XFM beamline. The samples were raster scanned in continuous mode with a monochromatic (18.5 keV) X-ray beam focused to 1.5 × 1.5 µm2. A 396-element Maia detector63,66 collected the X-ray fluorescence and scatter emitted with 1.9 millisec/pixel dwell and the data were processed using GeoPIXE II (ref.66).
Oxygen fugacity calculation and modelling
Oxygen fugacity was calculated using the GtfO2 software from ref.21. This software incorporates four independent oxybarometers and yields optimum log fO2 estimates by the method of least squares. The uncertainty reported in calculating the fO2 with this approach ranges from 0.6 to 0.9 log units21. The compositions of garnet, olivine and orthopyroxene used for the fO2 calculations are reported in Supplementary Table S2. The composition of olivine and orthopyroxene used for the calculation of the fO2 for SVT06 are from ref.18. The pressure (P) and temperature (T) used for the fO2 calculations on samples GSD07 and GSD08 are 3.5 GPa and 850 °C from ref.19, whereas P-T values for SVT06 of 5.5 GPa and 800 °C are from ref.67. For BDN02, P-T of 3 GPa and 800 °C are from ref.14 and are representative of the M3-1 and M3-2 metasomatic events. Given the majoritic nature of garnet in sample UGL03, pressure has been calculated using the geobarometer of ref.68 and temperature estimated using a lithospheric geotherm of 50.5 mW m−2. Oxygen fugacity values for BDN03 and UGL01 are from ref.13.
To model the fO2 of the unmetasomatised mantle end-member we started from the chemical composition in major oxides and calculated the composition of mineral phases in equilibrium at P ranging from 3 to 6 GPa along a 50 mW m−2 geotherm using THERMOCALC69 with the dataset of ref.70. Oxygen fugacity was calculated using the GtfO2 software of ref.21. The 50 mW m−2 geotherm was chosen because it provides a good fit with the P-T trend of most samples; ~90% are within a ±100 °C range. We have estimated that the changes in fO2 at the pressures of interest, for ~100 °C temperature variation, are smaller than the uncertainty on fO2 calculated for unknown samples21. Because the thermodynamic dataset of ref.70 does not allow for calculation at pressures higher than 6 GPa, we have extrapolated the fO2 at higher pressure from lines of best fit to the lower pressure results. It has been shown that the bulk Fe2O3 content of garnet peridotites is directly related to their MgO wt% content36, so we used this relationship to calculate the Fe2O3 for each mantle end-member. We chose the dataset of ref.36 because it provides Fe2O3 of a wide range of mantle rock compositions, from dunite to websterite, measured on whole rock samples by Mössbauer spectroscopy. We used a regression line fitting these samples (R2 = 0.8) in a diagram of Fe3+/Fetot vs MgO wt% to calculate the Fe3+/Fetot content of each mantle end-member described in the text. To investigate the speciation of carbon in equilibrium with depleted mantle as function of fO2, we used the equilibrium between carbonate and graphite/diamond in a mantle assemblage expressed as:
This reaction is referred to by the acronym EMOD/G (enstatite-magnesite-olivine-diamond/graphite)40. In Fig. 3 the EMOD/G buffer is normalised to APM.
References
Parkinson, I. J. & Arculus, J. R. The redox state of subduction zones: insights from arc-peridotites. Chemical Geology 160, 409–423 (1999).
Lee, C. T. et al. The redox state of arc mantle using Zn/Fe systematics. Nature 468, 681–685, https://doi.org/10.1038/nature09617 (2010).
Brounce, M. N., Kelley, K. A. & Cottrell, E. Variations in Fe3+/Fe of Mariana Arc Basalts and Mantle Wedge fO2. Journal of Petrology 55, 2513–2536, https://doi.org/10.1093/petrology/egu065 (2014).
Kasting, J. F., Eggler, D. H. & Raeburn, S. P. Mantle redox evolution and the oxidation state of the Archean atmosphere. Journal of Geology 101, 245–257 (1993).
Debret, B. & Sverjensky, D. Highly oxidising fluids generated during serpentinite breakdown in subduction zones. Scientific Reports 7 (2017).
Evans, K. A. The redox budget of subduction zones. Earth-Science Reviews 113, 11–32, https://doi.org/10.1016/j.earscirev.2012.03.003 (2012).
Evans, K. A. & Tomkins, A. G. The relationship between subduction zone redox budget and arc magma fertility. Earth and Planetary Science Letters 308, 401–409, https://doi.org/10.1016/j.epsl.2011.06.009 (2011).
Tomkins, A. G. & Evans, K. A. Separate zones of sulfate and sulfide release from subducted mafic oceanic crust. Earth and Planetary Science Letters 428, 73–83, https://doi.org/10.1016/j.epsl.2015.07.028 (2015).
Farquhar, J., Wu, N., Canfield, D. E. & Oduro, H. Connections between Sulfur Cycle Evolution, Sulfur Isotopes, Sediments, and Base Metal Sulfide Deposits. Economic Geology 105, 509–533 (2010).
Beyer, E. E., Griffin, W. L. & O’Reilly, S. Y. Transformation of Archaean Lithospheric Mantle by Refertilization: Evidence from Exposed Peridotites in the Western Gneiss Region, Norway. Journal of Petrology 47, 1611–1636, https://doi.org/10.1093/petrology/egl022 (2006).
Spengler, D. et al. Deep origin and hot melting of an Archaean orogenic peridotite massif in Norway. Nature 440, 913–917, https://doi.org/10.1038/nature04644 (2006).
Brueckner, K. H. Sinking intrusion model for the emplacement of garnet-bearing peridotites into continent collision orogens. geology 26, 631–634 (1998).
Rielli, A. et al. Evidece of sub-arc mantle oxidation by sulphur and carbon. Geochemical Perspectives Letters 3, 124–132, https://doi.org/10.7185/geochemlet.1713 (2017).
Malaspina, N., Scambelluri, M., Poli, S., Van Roermund, H. L. M. & Langenhorst, F. The oxidation state of mantle wedge majoritic garnet websterites metasomatised by C-bearing subduction fluids. Earth and Planetary Science Letters 298, 417–426, https://doi.org/10.1016/j.epsl.2010.08.022 (2010).
Brueckner, K. H., Carswell, D. A. & Griffin, W. L. Paleozoic diamonds within a Precambrian peridotite lens in UHP gneisses of the Norwegian Caledonides. Earth and Planetary Science Letters 203, 805–816 (2002).
Scambelluri, M., Van Roermund, H. L. M. & Pettke, T. Mantle wedge peridotites: Fossil reservoirs of deep subduction zone processes. Lithos 120, 186–201, https://doi.org/10.1016/j.lithos.2010.03.001 (2010).
Rielli, A. et al. Sulfur isotope and PGE systematics of metasomatised mantle wedge. Earth and Planetary Science Letters 497, 181–192 (2018).
Vrijmoed, J. C. et al. Metasomatism in the Ultrahigh-pressure Svartberget Garnet-peridotite (Western Gneiss Region, Norway): Implications for the Transport of Crust-derived Fluids within the Mantle. Journal of Petrology 54, 1815–1848, https://doi.org/10.1093/petrology/egt032 (2013).
Jamtveit, B., Carswell, D. A. & Mearns, E. W. Chronology of the high-pressure metamorphism of Norwegian garnet peridotites/pyroxenites. Journal of metamorphic geology 9, 125–139 (1991).
Medaris, L. G. A geothermobarometrie investigation of garnet peridotites in the Western Gneiss Region of Norway. Contributions to Mineralogy and Petrology 87, 72–86 (1984).
Miller, W. G. R., Holland, T. J. B. & Gibson, S. A. Garnet and Spinel Oxybarometers: New Internally Consistent Multi-equilibria Models with Applications to the Oxidation State of the Lithospheric Mantle. Journal of Petrology 57, 1199–1222, https://doi.org/10.1093/petrology/egw037 (2016).
Malaspina, N., Poli, S. & Fumagalli, P. The Oxidation State of Metasomatized Mantle Wedge: Insights from C-O-H-bearing Garnet Peridotite. Journal of Petrology 50, 1533–1552, https://doi.org/10.1093/petrology/egp040 (2009).
Yaxley, G., Berry, A., Rosenthal, A., Woodland, A. & Paterson, D. Redox preconditioning deep cratonic lithosphere for kimberlite genesis–evidence from the central Slave Craton. Scientific Reports 7, 30 (2017).
Yaxley, G. M. et al. An oxygen fugacity profile through the Siberian Craton — Fe K-edge XANES determinations of Fe3+/∑Fe in garnets in peridotite xenoliths from the Udachnaya East kimberlite. Lithos 140, 142–151, https://doi.org/10.1016/j.lithos.2012.01.016 (2012).
Lazarov, M., Woodland, A. B. & Brey, G. P. Thermal state and redox conditions of the Kaapvaal mantle: A study of xenoliths from the Finsch mine, South Africa. Lithos 112S, 913–923, https://doi.org/10.1016/j.lithos.2009.03.035 (2009).
Goncharov, A. G., Ionov, D. A., Doucet, L. S. & Pokhilenko, L. N. Thermal state, oxygen fugacity and C-O-H fluid speciation in cratonic lithospheric mantle: New data on peridotite xenoliths from the Udachnaya kimberlite, Siberia. Earth and Planetary Science Letters 357-358, 99–110, https://doi.org/10.1016/j.epsl.2012.09.016 (2012).
Woodland, A. & Koch, M. Variation in oxygen fugacity with depth in the upper mantle beneath the Kaapvaal craton, Southern Africa. Earth and Planetary Science Letters 214, 295–310 (2003).
Creighton, S. et al. Oxidation of the Kaapvaal lithospheric mantle driven by metasomatism. Contributions to Mineralogy and Petrology 157, 491–504, https://doi.org/10.1007/s00410-008-0348-3 (2009).
Creighton, S., Stachel, T., Eichenberg, D. & Luth, R. W. Oxidation state of the lithospheric mantle beneath Diavik diamond mine, central Slave craton, NWT, Canada. Contributions to Mineralogy and Petrology 159, 645–657, https://doi.org/10.1007/s00410-009-0446-x (2010).
McCammon, C. & Kopylova, M. G. A redox profile of the Slave mantle and oxygen fugacity control in the cratonic mantle. Contributions to Mineralogy and Petrology 148, 55–68, https://doi.org/10.1007/s00410-004-0583-1 (2004).
Frost, D. J. & McCammon, C. A. The Redox State of Earth’s Mantle. Annual Review of Earth and Planetary Sciences 36, 389–420, https://doi.org/10.1146/annurev.earth.36.031207.124322 (2008).
Beyer, E. E., Brueckner, H. K., Griffin, W. L., O’Reilly, S. Y. & Graham, S. Archean mantle fragments in Proterozoic crust, Western Gneiss Region, Norway. Geology 32, 609–612, https://doi.org/10.1130/G20366.1 (2004).
Griffin, W., O’reilly, S. Y., Afonso, J. C. & Begg, G. The composition and evolution of lithospheric mantle: a re-evaluation and its tectonic implications. Journal of Petrology 50, 1185–1204 (2008).
Begg, G. C. et al. The lithospheric architecture ofAfrica: Seismic tomography, mantle petrology, and tectonic evolution. Geosphere 5, 23–50, https://doi.org/10.1130/ges00179.1 (2009).
McCammon, C., Griffin, W. L., Shee, S. & O’Neill, H. S. C. Oxidation during metasomatism in ultramafic xenoliths from the Wesselton kimberlite, South Africa: implications for the survival of diamond. Contributions to Mineralogy and Petrology 141, 287 (2001).
Canil, D. et al. Ferric iron in peridotites and mantle oxidation states. Earth and Planetary Science Letters 123, 205–220 (1994).
Mungall, E. J. Roasting the mantle: Slab melting and the genesis of major Au and Au-rich Cu deposits. Geology 30, 915–918 (2002).
Debret, B. et al. Evolution of Fe redox state in serpentine during subduction. Earth and Planetary Science Letters 400, 206–218, https://doi.org/10.1016/j.epsl.2014.05.038 (2014).
Nebel, O. et al. Redox-variability and controls in subduction zones from an iron-isotope perspective. Earth and Planetary Science Letters 432, 142–151, https://doi.org/10.1016/j.epsl.2015.09.036 (2015).
Luth, R. W. Diamonds, Eclogites, and the oxidation state of the Earth’s mantle. Science 261, 66–68 (1993).
Pokrovski, G. S. & Dubrovinsky, L. S. The S3- Ion is stable in geological fluids at elevated temperatures and pressures. Science 331, 1052–1054 (2011).
Pokrovski, G. S. & Dubessy, J. Stability and abundance of the trisulfur radical ion S3- in hydrothermal fluids. Earth and Planetary Science Letters 411, 298–309, https://doi.org/10.1016/j.epsl.2014.11.035 (2015).
Hanor, J. S. B.-C. Geochemistry and Environments of Formation. Reviews in Mineralogy and Geochemistry 40, 193–275, https://doi.org/10.2138/rmg.2000.40.4 (2000).
Frezzotti, M. L., Selverstone, J., Sharp, Z. D. & Compagnoni, R. Carbonate dissolution during subduction revealed by diamond-bearing rocks from the Alps. Nature Geoscience 4, 703–706, https://doi.org/10.1038/ngeo1246 (2011).
Schmidt, M. & Poli, S. 4.19. Devolatilization during subduction. The Crust, Treatise onGeochemistry (eds Holland, H. D. & Turekian, K. K. Second Edition), Elsevier-Pergamon, Oxford, 669–701 (2014).
Rowe, M. C., Kent, A. J. R. & Nielsen, R. L. Subduction Influence on Oxygen Fugacity and Trace and Volatile Elements in Basalts Across the Cascade Volcanic Arc. Journal of Petrology 50, 61–91, https://doi.org/10.1093/petrology/egn072 (2009).
Wang, R. et al. Increasing magmatic oxidation state from paleocene to miocene in the eastern Gangdese Belt, Tibet: implication for collision-related porphyry Cu-Mo ± Au mineralization. Economic Geology 109, 1943–1965 (2014).
Stachel, T. et al. The trace element composition of silicate inclusions in diamonds: a review. Lithos 77, 1–19, https://doi.org/10.1016/j.lithos.2004.03.027 (2004).
Jugo, P. J., Wilke, M. & Botcharnikov, R. E. Sulfur K-edge XANES analysis of natural and synthetic basaltic glasses: Implications for S speciation and S content as function of oxygen fugacity. Geochimica et Cosmochimica Acta 74, 5926–5938, https://doi.org/10.1016/j.gca.2010.07.022 (2010).
Pokrovski, G. S. & Dubrovinsky, L. S. The S3–ion is stable in geological fluids at elevated temperatures and pressures. Science 331, 1052–1054 (2011).
Bell, D. R. et al. Silica and volatile-element metasomatism of Archean mantle: a xenolith-scale example from the Kaapvaal Craton. Contributions to Mineralogy and Petrology 150, 251 (2005).
Smart, K. A., Tappe, S., Stern, R. A., Webb, S. J. & Ashwal, L. D. Early Archaean tectonics and mantle redox recorded in Witwatersrand diamonds. Nature Geoscience 9, 255–259, https://doi.org/10.1038/ngeo2628 (2016).
Jacob, D., Jagoutz, E., Lowry, D., Mattey, D. & Kudrjavtseva, G. Diamondiferous eclogites from Siberia: remnants of Archean oceanic crust. Geochimica et Cosmochimica Acta 58, 5191–5207 (1994).
Aulbach, S., Pearson, N. J., O’reilly, S. Y. & Doyle, B. J. Origins of xenolithic eclogites and pyroxenites from the Central Slave Craton, Canada. Journal of Petrology 48, 1843–1873 (2007).
Richardson, S., Shirey, S., Harris, J. & Carlson, R. Archean subduction recorded by Re–Os isotopes in eclogitic sulfide inclusions in Kimberley diamonds. Earth and Planetary Science Letters 191, 257–266 (2001).
Heaman, L. M., Creaser, R. A., Cookenboo, H. O. & Chacko, T. Multi-stage modification of the Northern Slave mantle lithosphere: evidence from zircon-and diamond-bearing eclogite xenoliths entrained in Jericho kimberlite, Canada. Journal of Petrology 47, 821–858 (2006).
Pearson, D., Snyder, G., Shirey, S. & Taylor, L. Archaean Re-Os age for Siberian eclogites and constraints on Archaean tectonics. Nature 374, 711 (1995).
Bleeker, W. The late Archean record: a puzzle in ca. 35 pieces. Lithos 71, 99–134 (2003).
Evans, K., Reddy, S., Tomkins, A., Crossley, R. & Frost, B. Effects of geodynamic setting on the redox state of fluids released by subducted mantle lithosphere. Lithos 278, 26–42 (2017).
Frost, B. R. & Beard, J. S. On silica activity and serpentinization. Journal of petrology 48, 1351–1368 (2007).
Tomkins, A. G., Rebryna, K. C., Weinberg, R. F. & Schaefer, B. F. Magmatic sulfide formation by reduction of oxidized arc basalt. Journal of Petrology 53, 1537–1567 (2012).
Kesler, S. E. & Wilkinson, B. H. The role of exhumation in the temporal distribution of ore deposits. Economic Geology 101, 919–922 (2006).
Paterson, D. et al. The X-ray Fluorescence Microscopy Beamline at the Australian Synchrotron. 219–222, https://doi.org/10.1063/1.3625343 (2011).
Weng, T. C., Waldo, G. S. & Penner-Hahn, J. E. A method for normalization of X-ray absorption spectra. J Synchrotron Radiat 12, 506–510, https://doi.org/10.1107/S0909049504034193 (2005).
Berry, A. J., Yaxley, G. M., Woodland, A. B. & Foran, G. J. A XANES calibration for determining the oxidation state of iron in mantle garnet. Chemical Geology 278, 31–37, https://doi.org/10.1016/j.chemgeo.2010.08.019 (2010).
Ryan, C. G. et al. Maia X-ray fluorescence imaging: Capturing detail in complex natural samples. Journal of Physics: Conference Series 499, 1–12, https://doi.org/10.1088/1742-6596/499/1/012002 (2014).
Vrijmoed, J. C., Van Roermund, H. L. M. & Davies, G. R. Evidence for diamond-grade ultra-high pressure metamorphism and fluid interaction in the Svartberget Fe–Ti garnet peridotite–websterite body, Western Gneiss Region, Norway. Mineralogy and Petrology 88, 381–405, https://doi.org/10.1007/s00710-006-0160-6 (2006).
Wijbrans, C. H., Rohrbach, A. & Klemme, S. An experimental investigation of the stability of majoritic garnet in the Earth’s mantle and an improved majorite geobarometer. Contributions to Mineralogy and Petrology 171, https://doi.org/10.1007/s00410-016-1255-7 (2016).
Powell, R. & Holland, T. J. B. An internally consistent dataset with uncertainties and correlations: 3. Applications to geobarometry, worked examples and a computer program. Journal of metamorphic geology 6, 173–204 (1988).
Jennings, E. S. & Holland, T. J. B. A Simple Thermodynamic Model for Melting of Peridotite in the System NCFMASOCr. Journal of Petrology 56, 869–892, https://doi.org/10.1093/petrology/egv020 (2015).
Acknowledgements
We would like to thank G. Yaxley and J. Wykes for the assistance with the XANES measurements. A. Berry is thanked for provision of the garnet standard for the XANES experiment. The authors acknowledge use of facilities within the Monash Centre for Electron Microscopy. Part of this research was undertaken on the X-ray Fluorescence Microscopy beamline at the Australian Synchrotron, Victoria, Australia. A.R. acknowledges the Australian Government Research Training Program Scholarship and AusIMM Bicentennial Gold 88 Endowment award. The authors would like to thank two anonimous reviewer whose comments have greatly improved this manuscript and Kathryn Goodenough for editorial handling.
Author information
Authors and Affiliations
Contributions
A.R. conducted fieldwork, synchrotron analyses, EMP work, fO2 modelling and wrote the manuscript. A.G.T. supervised the project, conducted fieldwork and synchrotron analyses. O.N. co-supervised the project and assisted with the synchrotron work. J.B. and B.E. assisted with the synchrotron experiment and data reduction. D.P. assisted with the synchrotron experiment.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rielli, A., Tomkins, A.G., Nebel, O. et al. Garnet peridotites reveal spatial and temporal changes in the oxidation potential of subduction. Sci Rep 8, 16411 (2018). https://doi.org/10.1038/s41598-018-34669-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34669-0
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
