
Abstract
To obtain a comprehensive overview of the dynamic transcriptome during male floral bud development in Populus tomentosa, high-throughput RNA-seq was conducted during eight flowering-related stages. Among the 109,212 de novo assembled unigenes, 6,959 were differentially expressed during the eight stages. The overrepresented classed of genes identified by Gene Ontology (GO) enrichment included ‘response to environmental stimuli’ and ‘plant-type spore development’. One-third of the differentially expressed genes were transcription factors (TFs). Several genes and gene families were analyzed in depth, including MADS-box TFs, Squamosa promoter binding protein-like family, receptor-like kinases, FLOWERING LOCUS T/TERMINAL-FLOWER-LIKE 1 family, key genes involved in anther and tapetum development, as well as LEAFY, WUSCHEL and CONSTANS. The results provided new insights into the roles of these and other well known gene families during the annual flowering cycle. To explore the mechanisms regulating poplar flowering, a weighted gene co-expression network was constructed using 98 floral-related genes involved in flower meristem identity and flower development. Many modules of co-expressed genes and hub genes were identified, such as APETALA1 and HUA1. This work provides many new insights on the annual flowering cycle in a perennial plant, and a major new resource for plant biology and biotechnology.
Similar content being viewed by others

Introduction
Flowering is one of the most important developmental events during the life cycle of angiosperms. In recent years, the molecular and genetic regulation of flower development has been studied in detail in Arabidopsis 1. Flowering is mainly controlled by six different endogenous (autonomous, gibberellin, and age) and environmental (photoperiod, temperature, and vernalization) pathways that together form a complex regulatory network2. All of these pathways are integrated through the transcriptional regulation of two floral pathway integrators, FLOWERING LOCUS T (FT) and SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1). These activate the floral meristem identity genes, LEAFY (LFY) and APETALA1 (AP1). TERMINAL FLOWER 1 (TFL1, floral repressor) is involved in maintaining the vegetative and inflorescence meristems, by preventing the expression of AP1 and LFY. FT and SOC1 share an upstream regulator, CONSTANS (CO), which is a key component in the photoperiodic pathway, as well as FLOWERING LOCUS C (FLC), a potent floral repressor integrating autonomous and vernalization pathways2. Floral organ identity has been successfully described using the ABCDE model. Many ABCDE class genes are members of the MADS-box gene family and control the various processes of plant development3.
Unlike annuals, woody perennials have an extended juvenile phase. After they reach adulthood, flowering occurs annually and the life cycle of these trees alternates between growth and dormancy. Recent advances in plant genome studies have greatly facilitated investigation into the regulation of flowering and seasonal control of bud dormancy4,5,6,7. The Populus genome contains nearly all of the major orthologs regulating flowering in Arabidopsis 8,9. Gene expression analyses and functional studies in Populus have shown that flowering regulatory pathways are broadly conserved between Arabidopsis and Populus 8. However, the regulation of flowering in Populus also exhibits remarkably different, and sometimes unique, characteristics compared to annual plants. For example, Populus PTLF (the LFY ortholog in P. trichocarpa) was less effective at inducing early flowering10. The overexpression of PTAP1-1 (AP1 ortholog) does not induce early flowering in Populus 11. Unlike AP1 in Arabidopsis, ectopic expression of LAP1 (a tree ortholog of AP1) fails to induce early flowering in hybrid aspens; LAP1 is instead involved in short-day (SD)-mediated seasonal growth cessation12. In transgenic poplars, PtFT1 and PtFT2 (two very similar Populus FT-like paralogs) induce early flowering13,14. PtFT1 is induced by winter temperatures and determines the onset of reproduction, while PtFT2 is induced by long days and warm temperatures and then promotes vegetative growth13,15,16. PtFT2 is down-regulated in response to SD, resulting in seasonal growth cessation13,15, and is up-regulated during chilling-induced dormancy release15,16. Unlike CO in Arabidopsis, the overexpression of poplar CO1 and CO2 does not alter normal reproductive onset17. The down-regulation of PopCEN (CEN/TFL1 ortholog in Populus) by RNAi accelerates the onset of mature tree characteristics. PopCEN1 also regulates axillary meristem identity and may play a role in maintaining the indeterminacy of the inflorescence apex18.
P. tomentosa Carr., widely grown in northern China, an important native commercial tree species used in urban green spaces, as well as in timber and pulp production. However, its extended juvenile phase presents a substantial obstacle to study and breeding. Due to the trichomes of catkins of female poplars in every spring, more male P. tomentosa are planted in northern China. But, from the aspect of wood accumulation, in general, biomass in male poplars is less than in female poplars. As we know, there is a banlance between vegetative growth and reproductive growth. Flower production requires large inputs of resources and energy, with negative impacts on vegetative growth. So, studying the flowering and flower development in male P. tomentosa is valuable. Additionally, the allergenic properties of poplar pollen in male floral buds are potential health hazards for allergic person19,20. Although a few flowering-related Populus genes have been discovered and studied21, a genome-wide study of the molecular basis of flower development in poplar is lacking. In-depth studies of the molecular basis of Populus flower development will contribute to a shortened breeding cycle for helping productivity and an understanding of the complex regulatory mechanisms controlling flowering in poplar.
Transcriptome sequencing using RNA-seq is a powerful tool for studying gene expression, defining gene putative function, and elucidating the molecular basis of key developmental processes22. In this study, we used paraffin-embedded tissues to examine the morphogenesis of male floral buds in P. tomentosa, and high-throughput sequencing to obtain global gene expression profiles during floral bud development in poplar. Based on extensive data analyses, we identified the MADS-box, Squamosa promoter binding protein-like family (SPL), receptor-like kinase (RLK) genes, FT/TFL1, and CO-like families, and a number of homologs of well-known floral genes. Furthermore, the construction of co-expression networks suggested a link between floral genes and the identified hub genes.
Results and Discussion
Floral bud development in P. tomentosa
Before the floral transcriptome study, we examined the developmental process of floral buds in a morphological analysis. The growth curves of the floral buds had a sigmoid shape. The dry weights of the buds increased progressively and rapidly from stage 1 to 4. During stages 5–7, floral buds increased continuously and then entered dormancy before again increasing (Fig. 1). These results showed that flower development was associated with bud size.

Schematic diagram of developmental stages responsible for the seasonal flowering of Populus tomentosa. (A) Axillary floral buds and vegetative buds. (B) Poplar male floral buds throughout the year. (C) Dry weight of P. tomentosa during flower development. (D) Poplar male floral buds at different stages throughout the year. Floral buds at stage 1: floral induction; stage 2: primordia formation; stage 3: organogenesis; stage 4: enlargement; stage 5: archespore formation; stages 6 and 7: dormancy; and stage 8: microsporogenesis.
Floral buds embedded in paraffin were serially-sectioned to study their morphogenesis. Stage 1 buds are in the floral induction stage. The first morphological evidence of the transition from vegetative to reproductive growth in the axillary meristem is rapid elongation, bract formation, and the initiation of floral apices in the axils of each bract. Stage 2 buds are the smallest flower buds that can be distinguished from vegetative buds. During this stage, centripetal bract formation continues, the floral meristems are surrounded by the cup-like, reduced perianth developing in the axils of the bracts, and the bracts extend to completely enclose the inflorescence. In stage 3 buds, rounded stamen primordia emerge from the meristem, with organogenesis proceeding centrifugally. Tetrasporangiate anthers form and are surrounded by the reduced perianth cup in the axils of mature bracts. Each anther typically contains four microsporangia, with each microsporangium forming a locule. Microspore mother cells develop initially in each of the four locules. The microsporocytes are arranged compactly within the locule, at which stage the male buds over-winter. In February of the next year, the inner-wall layer of the microsporangium matures, with the tapetum serving as a nutritive layer. The microsporocytes form tetrads after meiosis. As the spores mature, the tapetal cells disintegrate and each microspore divides mitotically to form a pollen grain. After microsporogenesis, the mature pollen grains are released from the dehisced anther (Fig. 1D).
Global view of the P. tomentosa floral transcriptome
A mixed-reads pool obtained from all stages of buds generated 296,623 transcripts >200 bp in length and 109,212 unigenes with an average length of 640.77 bp. The N50 for transcripts and unigenes was 1,928 bp and 873 bp, respectively (Table 1). The size distribution of the transcripts and unigenes, and the results of the sequence similarity analysis against various databases, are showned in Fig. S1 and Table 2. Most of the annotated unigenes had highest similarity to P. trichocarpa (78.40%) (Fig. S2A).
Of the 35,140 Nr hits, 27,040 sequences had a COG classification (Table 2). Among the 25 COG categories, ‘General function prediction only’, ‘Posttranslational modification’, and ‘Signal transduction mechanisms’ were the largest groups (Fig. S2B). Among all 109,212 unigenes, 16,841 were successfully annotated with GO terms (Table 2). All leading GO terms at level 2 could be categorized into 52 groups (Fig. S2C), indicating an association between the identified genes and various biological processes. In addition to cellular and metabolic processes, many genes were assigned to ‘biological regulation’, ‘response to stimulus’, ‘pigmentation’, and ‘developmental process’. The prominence of ‘binding’ suggested an important role for TFs during flower development in P. tomentosa.
The relationships between the floral buds were investigated in a PCA of the whole-gene expression dataset. Stages 1–4 clustered closely, as did stages 5–7 (Fig. S3A). This indicated that similar transcriptional programs, albeit with distinct differences as well, were active within the floral buds at different stages. A count of the number of genes expressed at different levels in the eight samples showed medium-level expression (2 ≤ FPKM < 10) by the largest portion of transcripts, followed by high-level expression (FPKM ≥ 10). The proportions of genes at the three expression levels (high, medium, and low) were similar in all stages (Fig. S3B).
Changes in transcriptome profiles during flower development
Candidate genes involved in P. tomentosa flower development were identified in differential expression analyses between consecutive time points, resulting in the identification of 6,959 DEGs during flower development. Of these, 531, 83, 222, 1,498, 23, 176 and 1,638 genes were significantly up-regulated and 911, 84, 627, 1,482, 9, 280, 1,565 genes were significantly down-regulated in stages 2–8, respectively, when compared to the preceding stage (Fig. 2A). Notably, a particularly large number of genes was up- or down-regulated in stage 5 versus stage 4, and in stage 8 versus stage 7, which indicated a large shift in the transcriptional programs. This result was consistent with the PCA (Fig. S3A). Hierarchical and K-means clustering of all DEGs are shown in Fig. 2B,C.

Gene expression dynamics during the different stages of flower development in P. tomentosa. (A) The number of up- and down-regulated genes during the various stages of flower development with respect to the preceding stage. (B) Hierarchical cluster analysis. (C) K-means clustering of the DEGs during the eight developmental stages. Black numbers on the top are the number of genes for each cluster; the red numbers are the cluster labels.
GO enrichment
The major functional categories during floral development were identified in a GO enrichment analysis of 6,959 DEGs. Many significantly enriched GO terms were related to floral development, organ development, and secondary metabolite synthesis. In biological process, ‘response to abiotic stimulus’, ‘plant-type spore development’, and ‘cellular macromolecular complex subunit organization’ were highly represented. For molecular function, GO terms related to structural molecule activity and various aspects of ‘oxidoreductase activity’ were significantly enriched. In the category ‘cellular component’, significantly enriched genes were associated with organelles, membrane, and the cell wall (Fig. S4).
The enriched biological process GO terms of the up-regulated genes were analyzed for each of two consecutive time points. Although many of the enriched GO terms were common, a few were unique in different sets of genes. For example, the GO terms related to lipid and fatty acid biosynthesis were enriched in stages 2 and 3, RNA splicing in stage 3, amino acid catabolic processes in stage 4, DNA repair and signaling in stage 5, localization in stages 6 and 7, and defense responses and cell proliferation in stage 8 (Fig. S5).
Transcription factors
TFs are the key regulatory proteins, mediate the transcriptional regulation by binding to specific motifs in the promoter of target genes. TFs are known to play a significant role in floral development. Among the 6,959 DEGs, approximately one-third (2,231) of the DEGs were TFs. These genes fell into 54 diverse categories, covering nearly all TF families, with ERF, MYB, bHLH, NAC, B3, bZIP, and MIKC being the most highly represented. Hierarchical and K-means clustering analyses depicting the differential expression profiles of the TF genes during floral development are presented in Fig. S6.
Type II MADS genes
Many of the key regulatory genes involved in floral development and flowering time are members of the MADS-box family of TFs. In plants, MADS-box family proteins are divided into types I and II, with the latter further classified into MIKCC and MIKC*.
In Arabidopsis, the MIKC* group is involved in pollen maturation23. In peach, the expression of MIKC* genes during floral development is higher in pollen than in other tissues24. In apple, MIKC* genes are highly expressed at later stages of flower development25. Our results showed higher expression of close orthologs of these genes in P. tomentosa (PtAGL65.2, PtAGL65.3, and PtAGLS4) at later stages (Fig. S7), suggesting that MIKC* genes may be involved in pollen development in this species.
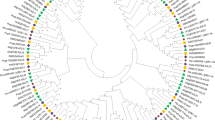
To determine the phylogenetic relationships among poplar MADS-box proteins, and then to group the respective genes within established subfamilies, we generated phylogenetic trees containing the full-length proteins from Arabidopsis, grapevine, and poplar. The MIKCC genes were divided into 13 subfamilies (Fig. 3). In this transcriptome, 37 MIKCC genes were identified. Their expression profiles in floral buds were compared by hierarchical clustering together with RT-qPCR to quantify the expression of MADS-box genes (Fig. 4). As a general rule, closely related genes within subfamilies display conserved expression patterns, although the expression levels of specific members may change during different stages. Hierarchical clustering showed that the MIKCC genes formed three clusters (Fig. 4A).

Phylogenetic tree of the MIKCc MADS-box proteins in poplar, Arabidopsis, and grapevine. The tree was generated after sequence alignment using the neighbor-joining method. Bootstrap values from 1,000 replicates were used to assess the robustness of the tree.

Expression profiles of MIKCc MADS-box genes in the floral transcriptome of P. tomentosa, and the identification of those genes by RT-qPCR. (A) Hierarchical cluster analysis of the identified MIKCc MADS-box genes in the floral transcriptome of P. tomentosa. (B) Schematic representation of the classical ABCDE model of flower development. Class A genes specify sepals; class A and B genes, petals; class B and C genes, stamens; class C genes, carpels; class D genes are specifically expressed in the ovules, and class E genes have partially redundant functions in different floral organs. (C) RT-qPCR validation of the expression levles of MIKCc MADS-box genes in floral buds. The blue and pink lines were derived from RNA-seq and RT-qPCR data, respectively. The values are means ± SD of three replicates.
Floral homeotic genes
MIKCC genes from the ABCDE model function as floral homeotic genes in Arabidopsis and other plants, where they specify floral meristem and floral organ identity26 (Fig. 4B).
The Arabidopsis A-class gene AP1 contributes to specifying floral meristem identity and participates in the development of sepals and petals26. In the present study, PtAP1 expression increased gradually during floral bud development (Fig. 4A,C). A similar expression pattern was reported for C. azalea floral buds27. In other species of Populus, PTAP1-1 and PTAP1-2 are expressed throughout the initiation of floral meristems8. In another dioecious plant of Salicaceae, willow, SAP1 is strongly expressed in all layers of the inflorescence meristem, in the developing flowers along the flanks of the inflorescence meristem, in the bract primordial, in young bracts, and in floral meristems28. In apple, AP1 orthologs are expressed at high levels during flower and fruit development25. The overexpression of apple AP1 orthologs causes early flowering in both tobacco and Arabidopsis 29,30, whereas the overexpression of PTAP1-1 does not induce early flowering in Populus 11. A recent study showed that, in hybrid aspen, LAP1 mediates photoperiodic control of seasonal growth cessation, acting downstream of the CO/FT module. Down-regulation of LAP1 is required for SD-mediated growth cessation. In contrast with AP1 targets in flowering, LAP1 connects the CO/FT module with the regulation of AINTEGUMENTA-like 1, which plays a role in SD-mediated growth cessation12.
B-class function is defined by AP3 and PI; these genes specify the identity of petals and stamens26. In the present study, PtAP3.1, PtAP3.2, and PtPI2 increased during flower development, reaching a peak during stage 8 (Fig. 4A,C). A similar pattern of PtAP3 and PdPI expression was observed by An et al.19 and Zhang et al.31, respectively. In Eucalyptus grandis, EgPI1-3 are also up-regulated during late compared to early flower development7. During microsporogenesis (stage 8), high-level expression of PtAP3 and PtPI indicated the close relationship of these genes to pollen maturation. AP3/PI genes are highly expressed in peach pollen24 and in the floral tissues of Japanese apricot32 and apple25. In black cottonwood, the AP3 ortholog is initially expressed in the inner whorl of both male and female floral meristems and maintained in the stamen primordia as the reproductive primordia begin to form33. The similar spatial expression pattern of AP3/PI clade genes in poplar, peach, Japanese apricot, and apple is consistent with their functional conservation in perennial woody plants.
The AG subfamily includes C- and D-class genes. The C-function is defined by AG, which specifies the stamen and carpel, and the D-function by SHP1&2 and STK, which shape the identity of the ovules26. In the RNA-seq data, PtAG1 was expressed first at low levels during early stages, and then at higher levels, whereas PtSTK2 was exclusively expressed during stage 8, suggesting its role in later developmental events (Fig. 4A,C). Similarly, the levels of EgAG and EgSTK expression increased during floral bud development in Eucalyptus 7. In P. trichocarpa, PTAG1&2 are expressed in the inner whorl of male and female flowers both before and after reproductive primordia emerge, indicating their function conservation in specifying male and female reproductive identity34. The rapid up-regulation of PtSTK2 during microsporogenesis suggested its importance in specifying male reproductive identity. Interestingly, in our data, we did not detect any homologs of Arabidopsis SHP1&2, as also reported for grapevine35. In other perennial trees, such as apple, peach, and Japanese apricot, AG homologs are highly expressed during flower and fruit development24,25,32.
SEP genes provide the E function and play redundant roles in floral organ and meristem identity in four whorls26. Tomato and strawberry SEP orthologs contribute to fruit development36,37. In this study, four SEP orthologs were identified. The expression of PtSEP1 and PtSEP3 increased gradually during floral bud development, indicating their increasingly important role from floral induction to organ development (Fig. 4A,C). Similarly, in P. tremuloides, the SEP-class genes PTM3/4 and 6 are expressed in all stages of male and female floral development, predominantly in the inner sexual whorl, within developing ovules of female flowers, and in the anther primordia of male flowers38. In apple, grape, and apricot, the high-level expression of SEP orthologs is maintained throughout development, from flower initiation to fruit development25,32,35.
In previous study, PtAP3 was not restricted to floral buds but was expressed at similar levels in the vegetative tissues of P. tomentosa 19. In addition to male floral buds and inflorescences of P. deltoides, PdPI is expressed in roots, female inflorescences, immature xylem, leaves, and apical buds31. In P. trichocarpa, the C-class gene PTAG is consistently expressed in vegetative tissues34. Expression of the P. tremuloides E-class genes PTM3&4 occurs in terminal buds, young leaves, and stems38. The broad expression of poplar BCE-class genes suggests their additional roles in vegetative development and is consistent with the long evolutionary history of angiosperms39.
SOC subfamily
In Arabidopsis, SOC1 functions as a flowering promoter, integrating signals from multiple pathways, together with AGL24, which activates downstream targets, such as the flowering promoter FT and the flower meristem identity genes AP1 and LFY 2. In our study, PtSOC1 and PtSOC3.1 were located to cluster 1, and PtSOC2-L to cluster 3. Cluster 1 transcripts were highly expressed in floral buds during the initiation and proliferation of inflorescence primordia (stages 1 and 2). PtSOC1 and PtSOC3.1 expression was highest in stage 1 (floral induction) and then decreased (except for the later increase of PtSOC1), which suggested that these genes play a crucial role in flowering induction (Fig. 4A,C). Similarly, in grape, the expression of VvSOC1.1 decreased during flower meristem initiation and flower development35. In apple, MdSOC1 expression gradually declined during flower induction40. In E. grandis, nearly half of the type II MADS-box genes belong to the SOC subfamily; its expansion has been attributed to tandem duplications7. By contrast, in our study, the P. tomentosa transcriptome contained only four members of the SOC family. Previous study similarly reported only five members in the SOC subfamily of P. trichocarpa 7. Thus, in poplar, the SOC subfamily did not undergo huge expansion that occurred in E. grandis.
FLC-like and SVP/AGL24 subfamilies
In Arabidopsis, the MIKCc genes regulating flowering transition mainly belong to the FLC, SVP, and SOC subfamilies. FLC act as an important floral repressor active in leaves and the apical meristem. Its expression is down-regulated after long-term cold during vernalization. In addition, FLC represses SOC1 and FT and interacts with SVP to repress of SOC1 and FT 26. We identified four FLC-like genes (PtFLC2–5), all of which were mainly expressed during later stages. Of these, PtFLC2 and PtFLC4 showed opposing expression pattern (Fig. 4A,C), as reported for two FLC homologs (VvFLC1&2) in the buds of grapevine6. PtFLC2 expression decreased in response to chilling during winter dormancy and increased after dormancy release (Fig. 4). Similar patterns of VvFLC1 expression in grape6, and of PEP1 (FLC homolog) in Arabis alpina 41, have been described. By contrast, the expression of PtFLC4 increased during dormancy and declined when growth resumed after the cold period. Similar results were reported for the FLC-like genes in apple25, Arabidopsis 42, and grape6. In P. tomentosa, divergence between the sequences of FLC-like genes and those of Arabidopsis and their different expression patterns may explain their functional divergence. Previous study in poplar showed that FLC-like assumed different functions in apical bud development and dormancy43.
In Arabidopsis, the SVP/AGL24 subfamily contains only two members: SVP acts as a floral repressor, through the negative regulation of FT, while AGL24 promotes flowering by positively regulating LFY 26. We identified five orthologs of SVP/AGL24 from the RNA-seq data. PtSVP expression was high during floral induction and initiation, then decreased gradually, without significant changes during dormancy (Fig. 4A,C), what would not justify their role in dormancy. In Eucalyptus, EgSVP1 was down-regulated in late versus early flower development7. Expression of the SVP ortholog in Camellia azalea was also reduced during floral bud development27. In Populus, low temperatures and long-days are required to break bud dormancy and they determine the specific flowering time16. In peach and leafy spurge, SVP orthologs, as dormancy-associated MADS-box genes (DAM),are important in growth cessation, bud set, and seasonal vegetative and floral bud dormancy44,45. Our data showed a weak reduction in PtSVP-L1 expression during dormancy release (Fig. 4A,C). Dormancy induction signals activate DAM expression, which then decreases with dormancy release in leafy spurge45, Japanese apricot46, raspberry47, and apple25.
In summary, MADS-box gene family play important roles in floral development in poplar. Here, MADS-box homologs were identified and their expression patterns at eight stages were studied. According to these results, their potential functions were speculated, but the exact function still needs further studies by genetic transformation, such as overexpression or silence (RNAi and CRISPR-Cas9). Previously, Song et al.48 also explored the floral development in P. tomentosa using transcriptome, as well as phytohormone and DNA methylation analysis. However, they focused on the differences between male and female poplars during floral development at transcriptional level. Relatively limited candidate genes (24 genes) were chosen for confirming their expression pattern during floral development in males and females. Although these genes might play important roles during flower development, they mainly involved in phytohormone synthesis and metabolism, but few genes were involved in tranditional floral pathways. Floral development is a dynamic developmental process and regulated by many related genes in complex network. Our study aimed to generate a global view of genome-wide transcriptome dynamics during floral bud development in P. tomentosa. Besides MADS-box genes, numerous other genes associated with floral development, such as SPL gene family and RLK genes etc., were identified and their expression patterns were also analyzed. All of these will be necessary and important to elucidate molecular basis of flower development in Populus.
SPL gene family
SPL family play crucial roles in plant growth and development processes, such as phase transition, flowering, fruit development and architecture6. In Arabidopsis, the SPL family has 16 members. AtSPL1, 7, 12, 14 and 16 are expressed constitutively, while the remaining genes are highly expressed in flowers49. Previous study in poplar identified 28 SPL family members50.
Ten of the sixteen AtSPLs (2–6, 9–11, 13 and 15) are post-transcriptionally regulated by the miR156 family, resulting in the incorporation of endogenous age/development signals into vegetative phase transition and flowering51,52. This regulatory mechanism is conserved in woody perennials6. AtSPL3–5 contain sequences complementary to miR156 in the 3′ UTR; all of those genes promote the juvenile-to-adult phase transition and flowering53. In our study, their orthologs, PtSPL16, 20, and 23–25,were highly expressed in the early stages of floral buds (Fig. 5). AtSPL2, 10, and 11 regulate the morphological changes in cauline leaves and flowers during the reproductive phase54. Their orthologs, PtSPL11, 19, and 29, decreased during floral bud development (Fig. 5). AtSPL9 and 15 act redundantly in controlling the juvenile-to-adult phase transition and the leaf initiation rate55. Orthologs PtSPL8, 17, and 27 were located close together in the phylogenetic tree (Fig. 5A).

Expression profiles of the squamosa promoter binding protein-like family (SPL) gene family in the floral transcriptome and their RT-qPCR validation. (A) Phylogenetic tree of SPL family proteins in poplar and Arabidopsis. The tree was generated after sequence alignment using the neighbor-joining method. Bootstrap values from 1,000 replicates were used to assess the robustness of the tree. (B) Hierarchical cluster analysis of the identified SPL genes in the floral transcriptome of P. tomentosa. (C) RT-qPCR validation of the SPL gene expression levels in floral buds. The blue and pink lines were derived from the RNA-seq and RT-qPCR data, respectively. Values are means ± SD.
Six AtSPLs are not targets of miR156/7 in Arabidopsis. Among them, AtSPL8 regulates pollen sac development56 and male fertility57. The expression of PtSPL26 (AtSPL8 ortholog) is restricted to the dormancy period (Fig. 5B). AtSPL14 regulates plant architecture and represses both the vegetative phase transition and flowering58. In the present study, the transcription of PtSPL9, a homolog of AtSPL14, was highest during the floral induction stage (Fig. 5).
PtSPL12&13 (AtSPL6 homologs) expression was highest during dormancy (Fig. 5) and the pattern was similar to that of PtFLC4; these SPL genes may be involved in dormancy maintenance. The expression of PtSPL12&13 decreased during dormancy (Fig. 5). SPL-like 3&6 have also been detected during the dormancy of other Populus species, with increases in SPL6-like and decreases in SPL3-like as dormancy progressed59. Further studies are needed to elucidate the roles of the SPLs in flowering and dormancy.
RLK genes
RLK play important roles in flower development and reproduction, including tapetum development and microspore maturation60, male-female interactions61, as well as pollen tube reception62. In this study, we identified 216 unigenes annotated as RLK. Their hierarchical clustering is shown in Fig. S8. The Arabidopsis ERECTA (ER) family receptor kinases ER-LIKE1 (ERL1), and ERL2 regulate inflorescence architecture, floral meristem organization, and floral organ identity63. In our study, PtERL1 and PtERL4 (comp650512_c0 and comp484228_c0) were expressed consistently during floral bud development (Fig. S8), indicating their roles in flower development. Arabidopsis somatic embryogenesis receptor-like kinases 1&2 (SERK1&2) are essential for tapetum development and microspore maturation60. In our study, PtSERK1 and PtSERK2 were down-regulated during floral bud development, while the transcripts of comp619813_c0 and comp636715_c0 (two other homologs of AtSERK 1&2) reached their highest level during stage 8 (Fig. S8), indicative of their different functions during floral organ development.
Other well-known genes
Some genes specifically involved in anther or tapetum development were clearly up-regulated during stage 8 but were undetected or expressed at extremely low levels during earlier stages. These genes included AMS, CYP703A2, TDF1, and ACOS5 (Fig. S9) and their expression coincided with active anther and stamen development. Similar pattern of up-regulation was observed in the late-stage of floral buds in Eucalyptus 7. In other species, these genes were shown to be associated with later stages of tapetum and pollen development, such as pollen wall formation64,65,66. The transcripts of other genes required for tapetum development, such as EMS1 and TPD1, reached their highest level during stage 5 and 6 (Fig. S9). In addition, genes involved in pollen tube growth, pollen hydration, and callose degradation during tetrad dissolution, such as POE1, SKU5-like, PME, OLE, and βGLU, were differentially expressed during floral bud development67 (Fig. S9A).
PtLFY was first up-regulated, then down-regulated, during floral bud development but increased during stage 8 (Fig. 6). In Arabidopsis, LFY is a floral meristem identity gene, regulates floral initiation and activates the floral homeotic genes2. As expected, PtLFY was up-regulated from floral induction to initiation and then down-regulated (Fig. 6). Previous study showed that PTLF is strongly expressed in developing inflorescences but is also detected in leaf primordia, young leaves, apical vegetative buds, and seedlings10. When overexpressing LFY constitutively in hybrid aspen, transgenic plants flowered in vitro within 7 months68, although this effect was later shown to be highly variable between poplar clones. The overexpression of LFY induces solitary flowers in male aspen clones; however, this occurs only infrequently in female clones10. As PTLF was less effective in accelerating flowering, the orthologous proteins may differ in their activities or regulatory interactions8. Interestingly, the catkins of PtLFY-RNAi poplars were small and lacked stigmas or ovules, while the trees had normal or robust vegetative growth69. PtLFY was also increased, along with that of PtSOC1, during dormancy release (Figs 4, 6). Arabidopsis SOC1 forms a positive feedback loop with AGL24. These two factors may form a complex that stimulates the up-regulation of LFY 2.

Expression profiles of several well-known floral-related genes and their RT-qPCR validation. (A) Heat map of gene expression. (B) Phylogenetic tree of the poplar, Arabidopsis, and grape FT/TFL1 gene family containing phosphatidylethanolamine-binding proteins (PEBPs), obtained using the neighbor-joining method. The unit for the scale bar indicates branch lengths (0.05 substitutions per site). (C) RT-qPCR validation of the expression profiles obtained by RNA-seq. The values are means ± SD.
WUSCHEL (WUS)-related homeobox (WOX) family genes play important roles in the maintenance and proliferation of the stem cell population in the shoot apical meristem; they also mediate floral organ development70. We identified four PtWUS and PtWOX genes; their expression patterns are shown in Fig. 6. In Arabidopsis floral meristem, WUS promotes the expression of AG, which then negatively feeds back on WUS, resulting in the down-regulation of stem cell proliferation and the promotion of determinacy71.
In Arabidopsis, CO initiates flowering via the up-regulation of FT 17. Similar to the Arabidopsis CO/FT regulon, the poplar CO2/FT1 regulon controls the timing of flowering and regulates both SD-induced growth cessation and fall bud set13. However, the overexpression of PtCO1&2 does not alter normal reproductive onset, spring bud break, or bud set in poplar11,17, although poplar CO1 could rescue the late-flowering phenotype of Arabidopsis co-1 mutants17. These results indicate that CO/FT was modified in poplar following the divergence of the Arabidopsis and poplar lineages. In our study, PtCO1 transcript was most abundant in stage 8. Hsu et al.17 also found that poplar CO1 was consistently expressed in reproductive buds, with the highest level in late winter (Fig. 6).
Members of the FT/TFL1 family are important regulators of flowering time and dormancy21,72. We identified four FT/TFL1 genes in our floral transcriptome (Fig. 6B). PtFT1 was up-regulated in the later stages of floral bud development, especially in response to winter temperatures, while PtCEN1 was mainly expressed in the floral induction stage, decreasing thereafter (Fig. 6A,C). In Arabidopsis, FT promotes flowering, while TFL1 represses its onset72. In Populus, the overexpression of PtFT1&2 induces early flowering13,14. PtFT1 acts as a strong promoter of precocious flowering and induces the formation of wild-type inflorescences13, whereas PtFT2 induces only the formation of individual flowers14. Subsequent studies showed that sub-functionalization of these two genes had occurred, they have distinct seasonal expression patterns15. PtFT1 is up-regulated in response to winter temperatures and determines reproductive onset, while PtFT2 is up-regulated in response to the long-days and warm temperatures of the growing season and promotes vegetative growth, as well as the inhibition of bud set13,15,16. In our study, the pattern of PtFT1 expression was consistent with those results (Fig. 6C). In Arabidopsis, SVP represses the expression of FT; likewise, our data showed the reciprocal expression of PtSVP and PtFT displayed (Figs 4 and 6). A reciprocal pattern of expression was also described for DAM and FT in leafy spurge45 and apple25. The down-regulation of PopCEN1&2 by RNAi accelerated the onset of mature tree characteristics and regulated axillary meristem identity18. In our study, PtCEN1 declined sharply from floral induction to initiation (Fig. 6C). Its low-level expression may allow the accumulation of transcripts of other floral promoters, resulting in floral initiation. Similar expression patterns were determined in the floral buds of apple40. Previous study showed that in PopCEN1-RNAi plants, the axillary inflorescences contained fewer flowers than did the wild-type, suggesting a role for PopCEN1 in maintaining the indeterminacy of the inflorescence apex. PopCEN1 also regulates the dormancy transition in Populus, while its overexpression alteres the chilling requirements and delays bud break18.
Co-expression networks
To search for the regulatory genes that participate in flowering in poplar, we selected 98 floral-related genes involved in flower meristem identity and flower development and used WGCNA to construct co-expression networks, in which the hub genes showed the densest connections (Fig. 7, Table S1). Many of the hub genes were MADS-box genes, including PtAP1, PtSVP-L3, PtSEP1, PtPI, and PtSVP1. In addition to PtAP1, the hub genes with the highest edge numbers were PtHUA1.1 and PtFWA.4. In Arabidopsis, HUA1 is required for floral determinacy and it facilitates AG pre-mRNA processing71. FLOWERING WAGENINGEN (FWA) encodes a homeodomain-containing TF that controls flowering73. Other highly connected hub genes were PtBRI1, PtUBC1.1, and PtAMP1.2. In Arabidopsis, BRASSINOSTEROID INSENSITIVE 1 (BRI1) is involved in tapetum cell differentiation in the anther wall, pollen exine formation, and the positive regulation of flower development74. UBIQUITIN CARRIER PROTEIN 1 (UBC1) is associated with the negative regulation of flower development75. ALTERED MERISTEM PROGRAM 1 (AMP1) alters flowering time and photomorphogenesis and increases cytokinin biosynthesis76.

Co-expression network of 98 floral-related genes associated with flower meristem identity and flower development in P. tomentosa. The threshold of the Pearson correlation coefficient was set at ≥0.7 (or ≤−0.7). The larger and more yellow the circles, the higher the connectivity.
Materials and Methods
Plant material and RNA extraction
Adult male P. tomentosa trees were grown in the nursery of Beijing Forestry University (Beijing, China) in a natural environment. The axillary floral buds were collected from June 2013 through February 2014, to cover eight representative stages of flower development including floral induction, floral initiation, and organ differentiation: stage 1, floral induction; stage 2, primordia formation; stage 3, organogenesis; stages 4, enlargement; stage 5, archespore formation; stages 6 and 7: dormancy; stage 8: microsporogenesis (Fig. 1). The collected samples were immediately frozen in liquid nitrogen and stored at −80 °C until further use. Total RNA was isolated from the pooled samples as described previously20. For every stage, equal amounts of total RNA from eight biological replicates were pooled to promote sample homogeneity and decrease bias. The quality and quantity of the total RNA samples was analyzed using a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Inc.) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Floral tissue microscopy
Histological sections were prepared by fixing the floral buds from the eight different developmental stages in formalin-acetic acid. The buds were then dehydrated in a graded ethanol series and embedded in paraplast. Serial sections were obtained using a Leica microtome, mounted on microscope slides, stained with 1% safranin, and observed with an Olympus BX-61 microscope (Olympus, Tokyo, Japan).
RNA deep sequencing
RNA samples for cDNA libraries and RNA-seq were prepared using the Illumina kit according to the manufacturer’s protocol (Illumina, San Diego, CA, USA). The eight libraries were sequenced separately with the Illumina HiSeqTM 2000 platform using paired-end technology. Contamination detection was assessed by comparing the randomly selected 500,000 reads from each raw sequencing dataset against the nucleotide database from NCBI. The files of raw fastq were also checked by FastQC. After removal of the adapter sequences, low-quality sequences and reads with >10% Q < 20 bases, all valid reads were pooled for de novo assembly using the Trinity program.
Annotation of unigenes
The longest transcript at each locus was regarded as a unigene. To understand the functions of the identified unigenes, they were annotated using BLASTx alignment against the NCBI non-redundant protein (Nr), Swiss-Prot protein, and Cluster of Orthologous Groups (COG) databases with an E-value cut-off of 10−5 77. Functional annotation by Gene Ontology (GO) terms was analyzed using Blast2GO78. Pathway annotation with the Kyoto Encyclopedia of Genes and Genomes (KEGG) was performed using the KAAS server79. Unigenes were also annotated against the TAIR10 database of Arabidopsis and the v3.0 database of P. trichocarpa.
Differential gene expression and enrichment analysis
Gene expression levels were estimated as fragments per kilobase of transcript per million mapped reads (FPKM)80. Differentially expressed genes (DEGs) were analyzed using the DESeq software81. The false discovery rate (FDR) was used to determine the p-value threshold in multiple tests82. Due to the time-series parameters of this study, seven sets of DEG analysis were separately performed. In each analysis, a criteria of |log2(Ratio)| ≥ 1 and an FDR of ≤ 0.001 between the two consecutive time points was used to identify DEGs. GO enrichment was performed based on a p-value < 0.05, after applying the Benjamini Hochberg correction using agriGO83. Hierarchical and k-means clustering were generated by MultiExperiment Viewer (MeV; ver. 4.6.2). A principal component analysis (PCA) was performed using MeV.
Quantitative reverse transcription PCR
Total RNA was treated with RQ1 DNase I (Promega, Madison, WI, USA) to remove contaminating genomic DNA. The first-strand cDNA was synthesized using 1 μg of RNA with oligo d(T)20 and a reverse transcription system (Promega). It was then diluted 1:10 with ddH2O and used as a template for RT-qPCR amplification on a 7500 Fast real-time PCR system platform (Applied Biosystems, Foster City, CA, USA) using SYBR® Premix Ex TaqTM (TaKaRa, Otsu, Japan). The gene-specific primers employed in the RT-qPCR analysis are listed in Table S2. Thermal cycling was performed at 95 °C for 30 s, followed by 40 cycles of amplification (95 °C for 5 s, 60 °C for 20 s, 72 °C for 15 s), with a final extension of 7 min at 72 °C. The plates were read every 0.2 °C for 1 s from 70 to 95 °C, to generate melting curves. PtACTIN 19 was used as the internal reference gene for normalization according to the 2−ΔΔCt method. Each reaction was performed in three replicates.
Annotation, phylogenetic analysis, and co-expression network of flower-related genes
Blastn searches of the Phytozome database, using Arabidopsis and P. trichocarpa genes as queries, were used to identify flower-related genes in P. tomentosa. Multiple sequence alignments and the phylogenetic analysis were performed as described previously84. Transcription factors (TFs) were identified by using the unigenes from the assembled sequences in a BLAST analysis against TFs from P. trichocarpa in the Plant Transcription Factor Database (PlantTFDB)85. A weighted genes co-expression network analysis (WGCNA) was performed using OmicsShare Tools.
References
O’Maoileidigh, D. S., Graciet, E. & Wellmer, F. Gene networks controlling Arabidopsis thaliana flower development. The New phytologist 201, 16–30, https://doi.org/10.1111/nph.12444 (2014).
Wellmer, F. & Riechmann, J. L. Gene networks controlling the initiation of flower development. Trends in genetics: TIG 26, 519–527, https://doi.org/10.1016/j.tig.2010.09.001 (2010).
Gregis, V., Sessa, A., Colombo, L. & Kater, M. M. AGL24, SHORT VEGETATIVE PHASE, and APETALA1 redundantly control AGAMOUS during early stages of flower development in Arabidopsis. The Plant cell 18, 1373–1382, https://doi.org/10.1105/tpc.106.041798 (2006).
Zhuang, W. B., Shi, T., Gao, Z. H., Zhang, Z. & Zhang, J. Y. Differential expression of proteins associated with seasonal bud dormancy at four critical stages in Japanese apricot. Plant biology 15, 233–242, https://doi.org/10.1111/j.1438-8677.2012.00589.x (2013).
Zhang, X. M., Zhao, L., Larson-Rabin, Z., Li, D. Z. & Guo, Z. H. De novo sequencing and characterization of the floral transcriptome of Dendrocalamus latiflorus (Poaceae: Bambusoideae). PloS one 7, e42082, https://doi.org/10.1371/journal.pone.0042082 (2012).
Diaz-Riquelme, J., Grimplet, J., Martinez-Zapater, J. M. & Carmona, M. J. Transcriptome variation along bud development in grapevine (Vitis vinifera L.). BMC plant biology 12, 181, https://doi.org/10.1186/1471-2229-12-181 (2012).
Vining, K. J. et al. The floral transcriptome of Eucalyptus grandis. The New phytologist 206, 1406–1422, https://doi.org/10.1111/nph.13077 (2015).
Brunner, A. M. Reproductive Development in Populus. (Springer New York, 2010).
Brunner, A. M. & Nilsson, O. Revisiting tree maturation and floral initiation in the poplar functional genomics era. New Phytologist 164, 43–51, https://doi.org/10.1111/j.1469-8137.2004.01165.x (2004).
Rottmann, W. H. et al. Diverse effects of overexpression of LEAFY and PTLF, a poplar (Populus) homolog of LEAFY/FLORICAULA, in transgenic poplar and Arabidopsis. The Plant journal: for cell and molecular biology 22, 235–245 (2000).
Strauss, S. H., Brunner, A. M., Busov, V. B., Ma, C. P. & Meilan, R. Ten lessons from 15 years of transgenic Populus research. Forestry 77, 455–465, https://doi.org/10.1093/forestry/77.5.455 (2004).
Azeez, A., Miskolczi, P., Tylewicz, S. & Bhalerao, R. P. A tree ortholog of APETALA1 mediates photoperiodic control of seasonal growth. Current biology: CB 24, 717–724, https://doi.org/10.1016/j.cub.2014.02.037 (2014).
Bohlenius, H. et al. CO/FT regulatory module controls timing of flowering and seasonal growth cessation in trees. Science 312, 1040–1043, https://doi.org/10.1126/science.1126038 (2006).
Hsu, C. Y., Liu, Y., Luthe, D. S. & Yuceer, C. Poplar FT2 shortens the juvenile phase and promotes seasonal flowering. The Plant cell 18, 1846–1861, https://doi.org/10.1105/tpc.106.041038 (2006).
Hsu, C. Y. et al. FLOWERING LOCUS T duplication coordinates reproductive and vegetative growth in perennial poplar. Proceedings of the National Academy of Sciences of the United States of America 108, 10756–10761, https://doi.org/10.1073/pnas.1104713108 (2011).
Rinne, P. L. et al. Chilling of dormant buds hyperinduces FLOWERING LOCUS T and recruits GA-inducible 1,3-beta-glucanases to reopen signal conduits and release dormancy in Populus. The Plant cell 23, 130–146, https://doi.org/10.1105/tpc.110.081307 (2011).
Hsu, C. Y. et al. Overexpression of CONSTANS homologs CO1 and CO2 fails to alter normal reproductive onset and fall bud set in woody perennial poplar. PloS one 7, e45448, https://doi.org/10.1371/journal.pone.0045448 (2012).
Mohamed, R. et al. Populus CEN/TFL1 regulates first onset of flowering, axillary meristem identity and dormancy release in Populus. The Plant journal: for cell and molecular biology 62, 674–688, https://doi.org/10.1111/j.1365-313X.2010.04185.x (2010).
An, X. M. et al. Isolation of a LEAFY homolog from Populus tomentosa: expression of PtLFY In P. tomentosa floral buds and PtLFY-IR-mediated gene silencing in tobacco (Nicotiana tabacum). Plant cell reports 30, 89–100, https://doi.org/10.1007/s00299-010-0947-0 (2011).
Ye, M. et al. Study of seed hair growth in Populus tomentosa, an important character of female floral bud development. BMC genomics 15, 475, https://doi.org/10.1186/1471-2164-15-475 (2014).
Cooke, J. E., Eriksson, M. E. & Junttila, O. The dynamic nature of bud dormancy in trees: environmental control and molecular mechanisms. Plant, cell & environment 35, 1707–1728, https://doi.org/10.1111/j.1365-3040.2012.02552.x (2012).
Ozsolak, F. & Milos, P. M. RNA sequencing: advances, challenges and opportunities. Nature reviews. Genetics 12, 87–98, https://doi.org/10.1038/nrg2934 (2011).
Liu, Y. et al. Functional conservation of MIKC*-Type MADS box genes in Arabidopsis and rice pollen maturation. The Plant cell 25, 1288–1303, https://doi.org/10.1105/tpc.113.110049 (2013).
Wells, C. E., Vendramin, E., Jimenez Tarodo, S., Verde, I. & Bielenberg, D. G. A genome-wide analysis of MADS-box genes in peach [Prunus persica (L.) Batsch]. BMC plant biology 15, 41, https://doi.org/10.1186/s12870-015-0436-2 (2015).
Kumar, G. et al. Comparative phylogenetic analysis and transcriptional profiling of MADS-box gene family identified DAM and FLC-like genes in apple (Malus x domestica). Scientific reports 6, 20695, https://doi.org/10.1038/srep20695 (2016).
Smaczniak, C., Immink, R. G., Angenent, G. C. & Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139, 3081–3098, https://doi.org/10.1242/dev.074674 (2012).
Fan, Z. et al. Genome-wide transcriptome profiling provides insights into floral bud development of summer-flowering Camellia azalea. Scientific reports 5 (2015).
Fernando, D. D. & Zhang, S. Constitutive expression of the SAP1 gene from willow (Salix discolor) causes early flowering in Arabidopsis thaliana. Development genes and evolution 216, 19–28, https://doi.org/10.1007/s00427-005-0026-7 (2006).
Kotoda, N. et al. Overexpression of MdMADS5, an APETALA1-like gene of apple, causes early flowering in transgenic Arabidopsis. Plant Science 162, 679–687 (2002).
Sung, S. K., Yu, G. H. & An, G. Characterization of MdMADS2, a member of the SQUAMOSA subfamily of genes, in apple. Plant physiology 120, 969–978 (1999).
Zhang, B., Su, X. & Zhou, X. A MADS-box gene of Populus deltoides expressed during flower development and in vegetative organs. Tree physiology 28, 929–934 (2008).
Xu, Z. et al. Genome-wide identification, characterisation and expression analysis of the MADS-box gene family in Prunus mume. Molecular genetics and genomics: MGG 289, 903–920, https://doi.org/10.1007/s00438-014-0863-z (2014).
Sheppard, L. A. et al. A DEFICIENS homolog from the dioecious tree black cottonwood is expressed in female and male floral meristems of the two-whorled, unisexual flowers. Plant physiology 124, 627–640 (2000).
Brunner, A. M. et al. Structure and expression of duplicate AGAMOUS orthologues in poplar. Plant molecular biology 44, 619–634 (2000).
Díaz-Riquelme, J., Lijavetzky, D., Martínez-Zapater, J. M. & Carmona, M. J. Genome-wide analysis of MIKCC-type MADS box genes in grapevine. Plant physiology 149, 354–369 (2009).
Seymour, G. B. et al. A SEPALLATA gene is involved in the development and ripening of strawberry (Fragaria x ananassa Duch.) fruit, a non-climacteric tissue. Journal of experimental botany 62, 1179–1188, https://doi.org/10.1093/jxb/erq360 (2011).
Vrebalov, J. et al. Fleshy fruit expansion and ripening are regulated by the Tomato SHATTERPROOF gene. TAGL1. The Plant cell 21, 3041–3062, https://doi.org/10.1105/tpc.109.066936 (2009).
Cseke, L. J. et al. SEP-class genes in Populus tremuloides and their likely role in reproductive survival of poplar trees. Gene 358, 1–16, https://doi.org/10.1016/j.gene.2005.05.035 (2005).
Yoo, M. J., Chanderbali, A. S., Altman, N. S., Soltis, P. S. & Soltis, D. E. Evolutionary trends in the floral transcriptome: insights from one of the basalmost angiosperms, the water lily Nuphar advena (Nymphaeaceae). The Plant journal: for cell and molecular biology 64, 687–698, https://doi.org/10.1111/j.1365-313X.2010.04357.x (2010).
Xing, L. B. et al. Transcription Profiles Reveal Sugar and Hormone Signaling Pathways Mediating Flower Induction in Apple (Malus domestica Borkh.). Plant & cell physiology 56, 2052–2068, https://doi.org/10.1093/pcp/pcv124 (2015).
Wang, R. et al. PEP1 regulates perennial flowering in Arabis alpina. Nature 459, 423–427, https://doi.org/10.1038/nature07988 (2009).
Deng, W. et al. FLOWERING LOCUS C (FLC) regulates development pathways throughout the life cycle of Arabidopsis. Proceedings of the National Academy of Sciences of the United States of America 108, 6680–6685, https://doi.org/10.1073/pnas.1103175108 (2011).
Chen, K. Y. Type. II MADS-box genes associated with poplar apical bud development and dormancy. Dissertations & Theses - Gradworks (2008).
Bielenberg, D. G. et al. Sequencing and annotation of the evergrowing locus in peach [Prunus persica (L.) Batsch] reveals a cluster of six MADS-box transcription factors as candidate genes for regulation of terminal bud formation. Tree Genetics & Genomes 4, 495–507 (2008).
Horvath, D. P., Sung, S., Kim, D., Chao, W. & Anderson, J. Characterization, expression and function of DORMANCY ASSOCIATED MADS-BOX genes from leafy spurge. Plant molecular biology 73, 169–179, https://doi.org/10.1007/s11103-009-9596-5 (2010).
Sasaki, R. et al. Functional and expressional analyses of PmDAM genes associated with endodormancy in Japanese apricot. Plant physiology 157, 485–497, https://doi.org/10.1104/pp.111.181982 (2011).
Mazzitelli, L. et al. Co-ordinated gene expression during phases of dormancy release in raspberry (Rubus idaeus L.) buds. Journal of experimental botany 58, 1035–1045, https://doi.org/10.1093/jxb/erl266 (2007).
Song, Y. et al. Sexual dimorphic floral development in dioecious plants revealed by transcriptome, phytohormone, and DNA methylation analysis in Populus tomentosa. Plant molecular biology 83, 559–576, https://doi.org/10.1007/s11103-013-0108-2 (2013).
Cardon, G. et al. Molecular characterisation of the Arabidopsis SBP-box genes. Gene 237, 91–104 (1999).
Li, C. & Lu, S. Molecular characterization of the SPL gene family in Populus trichocarpa. BMC plant biology 14, 131, https://doi.org/10.1186/1471-2229-14-131 (2014).
Guo, A. Y. et al. Genome-wide identification and evolutionary analysis of the plant specific SBP-box transcription factor family. Gene 418, 1–8, https://doi.org/10.1016/j.gene.2008.03.016 (2008).
Wu, G. & Poethig, R. S. Temporal regulation of shoot development in Arabidopsis thaliana by miR156 and its target SPL3. Development 133, 3539–3547, https://doi.org/10.1242/dev.02521 (2006).
Amasino, R. Seasonal and developmental timing of flowering. The Plant journal: for cell and molecular biology 61, 1001–1013, https://doi.org/10.1111/j.1365-313X.2010.04148.x (2010).
Shikata, M., Koyama, T., Mitsuda, N. & Ohme-Takagi, M. Arabidopsis SBP-box genes SPL10, SPL11 and SPL2 control morphological change in association with shoot maturation in the reproductive phase. Plant & cell physiology 50, 2133–2145, https://doi.org/10.1093/pcp/pcp148 (2009).
Schwarz, S., Grande, A. V., Bujdoso, N., Saedler, H. & Huijser, P. The microRNA regulated SBP-box genes SPL9 and SPL15 control shoot maturation in Arabidopsis. Plant molecular biology 67, 183–195, https://doi.org/10.1007/s11103-008-9310-z (2008).
Unte, U. S. et al. SPL8, an SBP-box gene that affects pollen sac development in Arabidopsis. The Plant cell 15, 1009–1019 (2003).
Xing, S., Salinas, M., Hohmann, S., Berndtgen, R. & Huijser, P. miR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. The Plant cell 22, 3935–3950, https://doi.org/10.1105/tpc.110.079343 (2010).
Stone, J. M., Liang, X., Nekl, E. R. & Stiers, J. J. Arabidopsis AtSPL14, a plant-specific SBP-domain transcription factor, participates in plant development and sensitivity to fumonisin B1. The Plant journal: for cell and molecular biology 41, 744–754, https://doi.org/10.1111/j.1365-313X.2005.02334.x (2005).
Ruttink, T. et al. A molecular timetable for apical bud formation and dormancy induction in poplar. The Plant cell 19, 2370–2390, https://doi.org/10.1105/tpc.107.052811 (2007).
Colcombet, J., Boisson-Dernier, A., Ros-Palau, R., Vera, C. E. & Schroeder, J. I. Arabidopsis SOMATIC EMBRYOGENESIS RECEPTOR KINASES1 and 2 are essential for tapetum development and microspore maturation. The Plant cell 17, 3350–3361, https://doi.org/10.1105/tpc.105.036731 (2005).
Kachroo, A., Schopfer, C. R., Nasrallah, M. E. & Nasrallah, J. B. Allele-specific receptor-ligand interactions in Brassica self-incompatibility. Science 293, 1824–1826, https://doi.org/10.1126/science.1062509 (2001).
Escobar-Restrepo, J. M. et al. The FERONIA receptor-like kinase mediates male-female interactions during pollen tube reception. Science 317, 656–660, https://doi.org/10.1126/science.1143562 (2007).
Shpak, E. D., Berthiaume, C. T., Hill, E. J. & Torii, K. U. Synergistic interaction of three ERECTA-family receptor-like kinases controls Arabidopsis organ growth and flower development by promoting cell proliferation. Development 131, 1491–1501, https://doi.org/10.1242/dev.01028 (2004).
Sorensen, A. M. et al. The Arabidopsis ABORTED MICROSPORES (AMS) gene encodes a MYC class transcription factor. The Plant journal: for cell and molecular biology 33, 413–423 (2003).
Zhu, J. et al. Defective in Tapetal development and function 1 is essential for anther development and tapetal function for microspore maturation in Arabidopsis. The Plant journal: for cell and molecular biology 55, 266–277, https://doi.org/10.1111/j.1365-313X.2008.03500.x (2008).
de Azevedo Souza, C. et al. A novel fatty Acyl-CoA Synthetase is required for pollen development and sporopollenin biosynthesis in Arabidopsis. The Plant cell 21, 507–525, https://doi.org/10.1105/tpc.108.062513 (2009).
Alagna, F. et al. Transcript Analysis and Regulative Events during Flower Development in Olive (Olea europaea L.). PloS one 11, e0152943, https://doi.org/10.1371/journal.pone.0152943 (2016).
Weigel, D. & Nilsson, O. A developmental switch sufficient for flower initiation in diverse plants. Nature 377, 495–500, https://doi.org/10.1038/377495a0 (1995).
Klocko, A. L. et al. Containment of transgenic trees by suppression of LEAFY. Nature biotechnology 34, 918–922, https://doi.org/10.1038/nbt.3636 (2016).
Lin, H. et al. Evolutionarily conserved repressive activity of WOX proteins mediates leaf blade outgrowth and floral organ development in plants. Proc Natl Acad Sci USA 110, 366–371, https://doi.org/10.1073/pnas.1215376110 (2013).
Irish, V. F. The flowering of Arabidopsis flower development. The Plant journal: for cell and molecular biology 61, 1014–1028, https://doi.org/10.1111/j.1365-313X.2009.04065.x (2010).
Pin, P. A. & Nilsson, O. The multifaceted roles of FLOWERING LOCUS T in plant development. Plant, cell & environment 35, 1742–1755, https://doi.org/10.1111/j.1365-3040.2012.02558.x (2012).
Soppe, W. J. et al. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Molecular cell 6, 791–802 (2000).
Karlova, R. et al. The Arabidopsis Somatic Embryogenesis Receptor-Like Kinase1 protein complex includes Brassinosteroid-Insensitive1. The Plant cell 18, 626–638, https://doi.org/10.1105/tpc.105.039412 (2006).
Cao, Y., Dai, Y., Cui, S. & Ma, L. Histone H2B monoubiquitination in the chromatin of Flowering Locus C regulates flowering time in Arabidopsis. The Plant cell 20, 2586–2602, https://doi.org/10.1105/tpc.108.062760 (2008).
Shi, Y. et al. The glutamate carboxypeptidase AMP1 mediates abscisic acid and abiotic stress responses in Arabidopsis. The New phytologist 199, 135–150, https://doi.org/10.1111/nph.12275 (2013).
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC bioinformatics 4, 41, https://doi.org/10.1186/1471-2105-4-41 (2003).
Conesa, A. & Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. International journal of plant genomics 2008, 619832, https://doi.org/10.1155/2008/619832 (2008).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic acids research 35, W182–185, https://doi.org/10.1093/nar/gkm321 (2007).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology 28, 511–515, https://doi.org/10.1038/nbt.1621 (2010).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome biology 11, R106, https://doi.org/10.1186/gb-2010-11-10-r106 (2010).
Benjamini, Y. & Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Annals of Statistics 29, 1165–1188 (2001).
Du, Z., Zhou, X., Ling, Y., Zhang, Z. & Su, Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic acids research 38, W64–70, https://doi.org/10.1093/nar/gkq310 (2010).
Chen, Z., Gao, K., Su, X., Rao, P. & An, X. Genome-Wide Identification of the Invertase Gene Family in Populus. PloS one 10, e0138540, https://doi.org/10.1371/journal.pone.0138540 (2015).
Jin, J., Zhang, H., Kong, L., Gao, G. & Luo, J. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic acids research 42, D1182–1187, https://doi.org/10.1093/nar/gkt1016 (2014).
Acknowledgements
We thank Prof. Steven H. Strauss from Oregon State University for the valuable comments and suggestions of this manuscript. This work was supported by the National Science and Technology Major Project of China (2018ZX08021001-002-004); the National Natural Science Foundation of China (31570661); and the National High Technology Research and Development Program (2013AA102703).
Author information
Authors and Affiliations
Contributions
X.A. and Z.C. planned and designed the research. Z.C., P.R., X.Y., T.Z., K.G. and X.Y. performed the experiments, Z.C. and X.S. analysed the data. Z.C. wrote the manuscript. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Z., Rao, P., Yang, X. et al. A Global View of Transcriptome Dynamics During Male Floral Bud Development in Populus tomentosa . Sci Rep 8, 722 (2018). https://doi.org/10.1038/s41598-017-18084-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18084-5
This article is cited by
-
Genome-wide analysis and molecular dissection of the SPL gene family in Fraxinus mandshurica
BMC Plant Biology (2022)
-
Time-series RNA-Seq transcriptome profiling reveals novel insights about cold acclimation and de-acclimation processes in an evergreen shrub of high altitude
Scientific Reports (2022)
-
Hybrid transcriptome sequencing approach improved assembly and gene annotation in Cynara cardunculus (L.)
BMC Genomics (2020)
-
Gene expression trajectories during male and female reproductive development in balsam poplar (Populus balsamifera L.)
Scientific Reports (2020)
-
Genome-wide analysis of the poplar NF-Y gene family and its expression in floral bud development of Populus tomentosa
Trees (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
