
Abstract
The earliest phases of floral development include a number of crucial processes that lay the foundation for the subsequent morphogenesis of floral organs and success in reproduction. Currently, key transcriptional changes during this developmental window have been characterized in the model species Arabidopsis thaliana, but little is known about how transcriptional dynamics change over the course of these developmental processes in other plant systems. Here, we have conducted the first in-depth transcriptome profiling of early floral development in Aquilegia at four finely dissected developmental stages, with eight biological replicates per stage. Using differential gene expression analysis and weighted gene co-expression network analysis, we identified both crucial genes whose expression changes mark the transitions between developmental stages and hub genes in co-expression modules. Our results support the potential functional conservation of key genes in early floral development that have been identified in other systems, but also reveal a number of previously unknown or overlooked loci that are worthy of further investigation. In addition, our results highlight not only the dynamics of transcriptional regulation during early floral development, but also the potential involvement of the complex, essential networks of small RNA and post-translational regulation to these developmental stages.
Similar content being viewed by others
Introduction
The earliest phase of floral meristem (FM) development requires exquisite coordination among many different developmental processes. These include the proper initiation and patterning of the floral organs, maintenance of the size of the FM during organ initiation, the eventual termination of the FM activity to ensure the correct number of whorls, and the overlay of the floral organ identity programs onto the primordia so that the boundaries of gene expression domains synchronize precisely with the physical boundaries between the primordia. Coordination of these processes is achieved through crosstalk between numerous regulatory frameworks at multiple levels, ranging from transcriptional regulation, to RNA stability, to epigenetic modification, and protein stability.
During the past 30 years, key regulatory mechanisms involved in early floral development have been characterized thanks to in-depth studies of model species such as Arabidopsis thaliana. These include the ABCE model of organ identity1,2, the WUSCHEL (WUS)-CLAVATA (CLV) feedback loop for meristem maintenance3, the AGAMOUS (AG)-KNUCKLES (KNU)-WUS pathway for FM termination4,5, the auxin signaling pathway for primordia initiation6, as well as genes specifying the boundaries between organs or gene expression domains7. However, novel genes and pathways contributing to these processes are constantly being discovered, and it is not yet clear how cross-regulation between different pathways and between different levels of the regulatory framework is controlled. Furthermore, although the orthologs of many genes have been studied in multiple plant model systems, relatively little is known about the degree of conservation of these programs across major lineages of flowering plants. To obtain a comprehensive view of the molecular basis of the early floral development, the conventional forward and reverse genetics approaches need to be coupled with global analysis at the transcriptional and genomic levels.
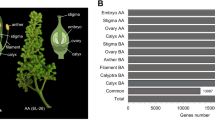
In this study, we have conducted the first in-depth transcriptional profiling of early floral development in Aquilegia coerulea at four finely-dissected stages (Fig. 1). The genus Aquilegia belongs to the basal eudicot buttercup family Ranunculaceae and is a model system for floral evolutionary developmental studies8. Analysis of floral ontogeny9 has shown that Aquilegia floral organ primordia initiate in whorls of five organs each, which are arranged in 10 orthostichies (vertical rows of organs), with alternate orthostichies either above the sepals or the petals (Fig. 1c). Aquilegia flowers all have multiple whorls of stamens, which is one of the major differences relative to other established model systems (e.g. A. thaliana, petunia, snapdragon) that all have only one whorl of stamens. The flowers of A. coerulea, as well as those of almost all other Aquilegia species, possess a fifth type of floral organ, the staminodes, which initiate in two whorls positioned between the stamens and carpels (Fig. 1). Unlike the syncarpous gynoecium of other model systems, Aquilegia is apocarpous with five distinct carpel primordia (Fig. 1). Using a candidate gene approach, previous studies have revealed the sub-functionalization of the B-class organ identity genes10,11,12 and that the JAGGED homolog is crucial for initiation of the floral organ primordia13, but genome scale studies to date have focused on late stage floral organ development14,15,16.

A. coerulea and the floral developmental stages used for RNA-seq. (a) Side view of a mature A. coerulea flower. (b) Front view of a mature A. coerulea flower. (c) Floral diagram of a typical A. coerulea flower. (d) Developmental stages, the number of biological replicates per stage, and the number of floral buds used for each biological replicate that were used for RNA-seq. Asterisks indicate staminodes and arrowheads indicate petals. Scale bar: a,b = 1 cm; d = 100 µm.
The current experiment was designed to obtain a broad characterization of the genetic pathways acting in early Aquilegia FM development. In order to ensure the power and accuracy of our sequencing results, we used eight biological replicates per developmental stage and obtained an average of 18× sequencing depth per sample. The developmental window we sequenced sampled stages that started with a late phase of stamen initiation, covered the period of FM termination, and ended with the initial stage of morphogenesis of the floral organs. Using differential expression (DE) analysis and weighted gene co-expression network analysis (WGCNA), we identified crucial genes marking the transitions between developmental stages, and hub genes in co-expression modules that are strongly associated with these developmental stages. Our results support the potential functional conservation of key genes in early floral development that have been identified in other systems, but also revealed a number of previously unknown or overlooked loci that are worthy of further investigation. In addition, our results highlight not only the dynamics of transcriptional regulation during early floral development, but also the potential involvement of the complex, essential networks of small RNA and post-translational regulation in this process.
Materials and methods
Plant materials: growing conditions and dissection
Seeds of Aquilegia x coerulea ‘Kiragami’ were purchased from Swallowtail Garden Seeds (Santa Rosa, CA, USA). Seeds were germinated in wet soil and seedlings were grown under conditions of 16 h daylight at 18 °C and 8 h dark at 13 °C. Once the plants developed approx. six true leaves, they were transferred into vernalization conditions (16 h daylight at 6 °C and 8 h dark at 6 °C) for three to 4 weeks, and subsequently moved back to the regular growth condition. Once the inflorescences started to develop, axillary FMs were quickly dissected on ice using surgical needles and flash frozen in liquid nitrogen. Three sepals were removed from every floral bud collected.
Scanning electron microscopy
Floral buds were dissected using the same method as above, fixed in FAA (10% formaldehyde, 50% ethanol, 5% acetic acid), and stored at 4 °C. Prior to imaging, samples were dehydrated through a graded ethanol series to 100% and then critical point dried with CO2 (Autosamdri-815, Tousimis, MD, USA). Images were taken with the JSM-6010 LC Scanning Electron Microscope (JEOL, MA, USA) at the Arnold Arboretum of Harvard University.
RNA extraction, library construction, and sequencing
The total RNA of all samples was extracted using the RNeasy Micro Kit (Qiagen, Netherlands). The integrity of extracted RNA was measured by using a 2200 TapeStation (Aligent Technologies, CA, USA). Subsequently, each RNA sample was diluted to a concentration of 1.25 ng/µl, measured by using Qubit RNA HS Assay Kit (Thermo Fisher Scientific, MA, USA). Sequencing libraries were prepared by the Bauer Core facility of Harvard University using the SMART-Seq v4 Ultra Low Input RNA Kit (Takara Bio USA, Inc., CA, USA) for cDNA synthesis and Illumina Nextera XT (Illumina) for library preparation. Libraries were fragmented to an average size of 350 bp. Library concentrations were examined using a Qubit 2.0 (Thermo Fisher Scientific, MA, USA) and qRT-PCR, and the insert sizes measured using a Bioanalyzer 2100 (Aligent Technologies, CA, USA). Libraries were then pooled and diluted to a final concentration of 3 nmol. Sequencing was conducted at Novogene (Beijing, China) using the 150 bp paired-end Illumina HiSeq4000 sequencing platform.
Mapping reads to the reference transcriptome
The raw sequencing reads were filtered by removing adapters and low-quality reads using Trim Galored v0.6.517 (quality Phred score cutoff = 20) and mapped to the Aquilegia × coerulea ‘Goldsmith’ v3.1 reference transcriptome (https://phytozome.jgi.doe.gov/) using Kallisto v0.46.118. Raw and processed reads are deposited under the GEO accession number GSE158507.
Differential expression (DE) analysis
Read counts and transcript per million reads (TPMs) were generated using the R package tximport v1.0.3 and the lengthScaledTPM method19. Lowly expressed transcripts were filtered based on analyzing the mean–variance trend, and transcripts with more than 1 counts per million reads in at least one of the 32 samples were retained. Reads were normalized using the variance stabilizing transformation (VST) method to perform principal component analysis (PCA). Differential gene expression analysis was conducted following the DESeq2 R package v1.28.120. For DE genes, the log2 fold change of gene abundance was calculated between pairwise groups, and the significance of expression changes was determined using the Student’s t-test. p values were adjusted with the Benjamini–Hochberg Procedure to correct for the false discovery rate21. A gene was considered to be significant if it had adjusted p value < 0.05 and log2 fold change ≥ 1. A heatmap of all DE genes was produced using the R package ComplexHeatmap v2.2.0. Hierarchical clustering was used to partition the DE genes into two clusters with Euclidean distance and ward.D2 clustering algorithm.
Gene ontogeny (GO) enrichment and kyoto encyclopedia of genes and genomes (KEGG) pathway analysis
Gene Ontogeny (GO) analysis for DE groups and WGCNA modules of interest was calculated using agriGO v2 (https://systemsbiology.cau.edu.cn/agriGOv2/)22,23 and the KEGG pathway analysis24,25 was performed using the DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/)26,27. For both analyses, the identifiers of the top-hit A. thaliana loci were obtained from the A. coerulea “Goldsmith” v3.1 reference genome annotation (https://phytozome.jgi.doe.gov/), all of the expressed genes in the Aquilegia RNA-seq data were used as the background reference. Enrichment in GO terms or pathways was calculated with Fisher’s exact test, and p values were adjusted for the false discovery rate using the Benjamini–Hochberg procedure. The minimum number of mapping entries for a GO category was set to be 5, and the count threshold for KEGG pathway analysis was set to be 2. Enrichment figures were produced using the R package ggplot2.
WGCNA
Weighted gene correlation network analysis (WGCNA)28 was constructed using the R package WGCNA v1.68 following the package tutorials; the illustrated workflow is shown in Fig. S1. To avoid noise from lowly expressed genes, all genes that had read counts less than 10 in more than 90% of the samples (i.e. 29 samples) were removed. The expression of the resultant 18,303 genes was then normalized using the varianceStabilizingTransformation function from the R package DESeq220. The adjacency matrix was calculated using a soft thresholding power of 5. Compared to a hard threshold, of which the correlation values above or below are considered to be connected or not, respectively; the soft threshold is used to raise the correlation to a power so that the difference between strong and weak correlations are exaggerated rather than defined into binary terms. The topological overlap matrix was calculated using a deepSplit value of 4, minModuleSize was set to 20, and mergeCutHeight was set to 0.2. Node and edge information of modules of interest were exported using the exportNetworkToCytoscape function of the WGCNA package. The node and edge files were then transformed into .json format using the R package jsonlite v1.6.1, and visualized using customized scripts modified based on the D3 JavaScript library.
Gene phylogenies
Neighbor-joining phylogenetic trees for genes that are discussed in the main text were constructed if the homologs in A. coerulea and A. thaliana were not each other’s reciprocal top BLAST hits. Potential homologs of the genes of interest were identified from the genomes of diverse species on Phytozome, all amino acid sequences were aligned using ClustalW29 and the neighbor-joining trees were constructed using MacVector v17.5.5(Gary, NC, USA).
Results and discussion
Developmental stages and sequencing information for transcriptome profiling
In order to finely dissect and capture the transcriptional dynamics during early phases of Aquilegia floral development, we defined four developmental stages for RNA sequencing (Fig. 1d). At stage 1 (s1), the dome of the FM is round and visible, and stamen primordia are in the progress of initiating. At stage 2 (s2), all the stamen and staminode primordia have completed initiation, the five carpel primordia are about to initiate, and the apex of the floral bud is star-shaped and flattened. At stage 3 (s3), the outer rim of each carpel primordium is elevated but the development of carpel primordia has not consumed all the cells of the apex, and the stamen and staminode primordia are still morphologically indistinguishable. At stage 4 (s4), the carpel primordia continue to develop and elongate, all the cells in the apex have been consumed by carpel development, and the staminode primordia have started to expand laterally, making them morphologically distinct from the stamen primordia.
A total of 72 floral buds were dissected from 30 individual plants for RNA extraction. For s1, s2, and s3, each biological replicate contained two floral buds from two individuals, while s4 contained one floral bud per biological replicate (Fig. 1d). The amount of total RNA per extraction varied from 20.2 to 138 ng. After quality assessment, eight samples at each stage with the best RNA qualities were used for subsequent library construction. All 32 selected samples had RNA integrity numbers higher than 8.9.
A total of 993.64 million raw reads (323.1G raw data) were generated from the 150 bp paired-end Illumina HiSeq4000 sequencing platform. The number of reads generated per sample ranged between 25 and 36 million (Fig. S2), and the average reads per developmental stage ranged from 29.3 to 32.9 million (Fig. S2). After filtering reads by quality (Phred score cutoff = 20), the fraction of reads retained ranged from 80.2 to 88.4% (Fig. S2). Retained reads were then mapped to the Aquilegia × coerulea ‘Goldsmith’ v3.1 reference transcriptome, generating on average 17.6× to 19.8× sequencing coverage per developmental stage (Fig. S2), and a total of 30,023 raw transcripts were mapped. Subsequently, lowly expressed transcripts were filtered if a transcript had less than 1 million read count in more than 31 samples, which resulted in 20,473 expressed genes in all samples. To conduct a preliminary exploration of the 32 samples, normalized reads of all samples were used to perform a principal component analysis (PCA; Fig. 2a). The two primary PCs explained 76% of the total variance among all samples and a clear clustering by developmental stages was observed (Fig. 2a).

PCA of normalized reads of all samples (a) and bar plot summary of DE genes between developmental stages (b).
Differential expression analysis between developmental stages
To identify genes that were significantly differentially expressed (DE) between developmental stages, the log2 fold change of the gene abundance was calculated between different stages. A gene was considered DE if it had an adjusted p value of < 0.05 and an absolute value of log2 ratio \(\ge \) 1. A total of 1944 DE genes were identified, with more genes upregulated at s4 compared to any other developmental stage (Fig. S3; Supp. Data 1). For each pairwise comparison, there were consistently more genes down-regulated at the earlier stage relative to the later stage (Fig. 2b; Supp. Data 1).
A small number of DE genes, 67 and 49, were identified when comparing s1 to s2, and s2 to s3, respectively. Among these 67 and 49 DE genes, only five loci (Table S1) appeared in both comparisons, suggesting that although s2 appeared to be transcriptionally similar to both s1 and s3, the similarities lay in different aspects in each comparison. One of the five genes in common, Aqcoe7G234000, is the homolog to the ARABIDOPSIS HISTIDINE PHOSPHOTRANSFER PROTEIN 6 (AHP6). It is also the top DE gene that is significantly up-regulated in s1 compared to s2, and among the top DE genes that are up-regulated at s2 compared to s3 (Table S1). In A. thaliana, AHP6 is one of the key genes mediating crosstalk between the auxin and cytokinin signaling pathways, and participates in a number of crucial developmental processes, including specifying the founder cells for lateral roots, protoxylem, and protocambium30. In shoot apical meristems (SAMs), inflorescence meristems (IMs), and FMs, AHP6 is involved in the patterning of phyllotaxy and organ initiation30,31,32. In particular, the AHP6 protein functions non-cell-autonomously to create a cytokinin-signaling inhibition field that contributes to the robustness of the auxin-signaling inhibition field for phyllotaxy and organ patterning31. We hypothesize that the AHP6 homolog in Aquilegia is functionally conserved since the expression level of AqAHP6 sharply declines as organ initiation ceases during the progression of s1 to s4 (Fig. S4).
Among the 24 genes that were found to be significantly up-regulated at s1 relative to s2, Aqcoe3G399500 is the top DE gene after AqAHP6 (Table S1). The A. thaliana homolog of this locus is PERIANTHIA (PAN), which has been shown to regulate floral architecture and directly activate the expression of the C-class gene AGAMOUS (AG)33. Homologs of other genes that specify meristem and organ primordia boundaries during early floral development also appeared to be significantly up-regulated at s1, including CUP SHAPED COTYLEDON334 (AqCUC3; Aqcoe6G165500) and HANABA TARANU35 (AqHAN; Aqcoe5G190300), as well as loci that are known to regulate floral organ size and growth, such as STERILE APETALA36 (AqSAP; Aqcoe1G384300) and AINTEGUMENTA-like 637 (ATL6/PLT3; Aqcoe6G092100).
Interestingly, two key genes controlling stomata development in various plant lineages, AqMUTE and AqEPF138,39, were both expressed at very low levels at s1, but significantly up-regulated in s2 followed by consistent high expression at later stages (Fig. S4). This may suggest that although s1 sepals have already achieved reasonable sizes, stomatal development does not initiate until s2. Other genes that were shown to be up-regulated in s2 compared to s1 include the adaxial identity gene AqCRC, which is consistent with previous studies showing that strong AqCRC expression coincident with the initiation of the carpel primordia15.
The top DE gene that is up-regulated in s2 relative to s3, Aqcoe7G055500, encodes a non-specific serine-threonine kinase with a predicted RNA-binding domain, which does not appear to be a member of any of the better-known serine-threonine classes40. Its homolog in A. thaliana, AT5G51800, has not been studied functionally but is highly expressed in the shoot apex, inflorescence meristem, developing carpels, and stigma (Arabidopsis eFP Browser 2.0). However, this gene is highly expressed at s2 in the current study, in which the FM has just completed organ primordia initiation, and it will, therefore, be interesting to examine its function in early Aquilegia floral development.
Gene ontogeny (GO) analysis of DE genes revealed a wide range of enrichment terms for every DE comparison except for [s1 vs. s2] and [s2 vs. s3], likely due to the small numbers of DE genes in these two comparisons, as mentioned above (Supp. Data 2). In general, there seems to be a large portion of overlap in GO terms in DE genes that were up- or down-regulated during early stages compared to later stages (Fig. 3a). For instance, GO terms that are related to organ formation, patterning, and development are enriched for DE genes that are up-regulated at s1 and s2. On the other hand, genes that are up-regulated at s3 and s4 are heavily enriched in metabolic and enzymatic activities, including active transmembrane transportation, carboxylesterase activity, oxidoreductase activity, and glucosyltransferase activity, all of which are involved in broader metabolic processes during plant development.

GO term and KEGG pathway enrichment analysis of DE genes. (a) Selected enriched GO terms from different DE comparisons. (b) Selected enriched KEGG pathways from different DE comparisons. (c) Enriched plant hormone signal transduction pathways from different DE comparisons. % in a and b were calculated as: number of genes that hit the specific GO term or KEGG pathway/total number of genes in the input DE list*100. Each DE comparison is noted as: earlier stage vs. later stage, up- (U) or down (D)-regulated at the earlier stage.; for instance: S1S2U means genes that are DE between s1 and s2 and are up-regulated at s1.
Subsequently, we conducted KEGG pathway enrichment analysis with the DE genes, and we observed a similar pattern for genes that are up-regulated in s4, in that they participate in various metabolic pathways (Fig. 3b). For instance, genes involved in nitrogen metabolic pathways are up-regulated and enriched in s4, indicating active amino acid biosynthesis and protein synthesis at s4 compared to earlier developmental stages. On the other hand, all the genes that are up-regulated at the earlier stages in each DE comparison are almost exclusively enriched in plant hormone signal transduction pathways (Fig. 3b).
We then examined the hormonal signaling pathways that were enriched for each DE list, which revealed several interesting points (Fig. 3c). Firstly, all DE lists contained genes in the auxin signaling pathways. The DE genes up-regulated in s1 compared to s4 included genes in all major components in auxin signaling, while all other DE lists only showed enrichment in a few components in the pathway. This indicates a gradual decrease in the number of highly expressed auxin signaling genes from s1 to s4. Secondly, as mentioned previously, AqAHP6 appeared to be differentially expressed at the earlier stages in all DE comparisons, which was also captured by the KEGG pathway analysis as enriched in AHP family members. Besides AqAHP6, homologs of the Arabidopsis Response Regulator (ARRs) proteins were shown to be up-regulated in s1 compared to s4, and ARRs are downstream of AHPs in the cytokinin signaling pathway, which emphasizes the decrease in cytokinin signaling from s1 to s4. Thirdly, genes that are up-regulated at s1 compared to s4 are detected from the abscisic acid, brassinosteroid, and salicylic acid signaling pathways, suggesting a complex interplay between multiple plant hormone pathways during the early stages of floral development.
We profiled the expression of type-II MADS-box genes41,42 and class-I KNOTTED-like homeodomain (KNOX) genes14, because many members of these families are known to play important roles during early floral development, along with additional homologs of genes involved in FM identity and maintenance (Fig. 4). Among the type-II MADS-box genes, the C-class gene AqAG2 and AGAMOUS-LIKE 12 (AqAGL12) showed a significant increase in the expression levels from s1 to s4. This might be expected for AqAG2, which is known to be strongly expressed in carpels10, but expression of AqAGL12 has not previously been investigated in Aquilegia. While the majority of the MADS-box genes showed a decrease in their expression levels from s1 to s4, this trend was only significant for SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (AqSOC1.1), FRUITFULL-LIKE 2 (AqFL2), SEPALLATA1 (AqSEP1) and APETALA3-3 (AqAP3-3) (Fig. 4). Similarly, most of the class-I KNOX genes and FM-related genes showed the highest expression levels at s1 with a decrease at subsequent stages, but AqKXL3 appeared to be expressed at the highest levels at s2 (Fig. 4).

Expression profiles of type-II MADS-box genes, class-I KNOX genes, and genes involved in FM maintenance and termination pathways. Expression levels of each gene over the developmental stages were scaled as the average expression of all biological replicates per stage subtracted by the average expression of all replicates of all stages, then divided by the standard deviation of the expression of all replicates of all stages. DE genes were indicated with symbols of each DE comparison.
Since the meristem stages we sampled captured the transition from organ primordia initiation to FM termination (i.e. the end of organogenesis), we also examined the expression of the homologs of genes in the known FM termination pathways4,5,43,44,45,46 (Fig. 4). Interestingly, several lines of evidence suggest limited conservation of these pathways, particularly the KNU-WUS pathway, between A. thaliana and A. coerulea. In A. thaliana, KNU has been shown to be essential in terminating the expression of WUS in the FM, and continuous expression of KNU appeared to be necessary to maintain the suppression of WUS via both transcriptional suppression and heterochromatinization44, but the expression of AqKNU (Figs. S4 and S6) decreased after s2. Furthermore, KNU and SPLAYED (SYD) function antagonistically by competing for a binding site in WUS promoter, but AqKNU and AqSYD showed similar, rather than the opposite, patterns of expression over s1 to s4 (Fig. S4). Additionally, FERTILIZATION-INDEPENDENT ENDOSPERM (FIE) is a key polycomb repressive complex2 component, which has been shown to physically interact with KNU to deposit repressive H3K27me3 marks and maintain the stable silencing of WUS in later developmental stages44. Unlike what was observed in silencing FIE expression in A. thaliana, however, a previous study has shown that the silencing of AqFIE did not lead to indeterminacy in the flowers47. MINI ZINC FINGER2 (MIF2) was shown to be a component of a protein complex together with KNUCKLES (KNU), TOPLESS (TPL) and HISTONE DEACETYLASE-like 19 (HDA19) to suppress the expression of WUS in the FM43, but the read counts of the MINI ZINC FINGER2 (MIF2) homolog were extremely low in all sample of all stages and this gene is considered as not expressed in our dataset. Besides the KNU-WUS pathway, TORNADO2 (TRN2) functions downstream of CRC in coordinating FM termination and carpel primordia formation in A. thaliana46, but phylogenetic analysis did not recover any ortholog of TRN2 the Aquilegia genome (Fig. S5). These findings suggest that the A. thaliana mechanisms controlling FM termination are not well conserved in Aquilegia.
Gene co-expression analysis, identification of module hub genes, and network construction of modules of interest
The DE analysis provided us with a good starting point of what genes might be functionally relevant to a certain developmental stage based on the changes in their expression levels, but a strong limitation for DE analysis is that each gene is considered in isolation while in reality, genes and gene products function in networks. In order to discover co-expressed genetic modules that are significantly associated with different stages during early floral development in Aquilegia, we conducted weight gene co-expression network analysis (WGCNA) using the RNA-seq data (refer to Fig. S1 for an illustrated workflow). Using a soft threshold power = 5 (Fig. S7), a total of 24 co-expression modules were constructed from 18,303 genes, with the smallest module (darkgrey) containing 21 genes and the largest module (turquoise) containing 6206 genes (Fig. 5a). Subsequently, we looked at the association between the modules and the four developmental stages by correlating the eigengene value (i.e. a value equivalent to the first component of the module; a singular value decomposition to summarize the expression levels of all genes in that module) of the modules with the stages. Among the 24 modules, we determined three modules to be of particular interest based on their correlation (Fig. 5a) and close distance in the hierarchical dendrogram clustering (Fig. 5b): the green module for its strong association with s1, and the brown and magenta modules for their strong association with s4. We will refer to the modules as green-s1, brown-s4, and magenta-s4 hereafter for clarity.

Associating gene co-expression modules with developmental stages. (a) Correlation between module eigengene values and the developmental stages. The first number in each cell represents the correlation value, and the second number in the parenthesis represents the p-value of the correlation. Arrowheads pointed to modules of interest. (b) Hierarchical clustering dendrogram of the eigengenes of modules and the developmental stages. (c) Selected GO term enriched from modules of interest. % was calculated as: number of genes hit the specific GO term/total number of genes in the input list*100.
To obtain a broad overview of genes in the three modules of interest, we performed GO and KEGG pathway analysis on these modules (Fig. 5c; Supp. Data 2). GO analysis revealed that although the green-s1 module has a moderate size (781 genes) compared to other modules, it has the largest number of enriched GO terms, most of which are specific to early floral developmental stages, such as stem cell/meristem maintenance and development, axis specification, and organ formation (Fig. 5c; Supp. Data 2). On the other hand, the brown-s4 module has a relatively large number of genes (1947 genes) but only two highly related cellular process GO terms are found to be significant, microtubule-based processes and movement (Fig. 5c; Supp. Data 2). The magenta-s4 module also shows strong and significant association with s4, but genes in this module are enriched in “floral whorl” and “floral organ development”, particularly in androecium, perianth, and gynoecium development (Fig. 5c; Supp. Data 2). This suggests that the WGCNA has identified modules that are significantly associated with different aspects of floral development at s4: genes in the brown-s4 module appear to be focused on the rapid growth of all floral organs in s4, in which active mobilization of the microtubules is necessary for rapid cell expansion; while genes in the magenta-s4 module are specific to the elaboration and maturation of each floral organ identity.
KEGG analysis revealed that the green-s1 and brown-s4 modules both contain genes that are involved in the starch and sucrose metabolism pathways (Supp. Data 2). In addition, genes in the green-s1 module are significantly enriched in the plant hormone signal transduction pathways, and genes in the brown-s4 module are significantly enriched in several sugar metabolic pathways, including fructose and mannose metabolism, and amino sugar and nucleotide sugar metabolism (Supp. Data 2). Sugar metabolism has been implicated in various aspects of plant development, especially the amino sugar and nucleotide sugar interconversion pathways that are required for cell wall biosynthesis48. Together with the GO enrichment in microtubule-based movement and processes for module brown-s4, this again emphasizes the initiation of rapid organ growth during s4 of floral development in Aquilegia. No pathway enrichment was found for genes in the magenta-s4 module, possibly due to the small module size.
Subsequently, we identified the potential hub genes of each module of interest based on the value of signed module membership (MM) and trait significance (TS). The former describes the correlation between a gene and the eigengene value of a module, while the latter describes the correlation between a gene and the trait of interest; the potential hub genes of a module should have significant and strong correlations with both MM and TS (Figs. S1 and S8). We, therefore, defined the hub genes as having their absolute values of MM and TS above the 90th percentile of the distribution of MM and TS of all the genes of the module. This approach identified 33, 56, and 14 hub genes for the green-s1, brown-s4, and magenta-s4 modules, respectively (Fig. 6; Table S2). Moreover, although every gene is connected to all other genes in a module, the strengths of the connection between gene pairs vary and the hub genes, by definition, should have the strongest values of connectivity. To better visualize the gene co-expression networks of each module, we only retained the edges that have the highest weight values (i.e. having the strongest connectivity) with the hub genes in the network for visualization (Figs. S8, 6, 7 and 8). To facilitate navigation among a group of highly interconnected genes (particularly in the case of the green-s1 and brown-s4 modules), we moved all the genes with the largest numbers of connections to the periphery of the visualized network and thus genes left in the center of the network had fewer connections compared to those of at the periphery.

Gene network of module green-s1. Color scale represents the value of MM for each gene. The “Aqcoe” prefix of a gene identifier was removed to facilitate the visualization. All the genes with the largest number of connections are at the periphery of the network (i.e., genes in the center have fewer connections compared to genes at the periphery). Genes that are presented as colored circles (instead of a solid dot) are genes that were discussed in the main text.

Gene network of module brown-s4. Color scale represents the value of MM for each gene. The “Aqcoe” prefix of a gene identifier was removed to facilitate the visualization. All the genes with the largest number of connections are at the periphery of the network (i.e. genes in the center have fewer connections compared to genes at the periphery). Genes that are presented as colored circles (instead of a solid dot) are genes that were discussed in the main text.

Gene network of module magenta-s4. Color scale represents the value of MM for each gene. The “Aqcoe” prefix of a gene identifier was removed to facilitate the visualization. Genes that are presented as colored circles (instead of a solid dot) are genes that were discussed in the main text.
Hub genes of the green module revealed new candidate genes associated with s1 floral development
The green-s1 module contains 33 potential hub genes and 17 of these encode transcription factors (TFs). The homologs of many of these TF-coding genes are involved in early FM and floral development, such as class-I KNOX genes (AqSTM1, AqSTM2, AqKXL2), AqAS1, AqSEP1, AqPAN, and the homologs of CUP SHAPED COTYLEDON3 (Aqcoe6G165500; AqCUC3), HOMEOBOX-3 (Aqcoe1G245800; AqHB3), PHABULOSA (Aqcoe1G178700; AqPHB), LATERAL SUPPRESSOR (Aqcoe1G411100; AqLAS), BLADE ON PETIOLE2 (Aqcoe2G033300; AqBOP), and STERILE APETALA (Aqcoe1G384300; AqSAP). Of these, AqPAN also showed the strongest association with s1 (Table S2).
Interestingly, although all the potential hub genes have similarly high MM and TS values (Table S2), the number of edges connected to the hub genes in the network appeared to be highly variable (Fig. 6), indicating a hierarchy of connectedness even among the hub genes. For instance, we have identified nine hub genes that have a strong negative association with the module and the s1 developmental stage (Table S2), but all of them except one (Aqcoe4G281800) have only a few edges connecting them with other genes in the network (and thus were not placed in the periphery of the network). Aqcoe4G281800 encodes a wax synthase that is involved in constructing cell walls, and also only has a moderate number of edges compared to the those associated with other periphery hub genes. By contrast, the five periphery hub genes with the largest number of edges are (Fig. 6): AqVRN1 (Aqcoe2G107800, homologous to VERNALIZATION1, VRN1, in A. thaliana); AqHCT (Aqcoe7G354200, codes for a hydroxycinnamoyl-Coenzyme A shikimate/quinate hydroxycinnamoyltransferase, HCT); AqUFO1(Aqcoe1G161900; UNUSUAL FLORAL ORGANS112), AqSAP; and AqEPFL4 (Aqcoe2G352600, homologous to EPIDERMAL PATTERNING FACTOR-LIKE4, EPFL4, in A. thaliana). The functional implications of some of these loci are unclear. For instance, A. thaliana VRN1 belongs to the AP2/B3-like transcriptional factor family, and its function is primarily understood to be the regulation of the vernalization locus FLOWERING LOCUS C (FLC)49. However, although Aquilegia does require vernalization to flower50, it lacks an FLC homolog51, and AqVRN1 is expressed in the early FM (Fig. S4), which differs considerably from the broad expression of AtVRN1 in various organs and developmental stages of A. thaliana49,52. The potential role for AqHCT is equally enigmatic, as the enzyme appears to function in phenylpropanoid biosynthesis, a pathway that supports highly diverse cellular functions53.
Luckily, the remaining hub genes are better understood. Homologs of UFO encode F-box proteins that generally function with LEAFY (LFY) homologs to promote FM identity and activate B-class gene expression across diverse dicot model systems54,55,56,57,58. In Aquilegia, the closely related paralogs AqUFO1 and AqUFO2 promote the transition from inflorescence to FM identity in conjunction with AqLFY, although there is no clear role in B gene activation12. In addition to the expected contribution to FM identity, the AqUFO1/2 paralogs also appear to promote the initiation of the stamen whorls, as whorl number dramatically decreases in silenced flowers from the usual 7–8 whorls to only 4–5. This function is intriguing for several reasons. First, the phenotype is only observed in AqUFO1/2-silenced flowers and not with AqLFY-silencing, suggesting that this function is specific to the F-box proteins and not a joint function with AqLFY. Second, it underscores a critical difference in Aquilegia floral development relative to other model systems in that, like many angiosperm flowers, Aquilegia has multiple whorls of stamens. The identification of AqUFO1 as a critical hub gene in early floral development is consistent with this novel role in promoting stamen initiation and/or continued FM proliferation and points to other loci that may function as novel co-factors for this role.
In A. thaliana, SAP plays a number of roles, including acting as a cadastral gene that prevents AG from entering the perianth whorls, and controlling both inflorescence architecture and ovule development59. Interestingly, similar to UFO, SAP also contains an F-box motif and was recently shown to be a component of an Skp1-cullin 1-F-box (SCF) E3 ubiquitin-protein ligase complex that controls lateral organ size by promoting cell proliferation, a function that has been demonstrated both in A. thaliana60 and cucumber61. To date, the only genes that have been identified as the direct targets of the SAP-mediated E3 ligase complex are PEAPOD1/2 (PPD1/2), which control organ growth36,60. The homolog of PPD1 and PPD2 in Aquilegia, Aqcoe5G048100, showed relatively stable expression from s1 to s4 (Fig. S4) and was assigned to the turquoise module (Fig. 5). It will be worth investigating whether this interaction is conserved in Aquilegia and what may be the other targets of the ligase complex during early floral development.
AqEFPL4 belongs to the EPFL gene family that encodes small, secreted cysteine-rich ligands. The best-studied members of the family are the EPIDERMAL PATTERNING FACTORS1/2, which are key regulators in stomata development38. As mentioned above, AqEPF1 was identified as one of the DE genes that is up-regulated at s2 compared to s1 (Table S1). In contrast, in A. thaliana the close homologs EPFL4 and EPFL6 have been shown to have distinct functions in inflorescence growth62,63. However, two very recent studies revealed that EPFL4 and EPFL6 are also expressed in the periphery of the shoot apical meristem (SAM) and function redundantly with EPFL1 and EPFL2 to regulate the meristem size and leaf initiation64,65. In particular, signaling downstream of EFPL ligands and their receptors confines the WUS-CLV3 pathway to the center of the SAM by inhibiting its expression in the meristem periphery65. In this regard, it is especially interesting that AqEPFL4 came out as one of the most connected hub genes in a module that is strongly associated with s1, suggesting that our results provide further evidence for the role of the EPFLs in the meristem homeostasis.
Perhaps the most surprising observation regarding the green-s1 module network is that one gene, AqAGO5a (Aqcoe5G237200, homologous to ARGONAUTE5, AtAGO5, in A. thaliana; Fig. S9), was not initially identified as a hub gene since neither its MM nor TS values were above the 90th percentile of distributions (Table S2), but it did come up as a potential hub gene in the network visualization due to its strong connections with all the other hub genes. AGO genes encode RNA slicers that are involved in post-transcriptional gene silencing by selectively recruiting microRNAs and siRNAs. In A. thaliana, AtAGO5 physically interacts with miRNA156 and regulates the phrase transition from the vegetative to reproductive state of the meristem66. Other AtAGO genes, such as AtAGO1 and AtAGO10, have also been shown to function in establishing polarity in floral organs and regulating meristematic activities in the FM by interacting with miRNA165/16667,68,69,70,71. Moreover, based on our gene phylogeny (Fig. S9a), the Aquilegia genome contains a close paralog of AqAGO5a, Aqcoe2G159700 (AqAGO5b), which shares 81.4% of similarity with AqAGO5a (Fig. S9b). The expression level of AqAGO5a was the highest at s1 and then gradually decreases from s2 to s4, while its paralog AqAGO5b, showed very stable expression from s1 to s4 (Fig. S9c). It will thus be intriguing to determine what are the targets of these two close paralogs and the potential functional divergence between them.
The brown and magenta modules captured different aspects of s4 floral development
Interestingly, among the 56 potential hub genes for the brown-s4 module, only three genes code for transcription factors (TFs), and there seemed to be an over-representation of genes coding for key components of the ubiquitination pathways (Table S2). Six out of the 22 hub genes that exhibit a negative association with s4 encode key components in the SCF E3 ligase complexes, including F-box family proteins (Aqcoe2G310400, Aqcoe7G317700, Aqcoe7G317600), RING/U-box family proteins (Aqcoe7G340300, Aqcoe3G311800), and HCP-like superfamily protein (Aqcoe5G357900). In addition, two hub genes that have positive associations with s4 code for a ubiquitin-related protein (Aqcoe3G298400) and a subunit of the COP9 complex (Aqcoe7G310200), respectively.
The gene network of the brown-s4 module revealed that most of the identified hub genes are highly interconnected to each other and thus are mostly placed at the periphery of the network (Fig. 7). One hub gene, Aqcoe7G340300, however, appeared to have strong connections not only to most of the other hub genes but also to a large number of additional module members. Aqcoe7G340300 codes for a RING/U-box type protein and its homologs, DA2 and GRAIN WIDTH AND WEIGHT2 (GW2) have been functionally studied in A. thaliana72 and rice73, respectively. Both DA2 and GW2 have E3 ubiquitin ligase activity and exhibit conserved functions in regulating organ size by restricting cell divisions in both systems74. The expression level of Aqcoe7G340300 steadily declined from s1 to s4 (Fig. S4), and if its function in suppressing cell division in lateral organs is conserved, the low expression level at s4 may indicate the onset of cell proliferation in the floral organs.
In contrast to what was observed for the brown-s4 module, in which most of the hub genes were directly connected to each other, the 14 hub genes for the magenta-s4 module were connected to each other via a number of common non-hub genes in the network (Fig. 8).
Among the magenta-s4 hub genes that have negative MMs, Aqcoe2G063200 encodes a small ubiquitin-like modifier (SUMO) and is homologous to SUM2 in A. thaliana. Sumoylation mediated by SUMOs is an important post-translational protein modification mechanism in plants and is involved in regulating diverse plant physiological and developmental processes, including flowering time75. Interestingly, another hub gene with negative MM, Aqcoe2G315500, codes for a copper/zinc superoxide dismutase (CSD) and the protein stability of its A. thaliana homolog CSD1 has been shown to be maintained via sumoylation76. This relationship was captured in our network analysis, making their function in early floral development an interesting target for further study. In addition, Aqcoe5G226900, the Aquilegia homolog of MONOPTEROS (AqMP) is another magenta-s4 hub gene with expression level that was highest at s1 and then steadily declining from s2 to s4. In A. thaliana, MP is critical to maintaining stem cell homeostasis in the SAM77 so this decline is consistent with the cessation of FM proliferation (Fig. 4). On the other hand, the homolog of Aqcoe2G426800, EXCESS MICROSPOROCYTES1 (EXS), is expressed during the differentiation of microsporocytes and tapetal cells and controls the somatic and reproductive cell fates in A. thaliana anthers78. If functional conservation is assumed, the sharp increase in the expression of Aqcoe2G426800 at s4 (Fig. S4) likely marks the beginning of microsporogenesis in Aquilegia.
Conclusions
The current study is the first in-depth transcriptome profiling of early Aquilegia floral development. Similar studies at finely dissected stages of early floral development have only been done in the model species A. thaliana79,80,81, although analyses have also been performed in tomato SAMs, and early inflorescences of model monocot species82,83. In contrast to the previous A. thaliana work, the relatively large size of Aquilegia FMs allowed us to visually confirm the developmental stage of dissected meristems and perform a large number of biological replicates. Overall, our results reveal similar themes to the A. thaliana analyses, with down-regulation of meristem maintenance corresponding to a shift towards primordium proliferation and differentiation. For the developmental window on which we chose to focus, the morphological changes between successive floral developmental stages are subtle, which helped us to tease apart the dynamic transcriptional changes between the stages. In particular, we have uncovered small numbers of DE genes when comparing s1 to s2 (67), and s2 to s3 (49), with only five genes appearing in common between both DE lists (Table S1). These two groups of DE genes mark two important processes during early floral development: the DE genes between s1/s2 appear to correspond to the termination of the FM and the end of organogenesis, while the DE genes between s2/s3 as associated with the onset of floral organ morphogenesis. Analyzing the homologs of known genes in the FM termination pathway (Fig. 4) provided evidence that the A. thaliana mechanisms are likely not conserved in Aquilegia. We have also identified a number of key genes that are likely of importance for different stages of early floral development in A. coerulea, including AqPAN, AqSAP, AqEFLP4, and AqAGO5, as well as many top DE genes and hub genes for which the homologs have not been studied in A. thaliana. Thus, our study is the third study to date to indicate that the EPFLs play a previously overlooked role in the meristem homeostasis (Fig. 6), as supported by its identification as one of the core hub genes for the green-s1 module.
In addition, understanding the mechanisms leading to functional divergence of close paralogs is an important component of understanding gene evolution in general. Utilizing DE analysis and WGCNA, we have uncovered evidence of potential functional divergence between the close paralogs of several key genes expressed during early floral development in Aquilegia.
These include the close paralogs of AqSTM1 and AqSTM2, AqUFO1 and AqUFO2, and AqAGO5a and AqAGO5b, of which only AqSTM2, AqUFO1, and AqAGO5a were recovered as core hub genes of the green-s1 module (Fig. 6; Table, S2). These observations help to direct future investigations on the mechanisms and consequences of the functional divergence between close paralogs; for instance, whether or not AqAGO5a and AqAGO5b function in different tissues or have the same miRNA targets.
Finally, our results highlight the dynamics and potential role of several post-transcriptional and post-translational regulatory mechanisms during early floral development in A. coerulea. In terms of post-transcriptional regulation, in our network analysis for the green-s1 module, AqAGO5a appeared as a hub gene due to its strong connections to all the other hub genes in the network, suggesting an important role in small RNA regulation in s1 (Fig. 6). The Argonaute protein family is a critical player in the RNA silencing process as an essential component of the RNA-induced silencing complex84. Homologs of the AGO1/5/10 clade have been studied in A. thaliana, rice, tomato, and tobacco, all of which participate in various important aspects in plant development, including regulation of SAM, FM, germ cell development, and stress responses84,85,86,87. Nonetheless, small RNA regulation has not yet been explored in Aquilegia, making AqAGO5a a good starting point for further exploration. It is also clear that post-translational regulation via protein stability plays a major role in the transition from primordium initiation to floral organ morphogenesis in the Aquilegia FM. This is supported by the ubiquitination-related hub genes of the brown-s4 module coding for RING/U-box proteins, as well as an abundance of hub genes for both the green-s1 and the brown-s4 modules coding for F-box proteins, which are the components of the SCF E3 ligase complex (Table S2). Some of these genes, such as UFO and SAP, are long known to participate in floral development in A. thaliana36,55,56,60, but the connection between other F-box coding genes and floral development still needs further investigation. For instance, only recently BOP2 was discovered to serve as the substrate adaptor in an SCF E3 ligase complex to regulate LFY post-transcriptionally88. Sumoylation is another important post-translational regulation mechanism that stabilizes proteins rather than promoting their turnover, and one of the hub genes for the magenta-s4 module codes for a SUMO (Fig. 8; Table S2). SUMOs have been studied in the context of environmental stress, nitrogen assimilation, and flowering time, but little is known about their role in floral development75. GO term enrichment analysis showed that genes in the magenta-s4 module are significantly involved in floral organ development (Fig. 5c; Supp. Data 2), including the SUMO hub gene, making it interesting to explore their connections.
References
Coen, E. S. & Meyerowitz, E. M. The war of the whorls: genetic interactions controlling flower development. Nature 353, 31–37 (1991).
Ditta, G., Pinyopich, A., Robles, P., Pelaz, S. & Yanofsky, M. F. The SEP4 gene of arabidopsis thaliana functions in floral organ and meristem identity. Curr. Biol. 14, 1935–1940 (2004).
Schoof, H. et al. The stem cell population of Arabidopsis shoot meristems is maintained by a regulatory loop between the CLAVATA and WUSCHEL genes. Cell 100, 635–644 (2000).
Sun, B., Xu, Y., Ng, K.-H. & Ito, T. A timing mechanism for stem cell maintenance and differentiation in the Arabidopsis floral meristem. Genes Dev. 23, 1791–1804 (2009).
Sun, B. et al. Timing mechanism dependent on cell division is invoked by polycomb eviction in plant stem cells. Science 343, 1248559–1248559 (2014).
Cheng, Y. & Zhao, Y. A role for auxin in flower development. J. Integr. Plant Biol. 49, 99–104 (2007).
Yu, H. & Huang, T. Molecular mechanisms of floral boundary formation in Arabidopsis. Int. J. Mol. Sci. 17, 317 (2016).
Kramer, E. M. Aquilegia : a new model for plant development, ecology, and evolution. Annu. Rev. Plant Biol. 60, 261–277 (2009).
Tucker, S. C. & Hodges, S. A. Floral ontogeny of Aquilegia, Semiaquilegia, and Enemion (Ranunculaceae). Int. J. Plant Sci. 166, 557–574 (2005).
Kramer, E. M. et al. Elaboration of B gene function to include the identity of novel floral organs in the lower eudicot Aquilegia. Plant Cell 19, 750–766 (2007).
Sharma, B. & Kramer, E. Sub- and neo-functionalization of APETALA3 paralogs have contributed to the evolution of novel floral organ identity in Aquilegia (columbine, Ranunculaceae). New Phytol. 197, 949–957 (2013).
Sharma, B. et al. Homologs of LEAFY and UNUSUAL FLORAL ORGANS promote the transition from inflorescence to floral meristem identity in the cymose Aquilegia coerulea. Front. Plant Sci. 10, 1218 (2019).
Min, Y. & Kramer, E. M. The Aquilegia JAGGED homolog promotes proliferation of adaxial cell types in both leaves and stems. New Phytol. 216, 536–548 (2017).
Yant, L., Collani, S., Puzey, J., Levy, C. & Kramer, E. M. Molecular basis for three-dimensional elaboration of the Aquilegia petal spur. Proc. R. Soc. B Biol. Sci. 282, 20142778 (2015).
Meaders, C., Min, Y., Freedberg, K. J. & Kramer, E. Developmental and molecular characterization of novel staminodes in Aquilegia. Ann. Bot. https://doi.org/10.1093/aob/mcaa029 (2020).
Ballerini, E. S., Kramer, E. M. & Hodges, S. A. Comparative transcriptomics of early petal development across four diverse species of Aquilegia reveal few genes consistently associated with nectar spur development. BMC Genom. 20, 668 (2019).
Krueger, F. FelixKrueger/TrimGalore. (2020).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research 4, 1521 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Benjamini, Y. & Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 1165–1188 (2001).
Du, Z., Zhou, X., Ling, Y., Zhang, Z. & Su, Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70 (2010).
Tian, T. et al. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 45, W122–W129 (2017).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinform. Oxf. Engl. 23, 2947–2948 (2007).
Chandler, J. W. & Werr, W. Cytokinin–auxin crosstalk in cell type specification. Trends Plant Sci. 20, 291–300 (2015).
Besnard, F. et al. Cytokinin signalling inhibitory fields provide robustness to phyllotaxis. Nature 505, 417–421 (2014).
Besnard, F., Rozier, F. & Vernoux, T. The AHP6 cytokinin signaling inhibitor mediates an auxin–cytokinin crosstalk that regulates the timing of organ initiation at the shoot apical meristem. Plant Signal. Behav. 9, e28788 (2014).
Maier, A. T. et al. Dual roles of the bZIP transcription factor PERIANTHIA in the control of floral architecture and homeotic gene expression. Development 136, 1613–1620 (2009).
Hibara, K. et al. Arabidopsis CUP-SHAPED COTYLEDON3 regulates postembryonic shoot meristem and organ boundary formation. Plant Cell 18, 2946–2957 (2006).
Ding, L. et al. HANABA TARANU (HAN) bridges meristem and organ primordia boundaries through PINHEAD, JAGGED, BLADE-ON-PETIOLE2 and CYTOKININ OXIDASE 3 during flower development in Arabidopsis. PLoS Genet. 11, e1005479 (2015).
Li, N. et al. STERILE APETALA modulates the stability of a repressor protein complex to control organ size in Arabidopsis thaliana. PLoS Genet. 14, e1007218 (2018).
Krizek, B. AINTEGUMENTA and AINTEGUMENTA-LIKE6 act redundantly to regulate Arabidopsis floral growth and patterning. Plant Physiol. 150, 1916–1929 (2009).
Torii, K. U. Stomatal differentiation: the beginning and the end. Curr. Opin. Plant Biol. 28, 16–22 (2015).
Hepworth, C., Caine, R. S., Harrison, E. L., Sloan, J. & Gray, J. E. Stomatal development: focusing on the grasses. Curr. Opin. Plant Biol. 41, 1–7 (2018).
Hardie, D. G. Plant protein serine/threonine kinases: classification and functions. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50, 97–131 (1999).
Sharma, B. & Kramer, E. M. The MADS-Box gene family of the basal eudicot and hybrid Aquilegia coerulea ‘Origami’ (Ranunculaceae)1. Ann. Mo. Bot. Gard. 99, 313–322 (2014).
Pabón-Mora, N., Sharma, B., Holappa, L. D., Kramer, E. M. & Litt, A. The Aquilegia FRUITFULL-like genes play key roles in leaf morphogenesis and inflorescence development. Plant J. 74, 197–212 (2013).
Bollier, N. et al. At-MINI ZINC FINGER2 and Sl-INHIBITOR OF MERISTEM ACTIVITY, a conserved missing link in the regulation of floral meristem termination in Arabidopsis and tomato. Plant Cell 30, 83–100 (2018).
Sun, B. et al. Integration of transcriptional repression and polycomb-mediated silencing of WUSCHEL in floral meristems. Plant Cell 31, 1488–1505 (2019).
Yamaguchi, N. et al. Chromatin-mediated feed-forward auxin biosynthesis in floral meristem determinacy. Nat. Commun. 9, 5290 (2018).
Yamaguchi, N., Huang, J., Xu, Y., Tanoi, K. & Ito, T. Fine-tuning of auxin homeostasis governs the transition from floral stem cell maintenance to gynoecium formation. Nat. Commun. 8, 1125 (2017).
Gleason, E. J. & Kramer, E. M. Conserved roles for polycomb repressive complex 2 in the regulation of lateral organ development in Aquilegia x coerulea ‘Origami’. BMC Plant Biol. 13, 185 (2013).
Seifert, G. J. Nucleotide sugar interconversions and cell wall biosynthesis: how to bring the inside to the outside. Curr. Opin. Plant Biol. 7, 277–284 (2004).
Bastow, R. et al. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 427, 164–167 (2004).
Ballerini, E. S. & Kramer, E. M. Environmental and molecular analysis of the floral transition in the lower eudicot Aquilegia formosa. EvoDevo 2, 4 (2011).
Sharma, B. et al. Developmental and molecular changes underlying the vernalization-induced transition to flowering in Aquilegia coerulea (James). Genes 10, 734 (2019).
Levy, Y. Y. Multiple roles of Arabidopsis VRN1 in vernalization and flowering time control. Science 297, 243–246 (2002).
Hoffmann, L. et al. Silencing of hydroxycinnamoyl-coenzyme A shikimate/quinate hydroxycinnamoyltransferase affects phenylpropanoid biosynthesis. Plant Cell 16, 1446–1465 (2004).
Meyerowitz, E. M. UFO: an Arabidopsis gene involved in both floral meristem and floral organ development. Plant Cell 7, 529–548 (1995).
Durfee, T. et al. The F-box-containing protein UFO and AGAMOUS participate in antagonistic pathways governing early petal development in Arabidopsis. Proc. Natl. Acad. Sci. 100, 8571–8576 (2003).
Samach, A. et al. The UNUSUAL FLORAL ORGANS gene of Arabidopsis thaliana is an F-box protein required for normal patterning and growth in the floral meristem. Plant J. 20, 433–445 (1999).
Lee, I., Wolfe, D. S., Nilsson, O. & Weigel, D. A LEAFY co-regulator encoded by unusual floral organs. Curr. Biol. 7, 95–104 (1997).
Wilkinson, M. D. & Haughn, W. UNUSUAL FLORAL ORGANS controls meristem identity and organ primordia fate in Arabidopsis. Plant Cell 7, 1485–1499 (1995).
Byzova, M. V. et al. Arabidopsis STERILE APETALA, a multifunctional gene regulating inflorescence, flower, and ovule development. Genes Dev. 13, 1002–1014 (1999).
Wang, Z. et al. SCFSAP controls organ size by targeting PPD proteins for degradation in Arabidopsis thaliana. Nat. Commun. 7, 11192 (2016).
Yang, L. et al. LITTLELEAF ( LL ) encodes a WD40 repeat domain-containing protein associated with organ size variation in cucumber. Plant J. 95, 834–847 (2018).
Abrash, E. B., Davies, K. A. & Bergmann, D. C. Generation of signaling specificity in Arabidopsis by spatially restricted buffering of ligand-receptor interactions. Plant Cell 23, 2864–2879 (2011).
Uchida, N. et al. Regulation of inflorescence architecture by intertissue layer ligand-receptor communication between endodermis and phloem. Proc. Natl. Acad. Sci. 109, 6337–6342 (2012).
Kosentka, P. Z., Overholt, A., Maradiaga, R., Mitoubsi, O. & Shpak, E. D. EPFL signals in the boundary region of the SAM restrict its size and promote leaf Initiation. Plant Physiol. 179, 265–279 (2019).
Zhang, L., DeGennaro, D., Lin, G., Chai, J. & Shpak, E. D. ERECTA family signaling constrains CLAVATA3 and WUSCHEL to the center of the shoot apical meristem. BioRxiv https://doi.org/10.1101/2020.02.24.962787 (2020).
Roussin-Léveillée, C., Silva-Martins, G. & Moffett, P. ARGONAUTE5 mediates fine-tuning of vegetative-to-reproductive phase transition through its interaction with miR156 in Arabidopsis. BioRxiv https://doi.org/10.1101/640680 (2019).
Kidner, C. A. & Martienssen, R. A. The role of ARGONAUTE1 (AGO1) in meristem formation and identity. Dev. Biol. 280, 504–517 (2005).
Tucker, M. R. et al. Vascular signalling mediated by ZWILLE potentiates WUSCHEL function during shoot meristem stem cell development in the Arabidopsis embryo. Development 135, 2839–2843 (2008).
Zhu, H. et al. Arabidopsis Argonaute10 specifically sequesters miR166/165 to regulate shoot apical meristem development. Cell 145, 242–256 (2011).
Ji, L. et al. ARGONAUTE10 and ARGONAUTE1 regulate the termination of floral stem cells through two microRNAs in Arabidopsis. PLoS Genet. 7, e1001358 (2011).
Roodbarkelari, F., Du, F., Truernit, E. & Laux, T. ZLL/AGO10 maintains shoot meristem stem cells during Arabidopsis embryogenesis by down-regulating ARF2-mediated auxin response. BMC Biol. 13, 74 (2015).
Xia, T. et al. The ubiquitin receptor DA1 interacts with the E3 ubiquitin ligase DA2 to regulate seed and organ size in Arabidopsis. Plant Cell 25, 3347–3359 (2013).
Song, X.-J., Huang, W., Shi, M., Zhu, M.-Z. & Lin, H.-X. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat. Genet. 39, 623–630 (2007).
Dong, H. et al. Ubiquitylation activates a peptidase that promotes cleavage and destabilization of its activating E3 ligases and diverse growth regulatory proteins to limit cell proliferation in Arabidopsis. Genes Dev. 31, 197–208 (2017).
Park, H. J. et al. SUMO and SUMOylation in plants. Mol. Cells 32, 305–316 (2011).
Chen, C.-C., Chen, Y.-Y. & Yeh, K.-C. Effect of Cu content on the activity of Cu/ZnSOD1 in the Arabidopsis SUMO E3 ligase siz1 mutant. Plant Signal. Behav. 6, 1428–1430 (2011).
Luo, L., Zeng, J., Wu, H., Tian, Z. & Zhao, Z. A molecular framework for auxin–controlled homeostasis of shoot stem cells in Arabidopsis. Mol. Plant 11, 899–913 (2018).
Canales, C., Bhatt, A. M., Scott, R. & Dickinson, H. EXS, a Putative LRR receptor kinase, regulates male germline cell number and tapetal identity and promotes seed development in Arabidopsis. Curr. Biol. 12, 1718–1727 (2002).
Wellmer, F., Alves-Ferreira, M., Dubois, A., Riechmann, J. L. & Meyerowitz, E. M. Genome-wide analysis of gene expression during early Arabidopsis flower development. PLOS Genet. 2, e117 (2006).
Ryan, P. T. et al. Patterns of gene expression during Arabidopsis flower development from the time of initiation to maturation. BMC Genom. 16, 488 (2015).
Klepikova, A. V., Kasianov, A. S., Gerasimov, E. S., Logacheva, M. D. & Penin, A. A. A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 88, 1058–1070 (2016).
Lemmon, Z. H. et al. The evolution of inflorescence diversity in the nightshades and heterochrony during meristem maturation. Genome Res. 26, 1676–1686 (2016).
Feng, N. et al. Transcriptome profiling of wheat inflorescence development from spikelet initiation to floral patterning identified stage-specific regulatory genes. Plant Physiol. 174, 1779–1794 (2017).
Fang, X. & Qi, Y. RNAi in plants: an argonaute-centered view. Plant Cell 28, 272–285 (2016).
Hendelman, A. et al. The developmental outcomes of P0-mediated ARGONAUTE destabilization in tomato. Planta 237, 363–377 (2013).
Zhang, H., Xia, R., Meyers, B. C. & Walbot, V. Evolution, functions, and mysteries of plant ARGONAUTE proteins. Curr. Opin. Plant Biol. 27, 84–90 (2015).
Jones, L., Keining, T., Eamens, A. & Vaistij, F. E. Virus-induced gene silencing of Argonaute genes in Nicotiana benthamiana demonstrates that extensive systemic silencing requires Argonaute1 -like and Argonaute4 -like genes. Plant Physiol. 141, 598–606 (2006).
Chahtane, H. et al. LEAFY activity is post-transcriptionally regulated by BLADE ON PETIOLE2 and CULLIN3 in Arabidopsis. New Phytol. 220, 579–592 (2018).
Acknowledgements
The authors would like to thank Pierre Baduel, the sequencing team at the Bauer Core Facility at the Harvard University and the Harvard FAS Informatics team for discussing analysis strategies, Dan Utter and Brian Arnold for help troubleshooting the Linux scripts, Weilin Meng for help troubleshooting and optimizing the network visualization, and the R package developers and bioinformaticians Michael Love, Kevin Blighe, and Peter Langfelder for actively responding to questions on online forums. We thank the Department of Organismic and Evolutionary Biology Ph.D. program at Harvard University for providing funding to support this project.
Author information
Authors and Affiliations
Contributions
The study was conceived of and designed by Y.M. and E.M.K. All experiments and data analysis were conducted by Y.M. with oversight provided by E.M.K. Manuscript was written by Y.M. with revisions by E.M.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Min, Y., Kramer, E.M. Transcriptome profiling and weighted gene co-expression network analysis of early floral development in Aquilegia coerulea. Sci Rep 10, 19637 (2020). https://doi.org/10.1038/s41598-020-76750-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-76750-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
