
Abstract
The diversity of fin morphology within and across fish taxa offers great, but still largely unexplored, opportunities to investigate the proximate mechanisms underlying fin shape variation. Relying on available genetic knowledge brought forth mainly by the comprehensive study of the zebrafish caudal fin, we explored candidate molecular mechanisms for the maintenance and formation of the conspicuously elongated filaments adorning the unpaired fins of the East African “princess cichlid” Neolamprologus brichardi. Via qPCR assays, we detected expression differences of candidate genes between elongated and short regions of intact and regenerating fins. The identified genes include skeletogenic and growth factors (igf2b, fgf3, bmp2 and bmp4), components of the WNT pathway (lef1, wnt5b and wnt10) and a regulatory network determining fin ray segment size and junction (cx43, esco2 and sema3d), as well as other genes with different roles (mmp9, msxb and pea3). Interestingly, some of these genes showed fin specific expression differences which are often neglected in studies of model fish that focus on the caudal fin. Moreover, while the observed expression patterns were generally consistent with zebrafish results, we also detected deviating expression correlations and gene functions.
Similar content being viewed by others

Introduction
The phenotypic diversity of fish is outstanding among vertebrates. One part of this immense diversity is variation in fin shape, which often distinguishes even closely related species (e.g. Xiphophorus platyfish and swordtails, Bodianus lipfish, various gourami and cichlid genera). Although fin shape is connected with swimming performance1, fins also serve important signal functions in competitive and sexual interactions. Both courtship and aggressive displays typically involve the presentation of erected fins, and the importance of fin shape in mate choice and male-male competition2,3,4 suggests that various selection pressures play a role in fin shape diversification.
The developmental mechanisms regulating fin growth and shape have mainly been studied in the zebrafish caudal fin5. The capability of fish to fully regenerate amputated fins makes them an ideal model in regeneration biology, not least because of the high degree of conservation in developmental patterning between fish fins and the appendages of other vertebrates5,6,7. The teleost fin is supported by segmented fin rays, which consist of an inner core of mesenchymal cells, vessels and nerves, shielded by bone (hemirays) and covered with epithelium. The fin rays grow by the addition of bony segments at their distal end, and altered regulation of fin ray growth, including cellular proliferation, differentiation and survival, can be an important source of fin shape variation8. Regeneration studies showed that after amputation, regeneration of the fin tissue is initiated by the establishment of highly proliferative tissue (blastema) immediately distal to each truncated fin ray. The blastema continues to elongate at the distal edge while the proximal part of the new tissue re-differentiates into the mature fin structures until the original size is restored5,9. At the molecular level, components of various developmental pathways, such as WNT, FGF, Hedgehog and retinoic acid (RA) signalling, as well as epigenetic, skeletogenic and structural remodelling factors have been identified to orchestrate both ontogenetic and regenerative fin growth8,9,10. Recently, candidate effectors (gene products and metabolites) of positional memory, a prerequisite for the regeneration of the proper appendage size and pattern, were described in the zebrafish caudal fin11.
Contrasting with the extensive work in fish models (mainly zebrafish), little use has so far been made of the natural diversity among fishes12,13,14. The resources created in zebrafish open countless possibilities to investigate genes, regulatory networks, signalling cascades, etc., for their role in fin shape diversification and to test for molecular convergence of fin shape determination across species. Moreover, as most research has focused on the caudal fin because of its particularly fast regeneration5, it is not yet clear whether the mechanisms controlling fin growth are consistent across fin types. In the current study, we take advantage of the fin shape characteristics of an East African cichlid fish Neolamprologus brichardi, namely the long filaments adorning the unpaired fins of the adult fish (Fig. 1A). We hypothesized that the formation of this local outgrowth is influenced by genes involved in fin ray organization and growth, and predicted differences in gene expression levels between elongated and short fin regions during the maintenance of the phenotype in adult fins and/or during their regeneration. Importantly, our study species allows us to compare gene expression levels between adjacent normal (short) and elongated regions within a fin type, and then compare patterns of gene expression differences across fin types. Furthermore, the availability of an annotated transcriptome15 for N. brichardi facilitates the utilisation of zebrafish genetic resources. The 40 candidate genes investigated in this study belong to different signaling pathways and functional groups (e.g. skeletogenesis, extracellular matrix formation and WNT signalling) associated with ontogenetic and regenerative fin growth in teleost fish, mainly zebrafish (Table 1). Comparisons between elongated and short fin regions and between regeneration stages of each fin type identified a number of differentially expressed genes, some of which showed significant expression correlations. With this work, we provide the first investigation of the molecular mechanisms underlying ornamental outgrowth of unpaired fins in a cichlid fish and lay the foundation for further comparative studies within and beyond the highly diverse cichlid family.

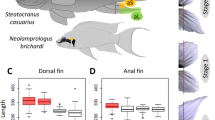
Fin dissection in Neolamprologus brichardi, fin ray segment length measurements and expression analysis of candidate reference genes. (A) An adult Lake Tanganyika cichlid fish, N. brichardi, displays regional elongation of its unpaired fins. Fins were amputated along the dashed red line, and tissue samples were taken from the elongated fin parts (green shadows) and from the short regions (yellow shadows) of the fins. (B) Biopsies were taken from the original (day 0) and regenerated (days 15 and 35) fin tissue, as shown here for the caudal fin (photos by Wolfgang Gessl (www.pisces.at)). The slight curve of the distal edge of the caudal fin on day 1 is due to incomplete spreading of the fin. (C) Fin ray segment length in L and S regions of each fin. P-values were obtained by linear mixed models. (D) Expression profiles of candidate reference genes in raw Cq values for all samples (all anal, dorsal and caudal fin samples of 3 cutting steps from 3 biological replicates). The middle line denotes the median and boxes indicate the 25/75 percentiles.
Results
Morphological characterization
The outgrowth of fin filaments, i.e. the elongations of the unpaired fins, was not associated with increases in the length of fin ray segments in these regions. In contrast, segments in the elongated fin regions were slightly shorter than in the short regions (Fig. 1C). We conclude that the elongation of fin rays is due to increases in the number, but not length, of fin ray segments.
Validation of reference genes
Gene expression analyses by qPCR rely on the validation of stably expressed reference genes16, and depending on species, tissue and experimental conditions, the expression stability of reference genes can be variable17. In order to identify suitable reference genes for our study, we examined the expression of 7 reference gene candidates in cDNA of the tissue samples from the intact and regenerating fins. The expression levels of these candidate genes from highest to lowest (lowest to highest Cq) are actb1 > rps11 > rps18 > hprt1 > tbp > elf1a > gapdh (Fig. 1D). Based on standard deviations of Cq values and the three algorithms implemented in BestKeeper, geNorm and NormFinder, rps18 and actb1 consistently achieved top ranks for expression stability (Table 2). Consequently, expression of rps18 and actb1 was used to normalize target gene expression for quantitative comparisons between fin regions.
Candidate target gene expression
Next, we assessed the expression levels of 40 candidate target genes in the elongated and short regions of the dorsal, anal and caudal fins. Gene expression was quantified in biopsies of the original fin tissue (stage 0) and at two stages during tissue regeneration. Since stage 0 tissue samples were taken from young adults (Supplementary Fig. 1), they represent growing fins. At regeneration stage 1 (day 15), the elongation of the fin tips had just become apparent, and at stage 2 (day 35), elongation was pronounced but fin regeneration not yet completed (Fig. 1B). Since fin types are known to regenerate at different speed5 and may also differ in their anatomical properties, comparisons of expression levels between elongated and short regions were carried out within each fin type separately (Supplementary data 2). We focus on 13 genes which showed significant expression differences between the elongated and short regions in at least two stages of two fins. For the presentation of the expression data, we classified these genes by their functional properties, i.e. growth and skeletogenic factors (Fig. 2), components of WNT signaling (Fig. 3), genes involved in fin ray segment formation (Fig. 4) and other genes with different functions (Fig. 5). Additionally, expression data for all of the investigated genes are shown in Supplementary data 2.

Expression levels of skeletogenic and growth factors bmp2, bmp4, fgf3 and igf2b. Means and standard deviations of RQ in three biological replicates are shown for the elongated (L) and short (S) regions of the caudal, dorsal and anal fin in original (stage 0) and regenerating tissue. See Fig. 1A for fin region codes; numbers 0 to 2 identify regeneration stages. Circles above bars indicate significantly elevated expression (P < 0.05 in paired t-tests) in comparisons between L and S tissue samples (i.e., compared to the bar matching the colour code of the circle); note that the analysis was restricted to comparisons within the same fin type and the same regeneration stage. Triangles above bars indicate significantly elevated expression (P < 0.05 in paired t-tests) in comparisons among regeneration stages; comparisons were restricted to corresponding fin types and regions.

Expression levels of genes encoding components of WNT signalling (lef1, wnt5b and wnt10a). Means and standard deviations of RQ in three biological replicates are shown for the elongated (L) and short (S) regions of the caudal, dorsal and anal fin in original (stage 0) and regenerating tissue. See Fig. 1A for fin region codes; numbers 0 to 2 identify regeneration stages. Circles above bars indicate significantly elevated expression (P < 0.05 in paired t-tests) in comparisons between L and S tissue samples (i.e., compared to the bar matching the colour code of the circle); note that the analysis was restricted to comparisons within the same fin type and the same regeneration stage. Triangles above bars indicate significantly elevated expression (P < 0.05 in paired t-tests) in comparisons among regeneration stages; comparisons were restricted to corresponding fin types and regions.

Expression levels of genes involved in fin ray formation and cell differentiation in the original and regenerating fin tissue (cx43, esco2 and sema3d). Means and standard deviations of RQ in three biological replicates are shown for the elongated (L) and short (S) regions of the caudal, dorsal and anal fin in original (stage 0) and regenerating tissue. See Fig. 1A for fin region codes; numbers 0 to 2 identify regeneration stages. Circles above bars indicate significantly elevated expression (P < 0.05 in paired t-tests) in comparisons between L and S tissue samples (i.e., compared to the bar matching the colour code of the circle); note that the analysis was restricted to comparisons within the same fin type and the same regeneration stage. Triangles above bars indicate significantly elevated expression (P < 0.05 in paired t-tests) in comparisons among regeneration stages; comparisons were restricted to corresponding fin types and regions.

Expression levels of candidate genes mmp9, msxb and pea3. Means and standard deviations of RQ in three biological replicates are shown for the elongated (L) and short (S) regions of the caudal, dorsal and anal fin in original (stage 0) and regenerating tissue. See Fig. 1A for fin region codes; numbers 0 to 2 identify regeneration stages. Circles above bars indicate significantly elevated expression (P < 0.05 in paired t-tests) in comparisons between L and S tissue samples (i.e., compared to the bar matching the colour code of the circle); note that the analysis was restricted to comparisons within the same fin type and the same regeneration stage. Triangles above bars indicate significantly elevated expression (P < 0.05 in paired t-tests) in comparisons among regeneration stages; comparisons were restricted to corresponding fin types and regions.
Expression differences between short and long fin regions
Several patterns emerged in the comparisons of relative gene expression levels between elongated and short fin regions. ‘Expression in the elongated region’ is abbreviated as ‘L-expression’ in the following text, and reported as ‘higher’ or ‘lower’ in comparison to expression in the short region (‘S-expression’). We note that the design of our study only allowed for the detection of pronounced differences, such that non-significant contrasts do not necessarily imply the absence of an expression difference. The direction of significant expression differences detected between tissue samples (i.e. higher or lower L-expression) was consistent within genes. More genes were found with signals of higher L-expression (n = 8 genes; bmp4, fgf3, igf2b, lef1, cx43, esco2, mmp9 and msxb) than lower L-expression (n = 5 genes; bmp2, wnt5b, wnt10a, sema3d and pea3) (Figs 2–5). Two genes (cx43 and mmp9) showed higher L-expression in all fins and at most stages. Higher L-expression of bmp4, fgf3, lef1 and msxb was detected in all fins, but not at all stages. Fin-specific differences were observed for igf2b and esco2, with higher L-expression only in caudal and dorsal fins. Lower L-expression across all fins and most stages was observed for sema3d and wnt10a. Interestingly, while wnt10a showed the strongest L/S expression differences in the caudal fin, lower L-expression of wnt5a was restricted to the dorsal and anal fins (Fig. 3). bmp2 and pea3 showed fin-specific lower L-expression (caudal and anal fins). Additionally, we note contrasting L/S expression differences for two skeletogenic genes, bmp2 (lower L-expression) and bmp4 (higher L-expression) (Fig. 1).
The gene expression analyses in the caudal fin consisted of two comparisons, (1) between the dorsal elongated (dcL) and the centre short (cS) region and (2) between the ventral elongated (vcL) and the cS region. For some genes, only one of these comparisons yielded a significant result. While these instances could represent dorso-ventral differences in gene expression levels, some of these genes showed dcL/cS and vcL/cS differences in the same direction, even though only one of the comparisons was significant (e.g. mmp9 at stage 2; Fig. 5). When we compared dcL and vcL expression levels to test for dorso-ventral differences, significant contrasts were observed for only few genes (Supplementary data 2). We also note that for some genes, L/S expression differences fell below statistical significance at individual regeneration stages (e.g. mmp9 at stage 2 in the dorsal fin; Fig. 5). We consider it likely that in some cases, the absence of consistent statistical test results in corresponding tissue comparisons can be ascribed to power limitations imposed by our study design.
Expression differences across regeneration stages
Gene expression levels in the short and elongated regions of each fin were compared between the original and the regenerated biopsies (i.e. stage 0 versus stage 1 and 2) in order to detect up- or downregulation of genes during regeneration, in particular differences in gene regulation across regeneration between short and elongated fin regions. We focus on the results for the genes reported above (Figs 2–5); complete data are given in the Supplementary data 2. Again, the design of our study only allowed for the detection of pronounced differences. Most of the variation in gene expression across stages concerned the upregulation of genes in regenerating tissue compared to the original tissue, and was not specific to either the short or the elongated fin regions (e.g. igf2b, msxb, cx43, fgf3). For two genes, we detected significant upregulation in the short region of one of the fins, whereas expression did not change across stages in the elongated region (dorsal fin, wnt10a; anal fin, sema3d).
Expression correlation analyses
The analysis of pairwise gene expression correlations can be an initial step towards identification of gene regulatory networks (GRNs) and coexpression modules for functional/transcriptional investigations18. In non-model teleost fish, analyses of gene expression correlations have frequently been used to identify GRNs potentially involved in morphological variation in skeletal structures19,20,21,22. In our study, pairwise comparisons of gene expression levels among the 13 genes with L/S expression differences revealed a large number of significant correlations. The majority of the significant results concerned positive correlations (blue shadings in Supplementary Fig. S2), many of which occurred in all three fins (framed cells) indicating potential co-regulatory mechanisms. Negative expression correlations (red shadings in Supplementary Fig. S2) were mainly restricted to the caudal fin. The strongest positive correlations detected in all fins for the genes with lower L-expression were observed between wnt5b-sema3d and bmp2-pea3. Strong correlations between genes with higher L-expression were found for, e.g., esco2-msxb, msxb-lef1, igf2b-msxb-cx43, fgf3-msxb-bmp4-cx43, and cx43-mmp9. Furthermore, bmp4, cx43 and msxb showed the highest number of positive expression correlations with other genes across the three fins, while expression of wnt5b and wnt10a was correlated with few other genes. None of the negative correlations were observed across fins.
Discussion
Given that ontogenetic and regenerative growth of fish fins depends on the expression of a vast number of genes9, it is plausible that some of these genes, belonging to different signaling pathways, are also involved in the determination of fin shape. Expression profiling studies in fish were mainly focused on the identification of genes that play a role in the general mechanisms of growth and regeneration of the caudal fin6,10,23,24. Hence, little is known about whether these gene regulatory networks determine fin shape traits, such as regional outgrowth, as well. In swordtail fish, where males grow a sword-like elongation on the ventral part of their caudal fin, sword-specific gene expression patterns were detected for a number of genes14,25,26.
We hypothesized that positional differences in gene expression levels underlie the extreme elongation of filaments at a specific region of a fin, as they occur in the unpaired fins of our study fish N. brichardi. Among the 40 candidate genes involved in zebrafish fin growth/regeneration that were screened for expression differences between elongated (L) and short (S) fin regions of N. brichardi, 13 genes showed repeated L/S expression differences. Most of these expression patterns were consistent with information on gene functions and interactions in the zebrafish, but we also noticed a few incongruences. Before discussing the 13 genes in more detail, we note that in our experiment, the elongated (L) and the short (S) tissue samples of the caudal fins also represent different positions along the proximo-distal axis (with L more distal than S), and L/S expression differences could therefore reflect expression gradients along this axis. A recent study described proximo-distal gene expression gradients along the zebrafish caudal fin for a number of genes11. None of the genes discussed on our study appeared in the ‘high confidence’ list of differentially expressed genes that was assembled from RNAseq and proteomics data, but some (bmp2, bmp4, lef1, wnt5b, sema3d, mmp9, igf2b) overlapped with the results based on RNAseq alone (Dataset S3 in Rabinowitz et al. 2017)11. However, the direction of the L/S difference in N. brichardi matched the proximo-distal gradient in zebrafish in only two cases (bmp4 and lef1). Unless proximo-distal expression gradients differ widely between zebrafish and N. brichardi, we may assume that our L/S comparison was not confounded by position along the proximo-distal axis.
Furthermore, the margins of the fins of N. brichardi are coloured white (Supplementary Fig. S1A), indicating the presence of light-reflecting iridophores and/or white leucophores, and expression differences between elongated and short regions could reflect different chromatophore compositions. We reduced this problem by using young adults, in which the white coloration is still less pronounced than in fully grown individuals, and removed most of the white margins from the tissue sample (Supplementary Fig. S1B). None of the genes showing expression differences between the long and short fin regions are enriched in the iridophore transcriptome of zebrafish27. However, since little is known about leucophore specific genes28, we cannot assess whether the possible presence of leucophores in elongated fin regions might contribute to the identified gene expression differences in this study.
Among the tested skeletogenic and growth factors, we found increased L-expression of genes encoding a fibroblast growth factor, fgf3, and an insulin growth factor, igf2b, in regenerating fins (Fig. 2). Both factors are known to be required for caudal fin regeneration in zebrafish through induction of the blastemal and wound epidermal markers (by igf2b) and promoting cell proliferation in the blastema (by fgf3)29,30. Intriguingly, the insulin/insulin-like pathway (ILS) has been proposed as a mechanism for tissue-specific exaggerated growth, linking the development of ornaments and weapons to individual condition31. Furthermore, a possible role of FGF signalling in fin shape determination has been demonstrated in the swordtail fish, where the growth of the sword-like extension of the male caudal fin involved the activation of the FGF pathway25. Interestingly, two genes coding for bone morphogenetic proteins (BMPs), bmp2 and bmp4, displayed opposite L/S expression differences, though the pattern was not repeated across all fins and stages (Fig. 2). BMP mediated signals are involved in induction of osteoblast differentiation and skeletogenesis during fin development and regeneration23,32,33,34,35. In the zebrafish caudal fin, expression of bmp4 is restricted to the distal blastemal, whereas bmp2 is expressed in newly differentiating scleroblasts and in the adjacent cells of the basal layer of the epidermis34. Possibly, the higher L-expression of bmp4 in our study may reflect elevated cell proliferation activity in the elongated fin regions, whereas higher S-expression of bmp2 may be due to a shift towards bone differentiation in most of the short fin tissue.
WNT/β-catenin signaling is known for its pivotal role in regulating different aspects of fin development and regeneration such as establishment of the distal blastema and specification of the lateral basal epidermal layer8,9,32,36,37,38,39. WNT signaling functions through complex interactions with other signals including BMP, Hedgehog (HH) and FGF pathways, and different WNT components can have opposing effects during epimorphic fin regeneration32,37,38. Notably, we found lower L-expression of two WNT ligands, wnt5b and wnt10a, across original and regenerating tissue of N. brichardi fins (Fig. 3). The decreased L-expression of wnt5b was in agreement with the findings in zebrafish where its overexpression reduced proliferation of the blastema mesenchyme and the overlying epithelium in the regenerating fin37,38. However, the decreased L-expression of wnt10a was in contrast to the zebrafish findings, since a wnt10a activated β-catenin-dependent signal is required for blastema formation and subsequent regeneration37. It should be noted that wnt5b activates a non-canonical WNT signalling pathway, but can also negatively regulate the canonical WNT signaling pathway during fin regeneration37. Importantly, the expression patterns of wnt5b and wnt10a displayed different profiles between the fins, i.e. while wnt5b L/S expression differences were detected only in the anal and dorsal fins, wnt10a had the most pronounced L/S expression differences in the caudal fin. This could indicate fin and/or axis specific functions of these ligands.
Another interesting set of target candidates encompassed components of a gene regulatory network (i.e. cx43, esco2, sema3d) involved in formation, growth and regeneration of fin ray segments and joints40,41,42,43,44. Two of these genes, cx43 and sema3d, displayed L/S expression differences indicating distinct activation of the network in these fin regions (Fig. 4). cx43, a gene encoding a subunit of the gap junction protein complex by which neighbouring cells exchange small molecules, was of particular interest because of its known role in regulation of fin ray length in zebrafish40 and its consistent higher L-expression in the present study. The length of fin rays depends on the number and size of bony segments8 and reduced expression of cx43 in zebrafish sof mutants has been shown to cause defects in the lengthening of bony fin ray segments40. The same study has identified cx43 expression in both the germinal region of newly growing segments and in the osteoblasts at segment boundaries and joints40. Further investigation in zebrafish revealed that overexpression of cx43 suppresses segment joint formation and increases cell proliferation resulting in elongated bony fin rays45,46. Opposite to the findings in zebrafish, increased expression of cx43 in L fin regions of N. brichardi was not accompanied by increased length of fin ray segments (Fig. 1C). On the contrary, we found a slight increase in segment length in S regions compared to L regions. This indicates that, first, the elongation of L regions does not involve increased length of fin ray segments. Secondly, overexpression of cx43 in L regions might also involve other mechanism(s), distinct from the one causing fin ray elongation in zebrafish, during fin morphogenesis in N. brichardi.
A possible reason for the discrepancy between zebrafish studies and our results could be the increased expression of sema3d in short fin regions, a gene encoding a conserved secreted ligand activating several cell surface receptors, which acts downstream of cx43 and mediates cx43-dependent cell proliferation and joint formation in zebrafish47. We found positive expression correlations between sema3d and wnt5b across all three fin types. This pattern could potentially be due to a regulatory link between these genes, and certainly invites further investigations. In contrast, the absence of an expression correlation between cx43 and sema3d and even contrasting L/S expression across the fins could suggest their regulatory decoupling and potential involvement of a distinct upstream effector for sema3d during fin morphogenesis in N. brichardi. It is worth emphasizing that although the expression levels of cx43 and sema3d appeared to be correlated in the zebrafish caudal fin, sema3d was not a direct target of cx43 and had a distinct expression pattern than cx43 during fin regeneration47. A recent study in zebrafish has demonstrated esco2, an enzyme establishing chromatid cohesion in mitosis and required for craniofacial and limb development in human, to be an upstream transcriptional regulator of cx43 and sema3d 44. In our results, except for cx43 in caudal and dorsal fins, we did not find consistent expression correlation between esco2 and these genes. We also note that, while cx43 downregulation was associated with upregulation of mmp9 in the zebrafish caudal fin46, both cx43 and mmp9 were more strongly expressed in L than in S tissue in our study. Such discordances between gene function in zebrafish and expression patterns observed in N. brichardi may be suggestive of divergence in regulatory mechanisms between taxa.
Finally, the other candidates showing differential L/S expression include a cell differentiation factor, msxb, a member of matrix metalloproteinase family, mmp9, and pea3, an Ets-related transcription factor and downstream target of FGF signalling (Fig. 5). In zebrafish, msxb regulates cell proliferation during caudal fin regeneration and is activated by FGF signalling48, which is consistent with the expression patterns observed in our study. Pronounced expression differences were observed for mmp9 with higher L-expression across all the fins and most stages and in fact mmp9 also showed a strong positive expression correlation with cx43 in all fins. In addition, the higher L-expression of both mmp9 and cx43 in the intact fins (day 0) could indicate their role in maintenance of the phenotype in adult N. brichardi. In the zebrafish caudal fin, a reduction of cx43 expression led to increased expression of evx1 and mmp9 (which acts downstream of evx1)46. This regulatory connection is not reflected in our observations, as evx1 showed no L/S expression differences, whereas both cx43 and mmp9 were more strongly expressed in the L region. Again, components of the regulatory pathway may have diverged between the taxa. Even within the zebrafish, different alleles of cx43 have been shown to cause different phenotypes49.
The existence of upstream regulator(s)/signaling pathway(s) that directly regulate both cx43 and mmp9 independent of evx1 is another potential scenario, suggesting that the identification of shared upstream regulator(s) of mmp9 and cx43 during fin growth and regeneration would be a promising agenda for future studies. The extracellular proteinase encoded by mmp9 participates in ECM remodelling by degrading specific types of collagens and has been shown to be highly expressed in populations of epidermal and mesodermal cells in regenerating fins10. In zebrafish, inhibition of mmp9 function during larval fin fold regeneration severely impaired this process through perturbing the reconfiguration of tissues (e.g. collagen fiber reorganization) but not cell proliferation and migration10,50. Furthermore, mmp9 is a member of a conserved co-expression gene network which acts downstream of several signaling pathways involved in craniofacial skeletal morphogenesis21. In contrast, little is known about its upstream regulator(s)/signaling pathways during fin morphogenesis. To our knowledge, the only signaling pathway shown to act upstream of mmp9 during fin regeneration is a signal transduced by aryl hydrocarbon receptor (AhR) which mediates the effects of a wide array of exogenous chemicals in different tissues51. The same pathway has been shown to disrupt cell communication through suppression of cx43 expression in human tissues52, though this has yet to be confirmed in regenerating fish fin. Interestingly, a receptor of the AhR pathway (ahr1) has a conserved genomic location close to evx1 gene in zebrafish and human, which might indicate a conserved functional and/or transcriptional link between the two genes53. Furthermore, activation AhR pathway has been shown to induce evx1 expression during zebrafish development54.
Conclusions
Our analysis identified expression differences between short and elongated fin regions in approximately one third of the tested candidate target genes. The differently expressed genes belong to different molecular pathways, including WNT, BMP, ILS and FGF signalling, as well as a regulatory network involved in fin ray growth and regeneration. Expression patterns were not fully concordant across fins, which is an important finding considering the almost exclusive focus on the caudal fin in previous studies of fin regeneration. Moreover, some of the observed patterns were not consistent with expression correlations and mutant phenotypes described in the zebrafish model. Fin- and taxon-specific patterns suggest that regulatory mechanisms are not uniform across unpaired fins and have diversified across taxa. Finally, the validation of stably expressed reference genes for the specific study conditions (species, tissue and laboratory protocol) is a technical asset of our study, which we recommend to incorporate in all qPCR analyses. While the expression differences observed in our study point us to genes that might be involved in the exaggerated growth of the elongated fin filaments, we cannot rule out the possibility that some signals also reflect axial patterns or concordant histological (e.g. pigmentation) differences between the fin regions. The phenotypic diversity among cichlids offers opportunities to address potentially confounding effects. For instance, genes identified in the present study can be tested in Lamprologini species with differently shaped fins and with differently coloured fin elongations for consistency with their role in regional fin outgrowth. The present study represents a first step towards the elucidation of the molecular mechanisms behind fin shape variation in cichlid fishes and lays the foundation for continuative studies.
Methods
Fin sampling and measurement of segment length
Fin biopsies were obtained from 24 captive bred young adult individuals of N. brichardi (12 males and 12 females) sized 5-7 cm (total length) and with intact fins. The elongated regions in unpaired fins of N. brichardi develop in juveniles of both sexes without any apparent sexual dimorphism in shape and length. Prior to the experiment, the fish were housed in pairs in twelve separate aquaria and fed on identical diets for one month. To obtain fin tissue biopsies, the fish were anesthetized in water containing 0.04 gram per litre of MS-222, and their dorsal, anal, and caudal fins were cut in front of the first ray bifurcation (branching) under a stereomicroscope (red dashed lines in Fig. 1A,B). Separate tissue samples were then taken from the elongated and the short region of each fin biopsy (green and yellow areas in Fig. 1A). Tissue samples representing elongated fin regions contained the last 6-7 fin ray segments of two elongated fin rays (red arrows in Supplementary Fig. S1B) and the connecting inter-ray tissues. Likewise, tissue samples representing the short fin regions contained 6-7 segments of two short fin rays (blue arrows in Supplementary Fig. S1B) and the connecting inter-ray tissues. In order to reduce a confounding effect of the accumulation of iridophores and/or leucophores along the margins of the elongated fin regions (white margins visible in Supplementary Fig. S1A), we did not include the fin rays at the edges of fins (green arrows in Supplementary Fig. S1B) in our samples. Each tissue sample was immersed in RNAlater (Qiagen) and stored frozen until RNA isolation.
We quantified gene expression in the original tissue (stage 0) and during regeneration. Therefore, we performed a second biopsy 15 days after the first cut, when the elongation of the fin tips had just become apparent (stage 1), followed by a third biopsy after 35 days, when elongation was pronounced but fin regeneration not yet completed (stage 2; Fig. 1B).
Throughout the paper, the investigated tissues types are identified by fin type (dorsal, caudal, anal), region (elongated, short) and stage (0, 1, 2), such that, for instance, aS-0 stands for the original tissue of the short region of the anal fin. In the caudal fin, we distinguish between the dorsal and the ventral elongated regions, such that dcL stands for the dorsal elongated region of the caudal fin, vcL for the ventral elongated region, and cS identifies the short region (Fig. 1A).
To compare the length of fin ray segments between elongated and short regions, we stained fin samples from 4 fish (two males and two females) with alizarin red using a modification (8 days clearing in 10% KOH) of the acid-free double staining protocol described by Walker and Kimmel (2007)55. Photographs taken with an Olympus OM-D EM-5 II camera, attached to Olympus SZX-ILLD2-200 binocular, were imported into ImageJ, where we measured the length of the five most distal, complete segments of two branches of the shortest and the longest rays per fin (or, for the caudal fin, of two branches of the longest dorsal and ventral rays). To account for possible correlations within rays and individual fins, linear mixed models (R package nlme, with ‘segment’ nested in ‘ray’ as grouping factors)56 were used to compare segment length between the elongated and short regions of each fin type.
Anaesthesia and fin biopsies were performed under permit number BMWFW-66.007/0024-wF/v/3b/2016 issued by the Federal Ministry of Science, Research and Economy of Austria (BMWFW). All methods were performed in accordance with the relevant guidelines and regulations of BMWFW.
RNA isolation and cDNA synthesis
Corresponding tissue samples from 8 fish (four males and four females) were pooled as biological replicates (n = 3 replicates) and transferred to tubes containing TRI Reagent (Sigma) and 1.4 mm ceramic spheres. The samples were homogenized by FastPrep-24 Instrument (MP Biomedicals, Santa Ana, CA, USA). RNA was extracted according to manufacturer’s Trizol protocol and dissolved in 40 µl RNase-free water. RNA samples were treated with DNase (New England Biolabs) to remove contaminating DNA. RNA concentration was measured by spectrophotometry using a Nanophotometer (IMPLEN GmbH, Munich, Germany). The quality of the RNA samples was evaluated in a R6K ScreenTape System on an Agilent 2200 TapeStation (Agilent Technologies) to ensure that the integrity number (RIN) of all samples was higher than 7. cDNA was prepared from 1000 ng of RNA using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems), according to the manufacturer’s protocol. Negative controls, i.e. reactions without addition of reverse transcriptase (-RT samples), were prepared to confirm the absence of genomic DNA. cDNA was diluted 1:3 times in nuclease-free water for further use in quantitative real-time PCR.
Gene selection, Primer design and real-time qPCR
To validate suitable reference genes for expression analysis, we screened 7 genes expressed in a variety tissues and frequently used as reference genes for qPCR studies of body compartments and skeletal structures in teleost fishes (Supplementary data 1)17,57,58,59. Additionally, we selected 40 candidate target genes that are known to be involved in fin development, morphogenesis and/or regeneration based on findings in zebrafish (Table 1 and Supplementary data 1).
The qPCR primers were designed based on recently released transcriptome data of Neolamprologus brichardi 15. The sequence alignment was performed using CLC Genomic Workbench, version 7.5 (CLC Bio, Aarhus, Denmark) and locations overlapping the exon boundaries of the genes were determined by the Nile Tilapia annotated genome sequences in the Ensembl database (http://www.ensembl.org/Oreochromis_niloticus). The qPCR Primers were designed on exon boundaries using Primer Express 3.0 software (Applied Biosystems, Foster City, CA, USA) and checked for self-annealing, hetero-dimers and hairpin structures with OligoAnalyzer 3.1 (Integrated DNA Technology) (Supplementary data 1).
Real-time PCR was performed in 96 well-PCR plates on an ABI 7500 real-time PCR System (Applied Biosystems) using Maxima SYBR Green/ROX qPCR Master Mix (2X) as recommended by the manufacturer (Thermo Fisher Scientific, St Leon-Rot, Germany). Each biological replicate was run in duplicate for each gene and the experimental set-up per run followed the preferred sample maximization method60. The qPCR was run with a 2 min hold at 50 °C and a 10 min hot start at 95 °C followed by the amplification step for 40 cycles of 15 sec denaturation at 95 °C and 1 min annealing/extension at 60 °C. A dissociation step (60 °C – 95 °C) was performed at the end of the amplification phase to identify a single, specific product for each primer set (Supplementary data 1). Primer efficiency values (E) were calculated with the LinRegPCR v11.0 programme (http://LinRegPCR.nl)61 and primer-pairs with E less than 0.9 were discarded and new primers designed (Supplementary data 1).
Data analysis
To identify the most stably expressed reference genes, three ranking algorithms were used; BestKeeper62, NormFinder63 and geNorm64. The standard deviation (SD) based on Cq values of the fin regions was calculated by BestKeeper to determine the expression variation for each reference gene. BestKeeper also determines the stability of reference genes through a correlation calculation or BestKeeper index (r). GeNorm calculates mean pairwise variation between each gene and other candidates (the expression stability or M value) in a stepwise manner and NormFinder identifies the most stable genes (lowest expression stability values) based on analysis of inter- and intra-group variation in expression levels17,58.
The mean Cq values of the two top-ranked reference genes was used as Cq reference and the difference between Cq values (ΔCq) of the target genes and the selected reference gene was calculated for each target gene; ΔCq target = Cq target − Cq reference. To obtain a ΔΔCq value, samples were normalized to the ΔCq value of a calibrator sample (ΔCq target − ΔCq calibrator). To this aim, one arbitrarily chosen biological replicate of dL-0, aL-0 and dcL-0 was used to calibrate samples of the dorsal, anal and caudal fin, respectively. Relative expression quantities (RQ) were calculated based on the expression level of the calibrator sample (E−ΔΔCq)65. For each target gene, differences in gene expression levels between elongated and short fin regions as well as among regeneration stages were tested by paired t-tests on log-transformed RQ data. To assess similarities in the expression patterns of the target genes, Pearson correlation coefficients (r) were calculated in R (http://www.r-project.org).
Data availability
All the data represented in this study are provided within the main manuscript or in the Supplementary materials.
Ethical approval
All experimental protocols related to the fishes used in this study were approved by the Federal Ministry of Science, Research and Economy of Austria. Please identify the approving body and license numbers in the methods section.
References
Blake, R. W. Fish functional design and swimming performance. J. Fish Biol. 65, 1193–1222 (2004).
Basolo, A. L. Female Preference Predates the Evolution of the Sword in Swordtail Fish. Science (80-.). 250, 808–810 (1990).
Benson, K. E. & Basolo, A. L. Male–male competition and the sword in male swordtails, Xiphophorus helleri. Anim. Behav. 71, 129–134 (2006).
Kozak, H. L., Cirino, L. A. & Ptacek, M. B. Female mating preferences for male morphological traits used in species and mate recognition in the Mexican sailfin mollies, Poecilia velifera and Poecilia petenensis. Behav. Ecol. 19, 463–474 (2008).
Pfefferli, C. & Jaźwińska, A. The art of fin regeneration in zebrafish. Regeneration 2, 72–83 (2015).
Nogueira, A. F. et al. Tetrapod limb and sarcopterygian fin regeneration share a core genetic programme. Nat. Commun. 7, 13364 (2016).
Nakamura, T., Gehrke, A. R., Lemberg, J., Szymaszek, J. & Shubin, N. H. Digits and fin rays share common developmental histories. Nature 537, 225–228 (2016).
Iovine, M. K. Conserved mechanisms regulate outgrowth in zebrafish fins. Nat. Chem. Biol. 3, 613–618 (2007).
Wehner, D. & Weidinger, G. Signaling networks organizing regenerative growth of the zebrafish fin. Trends Genet. 31, 336–343 (2015).
Yoshinari, N., Ishida, T., Kudo, A. & Kawakami, A. Gene expression and functional analysis of zebrafish larval fin fold regeneration. Dev. Biol. 325, 71–81 (2009).
Rabinowitz, J. S. et al. Transcriptomic, proteomic, and metabolomic landscape of positional memory in the caudal fin of zebrafish. Proc. Natl. Acad. Sci. 114, E717–E726 (2017).
Offen, N., Meyer, A. & Begemann, G. Identification of novel genes involved in the development of the sword and gonopodium in swordtail fish. Dev. Dyn. 238, 1674–1687 (2009).
Offen, N., Kang, J. H., Meyer, A., Begemann, G. & Irwin, R. Retinoic Acid Is Involved in the Metamorphosis of the Anal Fin into an Intromittent Organ, the Gonopodium, in the Green Swordtail (Xiphophorus hellerii). PLoS One 8, e77580 (2013).
Kang, J. H. et al. Transcriptomics of two evolutionary novelties: how to make a sperm-transfer organ out of an anal fin and a sexually selected ‘sword’ out of a caudal fin. Ecol. Evol. 5, 848–864 (2015).
Brawand, D. et al. The genomic substrate for adaptive radiation in African cichlid fish. Nature 513, 375–81 (2014).
Kubista, M. et al. The real-time polymerase chain reaction. Mol. Aspects Med. 27, 95–125 (2006).
Ahi, E. P. et al. Validation of Reference Genes for Expression Studies during Craniofacial Development in Arctic Charr. PLoS One 8, e66389 (2013).
Pfennig, D. W. & Ehrenreich, I. M. Towards a gene regulatory network perspective on phenotypic plasticity, genetic accommodation and genetic assimilation. Mol. Ecol. 23, 4438–4440 (2014).
Filteau, M., Pavey, S. A., St-Cyr, J. & Bernatchez, L. Gene Coexpression Networks Reveal Key Drivers of Phenotypic Divergence in Lake Whitefish. Mol. Biol. Evol. 30, 1384–1396 (2013).
Schneider, R. F., Li, Y., Meyer, A. & Gunter, H. M. Regulatory gene networks that shape the development of adaptive phenotypic plasticity in a cichlid fish. Mol. Ecol. 23, 4511–4526 (2014).
Ahi, E. P. et al. Transcriptional dynamics of a conserved gene expression network associated with craniofacial divergence in Arctic charr. Evodevo 5, 40 (2014).
Kratochwil, C. F., Geissler, L., Irisarri, I., Meyer, A. & S, K. Molecular Evolution of the Neural Crest Regulatory Network in Ray-Finned Fish. Genome Biol. Evol. 7, 3033–3046 (2015).
Schebesta, M., Lien, C.-L., Engel, F. B. & Keating, M. T. Transcriptional profiling of caudal fin regeneration in zebrafish. ScientificWorldJournal. 6(Suppl 1), 38–54 (2006).
Mathew, L. K. et al. Comparative expression profiling reveals an essential role for raldh2 in epimorphic regeneration. J. Biol. Chem. 284, 33642–53 (2009).
Offen, N., Blum, N., Meyer, A. & Begemann, G. Fgfr1 signalling in the development of a sexually selected trait in vertebrates, the sword of swordtail fish. BMC Dev. Biol. 8, 98 (2008).
Zauner, H., Begemann, G., Mari-Beffa, M. & Meyer, A. Differential regulation of msx genes in the development of the gonopodium, an intromittent organ, and of the “sword” a sexually selected trait of swordtail fishes (Xiphophorus). Evol. Dev. 5, 466–477 (2003).
Higdon, C. W., Mitra, R. D. & Johnson, S. L. Gene Expression Analysis of Zebrafish Melanocytes, Iridophores, and Retinal Pigmented Epithelium Reveals Indicators of Biological Function and Developmental Origin. PLoS One 8, e67801 (2013).
Kimura, T. et al. Leucophores are similar to xanthophores in their specification and differentiation processes in medaka. Proc. Natl. Acad. Sci. USA 111, 7343–8 (2014).
Chablais, F. & Jazwinska, A. IGF signaling between blastema and wound epidermis is required for fin regeneration. Development 137, 871–879 (2010).
Shibata, E. et al. Fgf signalling controls diverse aspects of fin regeneration. Development 143, 2920–9 (2016).
Warren, I. A., Gotoh, H., Dworkin, I. M., Emlen, D. J. & Lavine, L. C. A general mechanism for conditional expression of exaggerated sexually-selected traits. BioEssays 35, n/a–n/a (2013).
Stewart, S., Gomez, A. W., Armstrong, B. E., Henner, A. & Stankunas, K. Sequential and Opposing Activities of Wnt and BMP Coordinate Zebrafish Bone Regeneration. Cell Rep. 6, 482–498 (2014).
Quint, E. et al. Bone patterning is altered in the regenerating zebrafish caudal fin after ectopic expression of sonic hedgehog and bmp2b or exposure to cyclopamine. Proc. Natl. Acad. Sci. 99, 8713–8718 (2002).
Smith, A., Avaron, F., Guay, D., Padhi, B. K. & Akimenko, M. A. Inhibition of BMP signaling during zebrafish fin regeneration disrupts fin growth and scleroblast differentiation and function. Dev. Biol. 299, 438–454 (2006).
Kujawski, S. et al. Calcineurin Regulates Coordinated Outgrowth of Zebrafish Regenerating Fins. Dev. Cell 28, 573–587 (2014).
Poss, K. D., Shen, J. & Keating, M. T. Induction of lef1 during zebrafish fin regeneration. Dev. Dyn. 219, 282–286 (2000).
Stoick-Cooper, C. L. et al. Distinct Wnt signaling pathways have opposing roles in appendage regeneration. Development 134, 479–89 (2007).
Lee, Y. et al. Maintenance of blastemal proliferation by functionally diverse epidermis in regenerating zebrafish fins. Dev. Biol. 331, 270–280 (2009).
Wehner, D. et al. Wnt/β-Catenin Signaling Defines Organizing Centers that Orchestrate Growth and Differentiation of the Regenerating Zebrafish Caudal Fin. Cell Rep. 6, 467–481 (2014).
Iovine, M. K., Higgins, E. P., Hindes, A., Coblitz, B. & Johnson, S. L. Mutations inconnexin43 (GJA1) perturb bone growth in zebrafish fins. Dev. Biol. 278, 208–219 (2005).
Govindan, J. & Iovine, M. K. Hapln1a Is Required for Connexin43-Dependent Growth and Patterning in the Regenerating Fin Skeleton. PLoS One 9, e88574 (2014).
Govindan, J. & Iovine, M. K. Dynamic remodeling of the extra cellular matrix during zebrafish fin regeneration. Gene Expr. Patterns 19, 21–29 (2015).
Govindan, J., Tun, K. M. & Iovine, M. K. Cx43-Dependent Skeletal Phenotypes Are Mediated by Interactions between the Hapln1a-ECM and Sema3d during Fin Regeneration. PLoS One 11, e0148202 (2016).
Banerji, R., Eble, D. M., Iovine, M. K. & Skibbens, R. V. Esco2 regulates cx43 expression during skeletal regeneration in the zebrafish fin. Dev. Dyn. 245, 7–21 (2016).
Sims, K., Eble, D. M. & Iovine, M. K. Connexin43 regulates joint location in zebrafish fins. Dev. Biol. 327, 410–418 (2009).
Ton, Q. V. & Iovine, M. K. Identification of an evx1-Dependent Joint-Formation Pathway during FIN Regeneration. PLoS One 8, e81240 (2013).
Ton, Q. V. & Iovine, K. M. Semaphorin3d mediates Cx43-dependent phenotypes during fin regeneration. Dev. Biol. 366, 195–203 (2012).
Thummel, R. et al. Inhibition of zebrafish fin regeneration using in vivo electroporation of morpholinos againstfgfr1 andmsxb. Dev. Dyn. 235, 336–346 (2006).
Misu, A. et al. Two Different Functions of Connexin43 Confer Two Different Bone Phenotypes in Zebrafish. J. Biol. Chem. 291, 12601–12611 (2016).
LeBert, D. C. et al. Matrix metalloproteinase 9 modulates collagen matrices and wound repair. Development 142, 2136–2146 (2015).
Andreasen, E. A., Mathew, L. K., Löhr, C. V., Hasson, R. & Tanguay, R. L. Aryl hydrocarbon receptor activation impairs extracellular matrix remodeling during zebra fish fin regeneration. Toxicol. Sci. 95, 215–26 (2007).
Andrysík, Z. et al. Aryl hydrocarbon receptor-mediated disruption of contact inhibition is associated with connexin43 downregulation and inhibition of gap junctional intercellular communication. Arch. Toxicol. 87, 491–503 (2013).
Hahn, M. E. Dioxin Toxicology and the Aryl HydrocarbonReceptor: Insights from Fish and Other Non-traditional Models. Mar. Biotechnol. 3, S224–S238 (2001).
Goodale, B. C. et al. Structurally distinct polycyclic aromatic hydrocarbons induce differential transcriptional responses in developing zebrafish. Toxicol. Appl. Pharmacol. 272, 656–670 (2013).
Walker, M. & Kimmel, C. A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech. Histochem. 82, 23–28 (2007).
Pinheiro J, D, B., S, D., D, S. & Team, R. C. Linear and nonlinear mixed effects models. 1–128 (2016).
Yang, C. G. et al. Evaluation of reference genes for quantitative real-time RT-PCR analysis of gene expression in Nile tilapia (Oreochromis niloticus). Gene 527, 183–192 (2013).
Pashay Ahi, E., Walker, B. S., Lassiter, C. S. & Jónsson, Z. O. Investigation of the effects of estrogen on skeletal gene expression during zebrafish larval head development. PeerJ 4, e1878 (2016).
Ahi, E. & Sefc, K. A gene expression study of dorso-ventrally restricted pigment pattern in adult fins of Neolamprologus meeli, an African cichlid species. PeerJ 5:e2843 doi:10.7717/peerj.2843 (2017).
Hellemans, J., Mortier, G., De Paepe, A., Speleman, F. & Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 8, R19 (2007).
Ramakers, C., Ruijter, J. M., Deprez, R. H. L. & Moorman, A. F. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 339, (2003).
Pfaffl, M. W., Tichopad, A., Prgomet, C. & Neuvians, T. P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol. Lett. 26, 509–15 (2004).
Andersen, C. L., Jensen, J. L. & Ørntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 64, 5245–50 (2004).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034 (2002).
Pfaffl, M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45 (2001).
DeLaurier, A. et al. Zebrafishsp7: EGFP: a transgenic for studying otic vesicle formation, skeletogenesis, and bone regeneration. Genesis 48, 505–11 (2010).
Norton, W. H. J., Ledin, J., Grandel, H. & Neumann, C. J. HSPG synthesis by zebrafish Ext2 and Extl3 is required for Fgf10 signalling during limb development. Development 132, 4963–73 (2005).
Whitehead, G. G., Makino, S., Lien, C.-L. & Keating, M. T. fgf20 is essential for initiating zebrafish fin regeneration. Science 310, 1957–60 (2005).
Duszynski, R. J., Topczewski, J. & LeClair, E. E. Divergent requirements for fibroblast growth factor signaling in zebrafish maxillary barbel and caudal fin regeneration. Dev. Growth Differ. 55, 282–300 (2013).
Wills, A. A., Kidd, A. R., Lepilina, A., Poss, K. D. & Poss, K. D. Fgfs control homeostatic regeneration in adult zebrafish fins. Development 135, 3063–70 (2008).
Sharma, M. K. et al. Differential expression of the duplicated cellular retinoic acid-binding protein 2 genes (crabp2a and crabp2b) during zebrafish embryonic development. Gene Expr. Patterns 5, (2005).
Emoto, Y., Wada, H., Okamoto, H., Kudo, A. & Imai, Y. Retinoic acid-metabolizing enzyme Cyp26a1 is essential for determining territories of hindbrain and spinal cord in zebrafish. Dev. Biol. 278, 415–427 (2005).
Blum, N. & Begemann, G. Retinoic acid signaling controls the formation, proliferation and survival of the blastema during adult zebrafish fin regeneration. Development 139, 107–16 (2012).
Laforest, L. et al. Involvement of the sonic hedgehog, patched 1 and bmp2 genes in patterning of the zebrafish dermal fin rays. Development 125, 4175–84 (1998).
Pfefferli, C. et al. Specific NuRD components are required for fin regeneration in zebrafish. BMC Biol. 12, 30 (2014).
Borday, V. et al. evx1 transcription in bony fin rays segment boundaries leads to a reiterated pattern during zebrafish fin development and regeneration. Dev. Dyn. 220, 91–98 (2001).
Heude, É. et al. The dlx5a/dlx6a Genes Play Essential Roles in the Early Development of Zebrafish Median Fin and Pectoral Structures. PLoS One 9, e98505 (2014).
Ishida, T., Nakajima, T., Kudo, A. & Kawakami, A. Phosphorylation of Junb family proteins by the Jun N-terminal kinase supports tissue regeneration in zebrafish. Dev. Biol. 340, 468–479 (2010).
Poss, K. D., Nechiporuk, A., Hillam, A. M., Johnson, S. L. & Keating, M. T. Mps1 defines a proximal blastemal proliferative compartment essential for zebrafish fin regeneration. Development 129, 5141–9 (2002).
Nechiporuk, A. & Keating, M. T. A proliferation gradient between proximal and msxb-expressing distal blastema directs zebrafish fin regeneration. Development 129, 2607–17 (2002).
Münch, J., González-Rajal, A. & de la Pompa, J. L. Notch regulates blastema proliferation and prevents differentiation during adult zebrafish fin regeneration. Development 140, 1402–11 (2013).
Goldsmith, M. I., Fisher, S., Waterman, R. & Johnson, S. L. Saltatory control of isometric growth in the zebrafish caudal fin is disrupted in long fin and rapunzel mutants. Dev. Biol. 259, 303–317 (2003).
Green, J., Taylor, J. J., Hindes, A., Johnson, S. L. & Goldsmith, M. I. A gain of function mutation causing skeletal overgrowth in the rapunzel mutant. Dev. Biol. 334, 224–234 (2009).
Dufourcq, P. & Vriz, S. The chemokine SDF-1 regulates blastema formation during zebrafish fin regeneration. Dev. Genes Evol. 216, 635–639 (2006).
Bouzaffour, M., Dufourcq, P., Lecaudey, V., Haas, P. & Vriz, S. Fgf and Sdf-1 Pathways Interact during Zebrafish Fin Regeneration. PLoS One 4, e5824 (2009).
Mateus, R. et al. Control of tissue growth by Yap relies on cell density and F-actin in zebrafish fin regeneration. Development 142, 2752–2763 (2015).
Acknowledgements
The authors thank Wolfgang Gessl (www.pisces.at) for his responsible management of our fish facility and photography of the intact and regenerating fish fins. We also thank Holger Zimmermann and Stephan Koblmüller for sharing their precious knowledge on cichlid fishes of Lake Tanganyika, as well as Silke Werth and the Institute of Plant Sciences at University of Graz for technical assistance and access to their real-time PCR System. We are grateful to the reviewers for their constructive comments and suggestions that improved the quality and clarity of the paper. The authors acknowledge the financial support by the University of Graz.
Author information
Authors and Affiliations
Contributions
E.P.A. and K.M.S. designed the study, analysed the expression data and wrote the main manuscript text. E.P.A. has conducted the laboratory experiment and figure preparations for gene expression sections. F.R. conducted the staining, size measurements and data analysis for Fig. 1C. All authors reviewed the manuscript and approve its content.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ahi, E.P., Richter, F. & Sefc, K.M. A gene expression study of ornamental fin shape in Neolamprologus brichardi, an African cichlid species. Sci Rep 7, 17398 (2017). https://doi.org/10.1038/s41598-017-17778-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17778-0
This article is cited by
-
East African cichlid fishes
EvoDevo (2023)
-
Gene expression patterns associated with caudal fin shape in the cichlid Lamprologus tigripictilis
Hydrobiologia (2023)
-
Expression variations in ectodysplasin-A gene (eda) may contribute to morphological divergence of scales in haplochromine cichlids
BMC Ecology and Evolution (2022)
-
Transcriptomics unravels molecular players shaping dorsal lip hypertrophy in the vacuum cleaner cichlid, Gnathochromis permaxillaris
BMC Genomics (2021)
-
Divergence in larval jaw gene expression reflects differential trophic adaptation in haplochromine cichlids prior to foraging
BMC Evolutionary Biology (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
