
Abstract
Localized spins and itinerant electrons rarely coexist in geometrically-frustrated spinel lattices. They exhibit a complex interplay between localized spins and itinerant electrons. In this paper, we study the origin of the unusual spin structure of the spinel CoV2O4, which stands at the crossover from insulating to itinerant behavior using the first principle calculation and neutron diffraction measurement. In contrast to the expected paramagnetism, localized spins supported by enhanced exchange couplings are frustrated by the effects of delocalized electrons. This frustration produces a non-collinear spin state even without orbital orderings and may be responsible for macroscopic spin-glass behavior. Competing phases can be uncovered by external perturbations such as pressure or magnetic field, which enhances the frustration.
Similar content being viewed by others
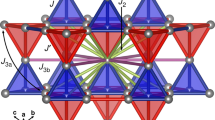
Introduction
Geometrically frustrated systems have been attracting attention because of their unusual magnetic properties induced by the suppressed conventional long-range magnetic orderings. The spin liquid state has been widely celebrated and theoretically predicted to exist in pyrochlore lattice and spinel systems with nearest-neighbor antiferromagnetic coupling1. One more ingredient which makes the playground of the geometrically frustrated systems wider and more interesting is the coupling between the localized spin and itinerant electron in the frustrated systems2. The interplay between localized spin and itinerant electron in geometrically frustrated systems is believed to be responsible for many intriguing phenomena such as a metallic spin-liquid state3, heavy-fermion behavior4, anomalous transport in spin-ice systems5, and exotic phases6,7,8. The effect of the interaction between localized spins and itinerant electrons has been investigated intensively on pyrochlores A 2 B 2O7 where A is rare-earth elements and B is transition metals and both A and B sublattices are frustrated3,4,5,6,7. However, it has rarely been explored on another strongly frustrated system, spinel AB 2O4, where the magnetic ion B is only frustrated.
In the spinels, the degree of the magnetic frustration and the electronic itinerancy on sublattice B is a function of the distance of nearest-neighbor B atoms (R B−B ). The shorter distance due to the chemical pressure from a smaller radius of A site cation or the physical pressure, the stronger both the electronic itinerancy and magnetic frustration are induced9. On the other hand, magnetic A site ions can relieve the frustration via the magnetic interactions between the spins on A site and the localized spins on sublattice B. Therefore, a rich phase diagram due to the strong interplay between localized spins and itinerant electrons can be anticipated when A sublattice is a small magnetic ion.
In the spinel vanadates AV2O4 (V3+, S = 1), the octahedral crystal field on the vanadium site with two electrons in t 2g levels cannot completely remove the degeneracy resulting in the non-quenched orbital angular momentum. The geometrical frustration in many spinel is relieved by lowering the crystal symmetry accompanying non-collinear spin ordering and/or orbital ordering (OO), as an example, from cubic-tetragonal structural transition, which modifies of exchange paths between spins10,11,12,13,14,15,16. Therefore, OO of partially-filled d-electrons on the V site has been observed in many spinel vanadates (AV2O4, A = Mn, Fe, Cd, Zn, Mg). Moreover, recent studies suggest that the spinel vanadates become itinerant when R V−V falls around a threshold value of 2.94 Å, which is close to R V−V when A = Co (2.97 Å)17,18. Since it also has the smallest magnetic A-site cation of any known spinel vanadates, CoV2O4 is the ideal system to study the interplay between itinerancy and localized spins.
The frequency dependence of ac susceptibility and the divergence between the zero-field cooling and field cooling susceptibility at the cubic-tetragonal structural transition indicate a glassy spin state19,20,21. Although the spin-glass behavior of Mn1−x Co x V2O4 is enhanced by Co-doping9, CoV2O4 has higher magnetic ordering temperatures than those of compounds Mn1−x Co x V2O4 with x < 1. Indeed, CoV2O4 has higher para-collinear (T CL ) and collinear-non-collinear (T NC ) spin transition temperatures than those of any other vanadate spinels. This stands in marked contrast to the pyrochlores6,7, where spin-glass phases have lower ordering temperatures with smaller A site atom doping. Bizarrely, no structural phase transition with OO has been observed for CoV2O4 9 even though the spin structure of CoV2O4 has been suggested to be non-collinear spin structure from the previous magnetization and neutron diffraction studies16,19,21,22. This is sharply contrast to the fact that the most spinels show the structural phase transition concurrent with the orbital ordering and non-collinear spin structure as stated above. Due to these unusual magnetic behaviors, the exotic spin states in CoV2O4 highly demand a detailed study.
This paper clarifies the origin of the possible non-collinear (NC) spin states of CoV2O4 without OO and structural phase transition by using density functional theory (DFT) and spin models to interpret neutron-scattering measurements on high-quality CoV2O4 single crystals. Chemically-driven pressure by Co enhances the itinerancy in CoV2O4. This itinerancy weakens the OO and strengthens the magnetic and structural isotropies. The frustration fostered by these isotropies23 may be responsible for macroscopic glassy behavior in a magnetic field9,19. Due to the enhanced frustration, external perturbations such as pressure or magnetic field could uncover novel magnetic phases in cubic CoV2O4.
Results
NC spin states in cubic phase CoV2O4
To resolve the noncollinear spin structure, we performed the neutron diffraction measurements on cubic CoV2O4 and tetragonal MnV2O4 samples. Figure 1 compares the temperature dependence of the (002), (220), and (111) Bragg peaks in cubic CoV2O4 and tetragonal MnV2O4. CoV2O4 and MnV2O4 show (111) and (220) Bragg peaks rapidly growing below 150 K and 60 K, respectively, denoted as T CL . (111) and (220) Bragg peaks are consistent with the crystal symmetry of these systems, and indicate that collinear ferrimagnetic (FIM) orderings are stabilized. Below 75 K for CoV2O4, and 57 K for MnV2O4 denoted as T NC , the intensity of (002) Bragg peaks begin to increase, which is consistent with the previous neutron power diffraction study21,22. The (002) peaks are forbidden by structural symmetry. Therefore, the observation of (002) peak indicates the formation of an additional antiferromagnetic (AFM) component in the ab-plane, thus a canted spin structure or noncollinear spin structure below T NC 24,25. In case of MnV2O4, T NC accompanies the structural phase transition (T S )14. However, no clear structural transition in CoV2O4 was observed by recent X-ray diffraction and heat capacity measurements9. The appearance of Bragg peak intensity at the (002) position is consistent with the recent neutron scattering measurements where it was associated with a two-in/two-out (TI/TO) structure on the V-sublattice25,26. Also our DFT calculations later in the paper justify this assumption. Based on the diffraction data, it is valid to assume that the spin configuration of CoV2O4 is similar to the spin configurations of MnV2O4 27, and FeV2O4 28.

NC spin states in cubic CoV2O4 compared to tetragonal MnV2O4. Temperature dependence of the (111) (triangles), (220) (circles), and (002) (squares) Bragg peak intensities for CoV2O4 (a) and MnV2O4 (b) measured by neutron diffraction at HB-3A. The peak intensities of (111) and (220) above the magnetic transition temperature are fully from the nuclear structure and were subtracted. The (002) peak is not allowed from the structural symmetry and fully originated from the magnetic scattering. The background was subtracted. All the magnetic peaks observed by our neutron diffraction are instrument resolution limited besides the peak broadening caused by the structural transition for MnV2O4, and thus indicate the long range ordered magnetic moments. The strongly-reduced intensity of (002) peak in CoV2O4 indicates that only tiny amount of V spin orders, which is caused by enhanced itinerancy.
When the inter-vanadium distance R V−V lies around the critical value (2.94 Å)29, the system is expected to remain in metallic state down to very low temperatures. Therefore, the effect of the itinerant electrons should be considered in CoV2O4. Indeed, the ordered magnetic moment refined from the Bragg peaks is 0.47(3)μ B /V in CoV2O4, which is significantly reduced from 0.95(4)μ B /V in MnV2O4 due to the increased itinerancy of electrons. In addition, T NC of CoV2O4 is higher than that of MnV2O4 because of a new exchange interaction originating from the itinerant electrons, which we will clarify later with the calculated band structures. An additional magnetic frustration among the localized spins may be indirectly induced by electronic itinerancy as well. Curiously, the structural phase transition associated with OO commonly observed in many vanadates is absent in CoV2O4, even though the TI/TO seems to emerge robustly. Since the TI/TO state originates from OO in tetragonal compounds27,28, the isosymmetric TI/TO state in cubic CoV2O4 without any OO must have a different origin associated with its itinerancy and frustration.
Single-ion anisotropy suppressed by itinerancy
First-principles calculations were used to explore the microscopic origin for the complex NC state in cubic CoV2O4. As shown in Fig. 2(a) and (c), the major magnetic anisotropy appears on the V3+ site with a magnitude two or three orders larger than that for the A-site (Co/Mn). Although the V3+ ions are surrounded by similar octahedra in both CoV2O4 and MnV2O4, the local [111] single-ion anisotropy (SIA) of V3+ is significantly reduced in CoV2O4 (−1.2 meV) compared to that in MnV2O4 (−4.8 meV) due to the melting of OO by the pressure-induced itinerancy in CoV2O4. Calculated by DFT, the SIA on the V sites totally disappears with an external pressure around 10 GPa in CoV2O4.

Reduced single-ion anisotropy (SIA) of V in CoV2O4 compared with that in MnV2O4. (a) Total energy versus angle and associated SIA (meV) of V3+ in ambient (circle) and 10 GPa (square) pressure for bulk CoV2O4 and MnV2O4 (diamond). (b) NC spin configurations of V3+ and Co2+/Mn2+ spins pointing along local [111] and global [001] directions, respectively. The round bold (dotted) arrows close to V spins depict the rotational flexibility in CoV2O4 (MnV2O4). (c) SIA of Co2+ in bulk CoV2O4 under 10 Gpa compared to SIA of Mn2+ in bulk MnV2O4. (d) Orbital occupation configuration of Mn2+ (d 5) and Co2+ (d 7). (e) R V−V in CoV2O4 and MnV2O4 are compared with R V−V in other vanadates from ref.29.
While the AFM V-V interaction in a pyrochlore lattice with local [111] SIA favors the all-in/all-out (AI/AO) spin structure, the disappearance of SIA fosters strong magnetic frustration23. OO relieves the frustration in MnV2O4 with structural phase transitions. However, the frustration reappears in CoV2O4 due to the melting of the OO and the suppression of the easy-axis anisotropy by itinerancy, as shown in Fig. 2(a,b). The recovered frustration might be responsible for the macroscopic spin-glass behavior9 below T NC due to the competing ground states30.
The SIA of A-site (Fig. 2c), (A = Co, Mn) is quite negligible compared to the SIA of V3+. While Mn2+ has a weak easy-plane axis because of the compressed tetragonal structure (c/a < 1), Co2+ does not exhibit anisotropy because of the isotropic cubic structure. The SIA of Co2+ is much less dependent on pressure than that of V3+ since Co2+ electronic states lie significantly below the Fermi energy (ε F ) and are thereby electronically encapsulated, as shown in Fig. 3(a). Only V3+ states cross ε F . Therefore the pressure-induced itinerancy will only affect the spins on V3+ sites. The moment of Co ion is not affected by itinerancy.

Origin of the enhanced magnetic ordering temperature in CoV2O4. Projected density-of-states of CoV2O4 (a) compared to MnV2O4 (b) in unit of eV−1. Dotted arrows denote the energy differences, Δ between V and Co/Mn for possible AFM super-exchange (J A−V ~ −t 2/Δ). t is the hopping parameter between orbitals.
Enhanced exchange couplings
As shown in Fig. 2 in Supplementary material, the Bragg peaks do not split or broaden with decreasing temperature below 100 K, indicating the absence of a structural transition. In agreement with this measurement, DFT calculations confirm the structural isotropy (c/a = 1) of CoV2O4. As shown in Fig. 3, the t 2g (d xy = d yz = d xz ) and e g (\({d}_{{z}^{2}}\) = \({d}_{{x}^{2}-{y}^{2}}\)) electronic levels become equally occupied and degenerate in cubic CoV2O4. The structural and electronic isotropies also produce the same exchange interactions \({J}_{{\rm{V}}-{\rm{V}}}^{{\rm{in}}}\) = \({J}_{{\rm{V}}-{\rm{V}}}^{{\rm{out}}}\) = −12 meV between all spins on the tetrahedron as calculated from first principles, Fig. 4, where \({J}_{{\rm{V}}-{\rm{V}}}^{{\rm{in}}}\) = \({J}_{{\rm{V}}-{\rm{V}}}^{{\rm{out}}}\) are in-plane and out-of-plane exchange interaction, which are not equivalent in tetragonal phase (the right panel of Fig. 3). These coupled structural, electronic, and magnetic isotropies foster frustration and the observed NC phase in Fig. 1.

Evolution of magnetic couplings and competing ground states driven by Co-doping and pressure. (a) Change of all magnetic interactions with Co doping and external pressure (GPa) calculated by LSDA + U for the ground states at zero temperature. Points represent DFT results and the connecting lines are a guide for eye. \({J}_{{\rm{V}}-{\rm{V}}}^{{\rm{in}}}\) and \({J}_{{\rm{V}}-{\rm{V}}}^{{\rm{out}}}\) are expected to be degenerate at x = 0.8 (cubic) in Mn1−x Co x V2O4 from experiments9,24. Bold (dotted) lines represent the exchange (J) and SIA (D) interactions. (b) The anisotropic J V−V prefers the TI/TO state and the antiferromagnetic J Mn−V also stabilizes TI/TO state in Mn-rich region. (c) The isotropic J V−V prefers the AI/AO state but the antiferromagnetic J Co−V tries to stabilizes the TI/TO state in Co-rich region. Thus, two states of the isosymmetric one-angle TI/TO state and the two-angle state that evolves from the AI/AO state compete with each other. (d) The energy landscape of CoV2O4 with the disappearance of SIA at high pressures. The massive degeneracies in the energy landscape of two angles (θ 1 and θ 2) may induce spin glass or liquid phases as explained in the text.
Comparing the densities-of-states of CoV2O4 and MnV2O4 reveals the origin of the enhanced magnetic ordering temperatures in CoV2O4.The large energy difference (~5 eV) between the occupied V and Mn d states weakens the exchange between Mn and V. By filling the e g minority spin levels as indicated in Figs 2(d) and 3(a), Co significantly lowers the t 2g unoccupied energy level and enhances the exchange interaction between Co and V. DFT calculations reveal that the magnitude of the AFM J A−V is twice as large in CoV2O4 (−2.5 meV) as in MnV2O4 (−1.2 meV). As reflected by the neutron-scattering measurements in Fig. 1, the enhanced J A−V causes T CL to more than double in CoV2O4 (150 K) compared to MnV2O4 (53 K).
Surprisingly, the induced itinerancy also increases the NC ordering temperature even without OO in CoV2O4. As shown in Fig. 1, T NC significantly increases in CoV2O4 (75 K) compared to MnV2O4 (57 K). Although it exhibits the higher NC ordering temperature, CoV2O4 also exhibits glassy behavior9,19. While the reduced SIA and induced isotropies foster frustration23, the enhanced exchange interaction relieves the frustration and enhances the ordering temperatures. In the series of Mn1−x Co x V2O4, the spin-wave gap (~2 meV) remains relatively unchanged with Co-doping (x)24 despite the enhanced magnetic ordering temperatures proportional to J A−V. Since the spin-wave gap is proportional to \(\sqrt{{D}_{{\rm{V}}}\times {J}_{{\rm{A}}-{\rm{V}}}}\), the increase in \(|{J}_{{\rm{A}}-{\rm{V}}}|\) is compensated by the reduction in the anisotropy D v in CoV2O4. By enhancing both competing effects (itinerancy-driven isotropies with reduced SIA and strengthened exchange), Co doping can foster various novel states in CoV2O4.
Novel phases induced by frustration
The comparison between CoV2O4 and MnV2O4 in Fig. 4 reveals the origin of the NC states in CoV2O4. The key handle to tune the magnetic couplings is the distance between the V atoms (R V−V along the x-axis) controlled by chemical doping and external pressure. In MnV2O4, the OO of the V ions relieves the magnetic frustration of the pyrochlore lattice and stabilizes the TI/TO NC spin state. The AFM Mn-V interactions increase the canting angle while maintaining this TI/TO state (Fig. 4(b)). By introducing itinerancy, Co doping promotes isotropic V-V interactions and favors the AI/AO spin state. Within the tetrahedron network, the AI/AO state has two distinct canting angles θ and π-θ compared to the one canting angle θ of the TI/TO state, where the canting angle θ is a measure of deviation of spin from the −z axis as shown in Fig. 4(b). Guided by the DFT parameters for CoV2O4, our model calculation indicates that the new two-angle state based on the AI/AO state lies within 0.1 meV/unit-cell of the TI/TO ground state. The isotropic exchange (\({J}_{{\rm{V}}-{\rm{V}}}^{{\rm{in}}}={J}_{V-V}^{{\rm{out}}}\)) fosters a new two-angle AI/AO structure that can be stabilized by a magnetic field.
External pressure may also increase the degree of frustration. For high external pressure9,17 ~10 GPa, the enhanced itinerancy fully suppresses the local SIA (D v ~ 0) of V as in Fig. 4(a) and revives the magnetic frustration of the pyrochlore lattice. Although AFM exchange between the Co and V sites then induces the observed isosymmetric TI/TO spin structure, the frustration fostered by itinerancy and the alternative states that compete with the TI/TO ground state are possibly responsible for the measured magnetic anomalies17 and spin-glass behavior9. Moreover, the absence of SIA (D v ~ 0 and D A ~ 0) at high pressure above 10 GPa (Figs 2(a) and 4(a)) preserves the rotational symmetry and may stabilize a continuum of degenerate states where the two angles (θ 1, θ 2) rotate without energy cost as obtained in the Appendix and shown in the energy landscape of Fig. 4(d). This massive degeneracy can induce spin-glass or spin-liquid-like behavior. Since neutron scattering is limited to relatively low pressures, these novel states should be studied with synchrotron magnetic X-ray scattering.
Magnetic field measurement
Figure 4(a) provides a guide to uncover the novel states produced by the regenerated frustration. Although all other magnetic couplings (isotropic J V−V , reduced D v) foster frustration (bold red line), the remnant AFM interaction J Co−V (dotted blue line in Fig. 4(a)) still relieves frustration. An external magnetic field (\(\overrightarrow{H}=H\hat{z}\)) can help restore frustration by weakening J Co−V, thereby inducing the novel two-angle state of CoV2O4, as shown in Fig. 5.

Prediction of novel phases driven by magnetic field in Mn0.2Co0.8V2O4. Experimental results for the field-dependent (020) (a) and (220) (b) Bragg peak intensities. (c) Field dependence of spin angle θ calculated by spin models using DFT parameters. First-order phase transition from TI/TO to AI/AO-derived state with magnetic field is indicated by the jump in θ. Black open squares are from the neutron scattering measurement up to 10 T. (d) One-angle state based on TI/TO (left) and two-angle state based on AI/AO (right). The latter is driven by the competition between J Co−V and magnetic energy.
To check the effect of a magnetic field on CoV2O4, we carried out further elastic neutron-scattering measurements on the Co-rich single-crystal spinel Co0.8Mn0.2V2O4, which preserves the cubic structural and magnetic isotropies as in Fig. 4(a) but exhibits stronger scattering intensity than CoV2O4 due to the larger size of the single crystal. The V3+ AFM components in the ab-plane increase with the magnetic field (H z > 3 T), as indicated by the increased intensity of (020) (see Fig. 5(a)). At H z < 3 T, the increased intensity of (220) reflects the reorientation of the magnetic domains; at H z > 3 T the (220) intensity is saturated, indicating that all magnetic domains are fully oriented and that the FIM components are constant. This is consistent with magnetization measurements on a polycrystalline sample, which provide a saturation field of ~2 T19. Note that the magnetic components for \({M}_{z}^{{\rm{Co}}}\) and \({M}_{z}^{{\rm{V}}}\) both contribute the magnetic scatterings at (220). Considering the Co magnetic moments saturate ferromagnetically at zero field, we assume \({M}_{z}^{{\rm{Co}}}\) does not change with the field. Therefore, no change of the (220) intensity at fields above 3 T indicates \({M}_{z}^{{\rm{V}}}\) stays almost constant at the field between 3 T and 10 T. The small change in \({\rm{\Delta }}{M}_{z}^{{\rm{V}}}={M}^{{\rm{V}}}\mathrm{(0.6}{\mu }_{B})\cos \,{21}^{\circ }({\rm{at3T}})-{M}^{{\rm{V}}}\mathrm{(0.6}{\mu }_{B})\cos \,{24}^{\circ }({\rm{at10T}}) \sim 0.01{\mu }_{B}\), which is negligible compared with \(\Delta {M}_{xy}^{{\rm{V}}}\) and the resolution limit of ~0.1 μB. So we can safely assume that both \({M}_{z}^{{\rm{V}}}\) and \({M}_{z}^{{\rm{C}}o}\) are constant above 3 T. Since the AFM components of V3+ in the ab-plane (\({M}_{ab}^{{\rm{V}}}\)) continue to grow above 3 T, the canting angle (\(\theta ={\tan }^{-1}[{M}_{ab}^{{\rm{V}}}/{M}_{z}^{{\rm{V}}}]\)) of the V3+ spins must increase with the magnetic field along [001].
Using the spin model (Eq. 1) combined with DFT parameters (Fig. 4(a)), we confirm the increase in the canting angle with magnetic field in Fig. 5(c). The two-angle AI/AO state has an energy within 0.1 meV/unit-cell of the the one-angle TI/TO ground state in the Co-rich region. We predict that this new state is stabilized by a large magnetic field of about 140 T, as shown in Fig. 5(c),(d). Although only the one-angle TI/TO state was previously reported in vanadate compounds (AV2O4, A = Zn, Mn, Fe), various competing states appear in CoV2O4 due to frustration. It is likely that those states can be revealed by a magnetic field or pressure.
Of course, the critical magnetic field (H Z = 140 T) is too large for neutron scattering measurements. However, the first-order phase transition from the one-angle to the two-angle state may be captured by magnetic susceptibility measurements. Moreover, various methods can be employed to reduce the critical field. Since external pressure suppresses SIA and revives frustration as discussed in the previous section, pressure may also reduce the critical magnetic field. Contrary to the usual expectation, a magnetic field may strengthen frustration and noncollinearity in CoV2O4 by competing with the only exchange coupling (J Co−V) that hampers frustration.
Discussion
It is natural to wonder if cations significantly smaller than Co2+ such as Be2+ can be substituted on the A-site to induce even more itinerancy and consequent frustration. However, a non-magnetic A-site reduces the magnetization of V so much that the system would become paramagnetic29. Because strong magnetic interactions between the A and B sites is required, Co is the only candidate A-site cation to support localized spins with enhanced J Co−V while also promoting itinerancy on the B site.
Compared to other vanadates (AV2O4), the frustration in magnetically and structurally isotropic CoV2O4 explains its NC and macroscopic spin-glass properties. Since the AFM interaction between Co and V is the only factor that relieves the magnetic frustration, weakening the AFM interaction by a magnetic field or further reducing the SIA by external pressure can rekindle the frustration and reveal alternative states. Among spinel vanadates, CoV2O4 is uniquely located at the crossover between localized and itinerant behavior. Consequently, many exotic properties and new phases can be produced by restoring the frustration of the pyrochlore lattice.
Method
Sample preparation
Single crystals of CoV2O4, Co0.8Mn0.2V2O4 and MnV2O4 were grown by the traveling-solvent floating-zone (TSFZ) technique. The feed and seed rods for the crystal growth were prepared by solid state reaction. Appropriate mixtures of MnO, CoCO3, and V2O3 were ground together and pressed into 6-mm-diameter 60-mm rods under 400 atm hydrostatic pressure, and then calcined in Ar at 1050 °C for 15 hours. The crystal growth was carried out in argon in an IR-heated image furnace (NEC) equipped with two halogen lamps and double ellipsoidal mirrors with feed and seed rods rotating in opposite directs at 25 rpm during crystal growth at a rate of 20 mm/h.
Neutron-scattering experiments
Single-crystal neutron diffraction was performed to determine the crystal and magnetic structures using the four-circle diffractometer (HB-3A) at the High Flux Isotope Reactor (HFIR) of the Oak Ridge National Laboratory (ORNL). A neutron wavelength of 1.003 Å was used from a bent perfect Si-331 monochromator31. High magnetic field single-crystal neutron diffraction experiments were performed on the cold neutron triple-axis spectrometer (CTAX) at HFIR, ORNL. The incident neutron energy was selected as 5.0 meV by a PG (002) monochromator, and the final neutron energy was also set as 5.0 meV by a PG (002) analyzer. The horizontal collimation was guide-open-80′-open. Contamination from higher-order beams was removed using a cooled Be filter. The scattering plane was set in the (H,K,0) plane and the magnetic field was applied perpendicular to the scattering plane. The nuclear and magnetic structures were refined with the program FULLPROF32. We examined more than 18 peaks of lattice and magnetic Bragg peaks to extract the precise nuclear and magnetic structural information as shown in the S2 in the Supplementary material. Due to the domain re-orientation effect, Bragg peak intensities of both (220) and (020) diffractions increase sharply in small magnetic fields, but the (220) diffraction is saturated above about 3 T. The Bragg peak intensity of the (020) diffraction, corresponding to the magnetic component of V in the ab-plane, inceases linearly with field.
First-principles calculations
First-principles calculations were performed using density-functional theory within the local spin-density approximation with a correction due to on-site Hubbard interaction (LSDA + U) as implemented in the Vienna ab initio simulation package (VASP-5.3)33. We used the Liechtenstein34 implementation with on-site Coulomb interaction U = 6.0 eV and on-site exchange interaction J H = 1.0 eV to treat the localized 3d electron states in Co, Mn, and V; this choice of U is close to that chosen in previous work on CoV2O4 35 and MnV2O4 36,37. The spin-orbit interaction was included. The projector augmented wave (PAW) potentials38,39 explicitly include 13 valenced electrons for Mn (\(3{p}^{6}3{d}^{5}4{s}^{2}\)), 9 for Co (\(3{d}^{8}4{s}^{1}\)), 13 for V (\(3{s}^{2}3{p}^{6}3{d}^{4}4{s}^{1}\)), and 6 for oxygen (\(2{s}^{2}2{p}^{4}\)). The wave functions were expanded in a plane-wave basis with an energy cutoff of 500 eV. To evaluate the on-site single-ion anisotropy (SIA) interaction D, only one cation of interest was kept while the surrounding magnetic atoms were replaced by neutral and isoelectronic Ca2+ and Al3+ cations for Co2+/Mn2+ and V3+, respectively. This is the same technique that was successfully used for BiFeO3 40 and CaMn7O12 41.
Microscopic spin model
Spin states in spinels can be described by the following model Hamiltonian,
which contains six inequivalent sublattices. Isotropic exchange constants \({J}_{{\rm{Co}}-{\rm{V}}}\) describe nearest-neighbor interactions between the Co and V sites. J Co−Co and J V−V describe nearest-neighbor interactions between Co-sites and V-sites, respectively. The easy-axis anisotropy is assumed to be zero for the Co-sites, while for the B-site spins, the easy-axis anisotropy D v is along the local <111> direction. The azimuthal directions of each vanadium spin is constrained, but the canting angle θ i , described in Fig. 4, is allowed to vary between 0 and 2π. Since θ i may have a unique value in adjacent planes, both the two-in-two-out and all-in-all-out configurations are possible. These angles are equal to the polar angle when θ i is between 0 and π, while the polar angles equals \(2\pi -{\theta }_{i}\) and the azimuthal angle changes by π when θ i is greater than π.
The ground state spin configuration was found by minimizing the classical energy for a given set of parameters. To avoid local minima, this was accomplished by calculating the classical energy on a grid with \({\theta }_{i}=0\) to 2π and finding the two angles with the lowest energy. This process was repeated for values of the external magnetic field ranging from 0 to 173 T.
The inelastic neutron cross section for undamped spin waves was calculated using the 1/S formalism outlined in ref.42 and the appendices of ref.43. For direct comparison with experimental intensities, the effects of the magnetic form factor and the instrumental resolution were included in the calculation. The coefficients for Co2+, and V3+ are from ref.44. The resolution function was approximated as a Gaussian in energy with a full width at half-maximum of 1.5 meV. Effects from finite resolution in Q were not considered.
While DFT can provide guidance for the values of the isotropic exchange interactions, LSDA + U overestimates the experimental moment (Mv = 0.47(4)μ B ) of CoV2O4 measured by neutron scattering. Our spin model uses the magnetic moment (Mv = 0.5 μ B ), which is within the experimental uncertainty. In addition, parameters calculated with DFT were adjusted to reproduce the measured canting angle of CoV2O4 (θ = 20.8 ± 1.7°) in zero field. Care was also taken to avoid a long-range spiral configuration45 that was not observed in our neutron diffraction measurements. The final set of parameters used for CoV2O4 are \({J}_{{\rm{Co}}-{\rm{Co}}}=0.5\,{\rm{meV}}\), \({J}_{{\rm{Co}}-{\rm{V}}}=-2.5\,{\rm{meV}}\), \({J}_{{\rm{V}}-{\rm{V}}}=-11.0\,{\rm{meV}}\), \({D}_{{\rm{V}}}=-2.7\,{\rm{meV}}\), \(red{M}_{{\rm{Co}}}=\) 3.00 μ B and \(red{M}_{{\rm{V}}}=0.5\,{\mu }_{B}\).
Canting angles of the new phase with D v = 0.0
At high pressures, we take D v = 0.0 in Eq.(1). Then, the total energy per unit magnetic unit cell is
where \(\alpha =\,\cos \,{\theta }_{1}+\,\cos \,{\theta }_{2}\). The total energy is at a minimum if
which limits the allowed combination of θ 1 and θ 2. When θ 1 = θ 2 and D Co = D v = 0.0, this condition is identical to the expression for θ in ref.36.
References
Moessner, R. & Chalker, J. T. Properties of a Classical Spin Liquid: The Heisenberg Pyrochlore Antiferromagnet. Phys. Rev. Lett. 80, 2929 (1998).
Lacroix, C., Mendels, P., & Mila, F. Introduction to Frustrated Magnetism: Materials, Experiments, Theory. Springer (2001).
Nakatsuji, S. et al. Metallic Spin-Liquid Behavior of the Geometrically Frustrated Kondo Lattice Pr2Ir2O7. Phys. Rev. Lett. 96, 087204 (2006).
Lee, S.-B., Paramekanti, A. & Kim, Y. B. RKKY Interactions and the Anomalous Hall Effect in Metallic Rare-Earth Pyrochlores. Phys. Rev. Lett. 111, 196601 (2013).
Udagawa, M., Ishizuka, H. & Motome, Y. Non-Kondo Mechanism for Resistivity Minimum in Spin Ice Conduction Systems. Phys. Rev. Lett. 108, 066406 (2012).
Hanasaki, N. et al. Nature of the Transition between a Ferromagnetic Metal and a Spin-Glass Insulator in Pyrochlore Molybdates. Phys. Rev. Lett. 99, 086401 (2007).
Iguchi, S. et al. Emergence of a Diffusive Metal State with No Magnetic Order near the Mott Transition in Frustrated Pyrochlore-Type Molybdates. Phys. Rev. Lett. 102, 136407 (2009).
Kumar, S. & van den Brink, J. Frustration-Induced Insulating Chiral Spin State in Itinerant Triangular-Lattice Magnets. Phys. Rev: Lett. 105, 216405 (2010).
Kiswandhi, A. et al. Chemical pressure effects on structural, magnetic, and transport properties of Mn1−x Co x V2O4. Phys. Rev. B. 84, 205138 (2011).
Kismarahardja, A. et al. Dielectric properties of single crystal spinels in the series FeV2O4, MnV2O4 and CoV2O4 in high magnetic fields. Phys. Rev: Lett.B 87, 054432 (2013).
Nishiguchi, N. & Onoda, M. A pseudotetramer in the geometrically frustrated spinel system CdV2O4. J. Phys. Condens Matter 14, L551 (2002).
Lee, S. H. et al. Orbital and Spin Chains in ZnV2O4. Phys. Rev: Lett. 93, 156407 (2004).
Wheeler, E. M. et al. Spin and orbital order in the vanadium spinel MgV2O4. Phys. Rev. B. 82, 140406(R) (2010).
Garlea, V. O. et al. Magnetic and Orbital Ordering in the Spinel MnV2O4. Phys. Rev. Lett. 100, 066404 (2008).
MacDougall, G. J. et al. Magnons and a two-component spin gap in FeV2O4. Phys. Rev. B. 89, 224404 (2014).
Katsufuji, T. et al. Structural and magnetic properties of spinel FeV2O4 with two ions having orbital degrees of freedom. J. Phys. Soc. Jpn. 77, 053708 (2008).
Kismarahardja, A. et al. CoV2O4: A Spinel Approaching the Itinerant Electron Limit. Phys. Rev: Lett. 106, 056602 (2011).
Goodenough, J. B. In Metallic Oxides byH. Reiss, Progress in Solid State Chemistry Vol. 5 (Pergamon, New York, 1972).
Huang, Y., Yang, Z. & Zhang, Y. Magnetic, structural, and thermal properties of CoV2O4. J. Phys. Condens. Matter 24, 056003 (2012).
Kim Myung-Whun, J. S., Kim, T., Katsufuji & Kremer, R. K. Magnetic susceptibility and specific heat of a spinel MnV2O4 single crystal. Phys. Rev. B 83, 024403 (2011).
Reig-i-Plessis, D. et al. Structural transition and orbital glass physics in near-itinerant CoV2O4. Phys. Rev. B 93, 014437 (2016).
Dwight, K., Menyuk, N., Rogers, D. B., Wold, A. Proc. Int. Conf. on Magnetism 1964 (Nottingham) p538.
Bramwell, S. T. & Harris, M. J. Frustration in Ising-type spin models on the pyrochlore lattice. J. Phys. Condens. Matter 10, L215–L220 (1998).
Ma, J. et al. Strong competition between orbital ordering and itinerancy in a frustrated spinel vanadate. Phys. Rev. B 91, 020407 (R) (2015).
Dun, Z. L. et al. Competition between the inter- and intra-sublattice interactions in Yb2V2O7. Phys. Rev. B 91, 064425 (2015).
Koborinai, R. et al. Phys. Rev: Lett. 116, 037201 (2016).
Magee, A. J. Spin correlations in frustrated magnets with orbital ordering Ch. 4(Ph.D. thesis, Royal Holloway, University of London 2010).
MacDougall, G. J. et al. Magnetic order and ice rules in the multiferroic spinel FeV2O4. Phys. Rev. B 86, 060414(R) (2012).
Blanco-Canosa, S. et al. Enhanced Pressure Dependence of Magnetic Exchange in A2+[V2]O4 Spinels Approaching the Itinerant Electron Limit. Phys. Rev. Lett. 99, 187201 (2007).
Mydosh, J. A. In Spin Glasses: An Experimental Introduction by J. A. Mydosh (Talylor & Francis, 1993).
Chakoumakos, B. C. et al. Four-circle single-crystal neutron diffractometer at the High Flux IsotopeReactor. J. Appl. Crystallogr. 44, 655 (2011).
Rodrguez-Carvajal, J. Recent advances in magnetic structure determination by neutron powder diffraction. Physica B 192, 55 (1993).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Liechtenstein, A. I., Anisimov, V. I. & Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 52, R5467 (1995).
Kaur, R., Maitra, T. & Nautiyal, T. The nature of itineracy in CoV2O4: a first-principles study. J. Phys.: Condens. Matter 26, 045505 (2014).
Nanguneri, R. & Savrasov, S. Y. Exchange constants and spin waves of the orbital-ordered noncollinear spinel MnV2O4. Phys. Rev. Lett. 86, 085138 (2012).
Sarkar, S., Maitra, T., Valentí, R. & Saha-Dasgupta, T. Proposed Orbital Ordering in MnV2O4 from First-Principles Calculations. Phys. Rev. Lett. 102, 216405 (2009).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Weingart, C., Spaldin, N. & Bousquet, E. Noncollinear magnetism and single-ion anisotropy in multiferroic perovskites. Phys. Rev. B 86, 094413 (2012).
Zhang, J. T. et al. Magnetic properties and origins of ferroelectric polarization in multiferroic CaMn7O12. Phys. Rev. B 87, 075127 (2013).
Haraldsen, J. T. & Fishman, R. S. Spin rotation technique for non-collinear magnetic systems: application to the generalized Villain model. J. Phys.: Condens. Matter 21, 216001 (2009).
Fishman, R. S., Haraldsen, J. T., Furukawa, N. & Miyahara, S. Spin state and spectroscopic modes of multiferroic BiFeO3. Phys. Rev. B 87, 134416 (2013).
Dianoux, A. J. & Lander, G. Neutron Data Booklet (OCPScience, Philadelphia, 2003).
Tomiyasu, K., Fukunaga, J. & Suzuki, H. Magnetic short-range order and reentrant-spin-glass-like behavior in CoCr2O4 and MnCr2O4 by means of neutron scattering and magnetization measurements. Phys. Rev. B 70, 214434 (2004).
Acknowledgements
The work at UNIST (J.H.L., H.-J.L., M.L.) was supportedby National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP)(2015R1C1A1A01055760), Basic Research Laboratory (NRF-2017R1A4A1015323), and Creative MaterialsDiscovery Program through the NRF funded by the Ministry of Science and ICT(2017M3D1A1040828) . J.M. thanks the support of the Ministry of Science and Technology of China (2016YFA0300500). The research at ORNL were sponsored by Department of Energy, Office of Sciences, Basic Energy Sciences, Materials Sciences and Engineering Division (S.O., R.F.) and Scientific User Facilities Division (J.M., S.E.H., M.M.). S.E.H. acknowledges support by the Laboratory’s Director’s fund, ORNL. H.D.Z thanks the support from NSF with grant NSF-DMR-1350002. This research used resources of the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The authors acknowledge valuable discussions with G. MacDougall.
Author information
Authors and Affiliations
Contributions
J.H.L. conceived the original idea. J.H.L. and M.S.Y. carried out first-principles calculations. S.E.H. and R.S.F. performed spin-wave simulations. J.M., H.C., T.H., and M.M. measured and analyzed neutron scattering Bragg peaks. H.D.Z. synthesized the samples. J.H.L., S.E.H., J.M., H.B.C., M.L., T.H., H.-J.L., M.S.Y., S.O., M.M., and R.S.F. discussed the results. J.H.L. and R.F. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, J.H., Ma, J., Hahn, S.E. et al. Magnetic Frustration Driven by Itinerancy in Spinel CoV2O4 . Sci Rep 7, 17129 (2017). https://doi.org/10.1038/s41598-017-17160-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-17160-0
This article is cited by
-
First Principles Investigation of Electronic, Optical, and Magnetic Properties of MgYb2X4 (X = S, Se, Te)
Journal of Superconductivity and Novel Magnetism (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
