
Abstract
Rhodotorula mucilaginosa was isolated from a patient with onychomycosis, and identification was confirmed by morphological and cultural characteristics as well as by DNA molecular analysis. Antifungal agents naftifine (10 mg/mL, active substance in Exoderil) and bifonazole (10 mg/mL, active substance in Canespor) were tested in different concentrations to assess in vitro effects on fungal growth and carotenoid synthesis. The antifungal mechanisms of action of naftifine and bifonazole against R. mucilaginosa isolates were similar and affected the biosynthetic pathway of ergosterol. For the first time, this research demonstrates that naftifine affects the carotenoid biosynthetic pathway, producing depigmentation of R. mucilaginosa in solid and liquid media. Furthermore, depigmentation was a reversible process; naftifine-treated yeast cells that were depigmented resumed carotenoid production upon transfer to fresh media. Raman and UV-vis spectrophotometry in conjunction with chromatographic analysis detected changes in carotenoids in yeast cells, with torulene decreasing and B-carotene increasing after repigmentation. Transmission electron micrographs revealed critical ultrastructural modifications in the depigmented cells after naftifine treatment, i.e., a low-electron-density cell wall without visible mucilage or lamellate structure.
Similar content being viewed by others

Introduction
Onychomycosis is a multifactorial nail fungal infection1,2,3. It is an important public health problem because of its high prevalence, high rates of recurrence and progression to chronic lesions4. Onychomycosis accounts for 50% of all nail diseases. It affects 2–13% of the general population, and this percentage increases with age, reaching up to 40% in the elderly5. Thickened, discoloured, deformed nails without pain are the most common symptoms of a fungal nail infection. Although the clinical picture can be very suggestive, the diagnosis should be confirmed by a routine method before starting treatment5. The most commonly used methods are direct potassium hydroxide (KOH) examination, culture, and, to a lesser extent, nail biopsy. The diagnosis of onychomycosis is made when one or more of the three diagnostic tests are positive. Other tests are expensive and require the use of specialized equipment and materials6.
Common pathogens in onychomycosis are dermatophytes, nondermatophyte moulds (NDMs) and yeasts1, 3, 7. The nomenclature of fungal infections proposed by the International Society for Human and Animal Mycology suggests that the term onychomycosis should be replaced by tinea unguium when the aetiological agent is a dermatophyte; by onyxis when yeasts are the cause; by ungual candidiasis when the agent is Candida; and by ungual mycosis when the causal agent is an NDM8.
Most cases of tinea unguium are caused by dermatophytes that belong to three genera: Trichophyton, Microsporum, and Epidermophyton. The fungi Trichophyton rubrum, T. mentagrophytes, and Epidermophyton floccosum are the most common aetiologic agents of onychomycosis worldwide. The most commonly described species of NDM are Scopulariopsis brevicaulis, Fusarium spp., Acremonium spp., Aspergillus spp., Scytalidium spp., and Onychocola canadienses 8. Candida species are the most frequent among the yeasts7. Other yeasts accepted in recent years as causative agents of onychomycosis are Rhodotorula mucilaginosa 9, R. minuta 10 and R. glutinis 11. Several authors have isolated Rhodotorula from ecosystems and environments12,13,14,15, humans16,17,18 and animals13. During the last few decades, they have emerged as opportunistic pathogens, particularly in immunocompromised patients19.
Because onychomycosis aetiologic agents have variable susceptibility to antifungal drugs, laboratory diagnosis is a necessary tool for ascertaining the best therapeutic option4. Precise and correct identification of the species responsible for onychomycosis in a given patient is required to select the most appropriate treatment as well as to avoid the misuse of antifungals9.
Thus, in vitro susceptibility tests demonstrated that R. mucilaginosa is sensitive to low concentrations of amphotericin B and 5-flucytosine and resistant to high concentrations of fluconazole, itraconazole, voriconazole and terbinafine9. Additionally, in the case of R. glutinis, minimum inhibitory concentrations (MIC) vary depending on the antifungal drug: 0.125 μg/mL for itraconazole; 0.5 μg/mL for amphotericin B, anidulafungin and posaconazole; 1 μg/mL for voriconazole; 16 μg/mL for caspofungin and 128 μg/mL for fluconazole11. Overall, for humans the use of antifungals against Rhodotorula infections is still controversial because in vitro some are inactive against the majority of clinical isolates of Rhodotorula spp.19.
R. mucilaginosa is a basidiomycetous yeast. It presents only spheroidal to oval budding cells (2.5–6.5 × 6.5–14.0 µm) with carotenoid pigments12, 20 and without the rudimentary formation of hyphae21, 22. This species is characterized by yeast-like colonies that are coral pink and moist to mucoid on Sabouraud Dextrose Agar (SDA), by the inability to assimilate inositol and by the absence of fermentation23. Carotenoids can act as vitamin A precursors, having colouring and antioxidant properties24. Carotenoids are widely produced pigments found in algae, yeasts and plants. Their main functions are reactive oxygen species scavenging and protection against photooxidative damage. Four main pigments, torularhodin, torulene, beta-carotene and gamma-carotene, are synthesized by R. mucilaginosa species24, 25, and carotenoid production is usually enhanced by stress factors such as UV exposure, oxidative stress or osmotic stress26. Torularhodin was shown to be correlated with the survival of the cells under UV-B light27. Beta-carotene and torularhodin also showed antioxidant activity, preventing hyperoxia-induced cytotoxicity28.
Carotenoid synthesis starts from acetyl-CoA that is transformed into isopentenyl pyrophosphate and further to lycopene, which is the source of many other carotenoids29. Cases of depigmentation have previously been reported and are thought to be induced by chemicals interfering with the biosynthetic process. Naftifine, a topical allylamine, has been shown to interfere with the pigmentation process of Staphylococcus aureus colonies by specifically and strongly inhibiting isoprenoid synthesis30.
The study aimed first to find onychomycosis etiology and sensitivity to naftifine and bifonazole. Secondly, R. mucilaginosa growth, carotenoids metabolism and structural changes were analysed.
Results and Discussion
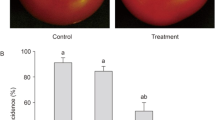
The results indicated that R. mucilaginosa was a causative agent in this specific case of onychomycosis in an aged patient with chronic HBV hepatitis (Fig. 1A). After incubation of all toenail samples in SDA control media, R. mucilaginosa isolates were identified by morphological and cultural characteristics. Control colonies of R. mucilaginosa obtained on SDA were mucoid, with red colour (Fig. 1B) and a diameter of 12-13 mm 3 days after inoculation. Molecular analysis was performed for species identity confirmation14, 23. The DNA sequence was run against the BLAST-NCBI nucleotide database31 as a query and matched identically with R. mucilaginosa (GenBank: KU052792.1) species.

(A) Toenails affected by onychomycosis; (B). Rhodotorula mucilaginosa control colonies on Sabouraud Dextrose Agar (SDA); (C). R. mucilaginosa control colonies (a) and depigmented colonies obtained by including the fungal inoculum in 1 mg/mL naftifine solution for 10 minutes before inoculation (b), depigmented colonies obtained on SDA with 0.1 mg/mL naftifine (c) and 0.2 mg/mL naftifine (d); (D). Transfer and growth of depigmented yeast cells to fresh SDA results in repigmentation.
Then, by exposing a control R. mucilaginosa isolate for 10 minutes to 1 mg/mL naftifine solution, depigmented fungal colonies were obtained on SDA control media. The depigmented colonies were mucoid and had similar diameters to the red colonies but showed a milky colour. Moreover, by including small concentrations of naftifine (0.1 mg/mL, 0.2 mg/mL) in SDA before inoculation of R. mucilaginosa isolate, an inhibitory effect was obtained compared to the control colonies (Fig. 1C). These depigmented colonies had a diameter of 7-8 mm on SDA with 0.1 mg/mL naftifine and of 5-6 mm on SDA with 0.2 mg/mL naftifine, showing a small dose-dependent inhibitory effect of the drug (Fig. 1C). Furthermore, by inoculating depigmented R. mucilaginosa isolate into SDA control media, colonies regained pigment (Fig. 1D). Considering that carotenoids are important substances against reactive oxygen species (ROS), depigmentation may be correlated with increased oxidative stress and fungal injury. Repigmentation suggested that the carotenoid metabolism changes induced in the fungi by naftifine were reversible. These surprising results suggested further analysis regarding R. mucilaginosa carotenoid metabolism and related structural changes.
Preliminary tests of R. mucilaginosa growth under our conditions indicated that the stationary phase was reached after 60–70 h. However, in the initial exponential stage the cells appeared mostly colourless (low carotenoid content), and only after approximately 50 h could the pigmentation be observed. This monitoring was performed at various pH values (pH range 3.5–7.5), especially for tracking carotenoid formation. The growth was pH-dependent only in the initial exponential phase (Fig. S1A). Although the pH profile change in time was dependent on the initial pH (Fig. S1B), indicating a pH-dependent metabolism for R. mucilaginosa, carotenoid formation in terms of both rate and composition did not depend on pH significantly, as observed by monitoring the intensity and spectral features from the resonant Raman spectra (Fig. S2). We aimed to further understand the factors that influenced and controlled carotenoid formation in Rhodotorula spp. beyond the factors that interfered with basic metabolism, such as type of C and N sources and their ratio26, compounds inducing ROS stress (e.g., duroquinone, methylene blue). ROS upregulated carotenoid content in R. mucilaginosa, indicating that the carotenoids were a response of the cells to the negative effects of ROS agents (e.g., superoxide, singlet oxygen) as previously shown28, 32. Other chemicals, such as amphotericin and diphenylamine, not only inhibited carotenoid formation but also cell development (Fig. S3). In addition, we hypothesized that the photosensitizers hypericin and hyperforin could also induce carotenoid formation; however, this was not the case (Fig. S3), most likely due to the low level of the induced potential stress.
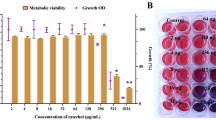
During these initial experiments, we observed that one of the two tested antifungal agents, namely, naftifine (an allylamine antifungal drug), did not inhibit cell growth at lower concentrations, but rather induced depigmentation of the cells when growing on solid medium (Fig. 1C). The same results were obtained in liquid media, in controlled conditions. In addition to determination of naftifine MIC50 (55 ± 14 mg/L) for its antifungal activity (Fig. 2A), quantified by OD600nm, the effect of naftifine on depigmentation was assessed by OD512nm and a corresponding MIC50 (0.088 ± 0.02 mg/L) was determined (Fig. 2B). Depigmentation did not affect the cell growth at these naftifine concentrations, as could be observed by the optical density values. In contrast, bifonazole (an imidazole antifungal drug) did not produce any depigmentation in R. mucilaginosa, although its MIC50 for antifungal activity was one order of magnitude lower, 4.4 ± 0.8 mg/L (Fig. 2C and D).

(A) Determination of MIC50 values for naftifine content in the pharmaceutical product Exoderil by monitoring the OD at 600 nm and (B) by absorbance at 512 nm after cell disruption with DMSO. (C) Determination of MIC50 values for bifonazole content in the pharmaceutical product Canespor by monitoring the OD at 600 nm and (D) by absorbance at 512 nm after cell disruption with DMSO.
Similar depigmentation was observed by resonant Raman spectroscopy (Fig. 3A), where besides the expected decrease in signal intensities of the main peaks 999 cm−1 (C = CH deformation), 1150 cm−1 (symmetrical C-C stretching), 1505 cm−1 (symmetrical C = C stretching) due to carotenoid content, subtle changes could be observed in the 1200–1400 cm−1 region. Minor Raman shifts were recorded for the secondary peak at 1283 cm−1 (torulene), ascribed to the CH2 deformation in carotenoids33, 34. This band was shifted in the case of torularhodin at 1284 cm−1, at 1286 cm−1 for β-carotene and at 1290 cm−1 for unidentified carotenoid (Cnd), indicating a possible change of the carotenoid composition in the cells, thus a qualitative change. This observation was further supported by the UV-vis spectra, where in addition to an absorbance decrease with increased naftifine concentrations, the spectral profiles also changed (i.e., the ratio of the three main spectral features) (Fig. 3B).

(A) Resonant Raman spectra of R. mucilaginosa cells grown in media containing various naftifine concentrations (indicated to the right of each spectrum). (B) Concentrationdependent UV-vis spectra of DMSO-disrupted R. mucilaginosa cells after growth in naftifinecontaining media. On the right, an image of the cell pellets at increasing naftifine concentrations after washing and before disruption is shown.
Next, for more in-depth investigations, chromatographic analysis of the extracted carotenoids was performed. From a variety of solvents or conditions for cell disruption available in the literature, we chose simple DMSO treatment of the washed cells. The advantages of this procedure are the high yield of extraction (only a colourless pellet remained, indicating a quantitative extraction) and the good solubility of ergosterol in this solvent, which allowed a simultaneous extraction of both type of constituents. The High-Performance Liquid Chromatography (HPLC) analysis allowed the separation of four main carotenoids including torularhodin, torulene and β-carotene (C1, C2 and C3, respectively), in good agreement with previous data27, 32 (Fig. 4A), while the other compound (Cnd) could not be identified. The identification was done using a pure standard for β-carotene and retention times, as well as using highly specific spectral features of the other two compounds, which were in very good agreement with already known data (Fig. 4B, Table 1). The unidentified compound presented torulene-like carotenoid absorption spectral features and had a chromatographic behaviour (lower retention time allowing assignment as hydroxy-torulene) resembling previously detected carotenoids in other Rhodotorula spp.35. Additionally, ergosterol was also separated using the same method and identified based on its chromatographic and highly specific spectral characteristics (Fig. 4A). Moreover, using a dedicated method for ergosterol extraction, a highly similar spectrum was obtained. Therefore, all these related components could be quantitatively evaluated, allowing a comparative analysis for cells growing in different conditions. Thin-Layer Chromatography (TLC) was also used for carotenoid separation after DMSO removal using extraction cartridges. Using this method, the same three components were separated and identified, followed by their Raman detection directly on the plate. The contour map of the detected signal is presented in Fig. 4C, and the individual Raman signals are shown in Fig. 4D. As expected, the spectra had a high degree of similarity due to highly similar chemical structures (Fig. 4E); nevertheless, subtle changes could be observed at approximately 1280 cm−1, and a slight spectral shift appeared at 1505 cm−1 band (Table 1, Fig. 4D). In addition to torularhodin, torulene and β-carotene, two unidentified carotenoids were also observed (probably different than the one from HPLC analysis, due to retention time analysis). This may have occurred because in the introduction of an extra sample preparation step (cartridge extraction for DMSO removal), analyte alteration could occur, especially for those present in lower quantities.

(A) HPLC-DAD chromatograms of the DMSO extracts of R. mucilaginosa (control, grown in the presence of 0.1 mg/L naftifine and repigmented) as well as β-carotene standards. The major peaks are indicated by C1-C4 labels and Cnd (unidentified compound). The chromatogram was monitored by mAbs at 490 nm for 25 minutes (carotenoid detection) and 254 nm from that point on. In the inset, the spectrum of C4 (ergosterol) is shown. (B) UV-vis spectra of the identified compounds. The arrows indicate the corresponding y-axis. (C) Raman signal contour map obtained by mapping the HPTLC chromatogram containing the carotenoid-specific components (indicated on the right along with the intensity colour code scale). (D) Individual Raman signals of the HPTLC separated components. (E) Chemical structures of the identified compounds.
A Principal Component Analysis (PCA) was applied to the absorbance spectra of the DMSO extracts of the cells grown under various naftifine concentrations, and the plot of the scores corresponding to the first two principal components are shown in Fig. 5A. Surprisingly, two different clusters were noted, one with samples at low concentrations and one with samples at high concentrations, together with a transition zone. Such a grouping indicated different compositions of carotenoids with increasing naftifine concentration. To eliminate the concentration effect (decrease in intensity as the carotenoid content is diminished), for PCA analysis normalized spectra were used, thus allowing a qualitative-only analysis (relative content of carotenoids). The normalized spectra are presented in Fig. 5B. A shift of the spectral maxima towards the UV region was observed (the solutions are in DMSO, and thus the maxima were different from those in the HPLC data, where the solvent was different). This shifting of the maxima clearly indicates an increase of β-carotene relative content and decrease of torularhodin relative content as the naftifine concentration was increased. This was in very good agreement with the previously presented chromatographic results (Fig. 4A). Of note, the same approach applied to the bifonazole data did lead to different results. In this case, the decrease in the intensity of carotenoid spectra and increase in antifungal agent concentration were correlated solely with OD decrease (population size of the cells), and no change of the relative concentrations of the various carotenoids was observed. Therefore, the normalized spectra were identical over the entire bifonazole concentration range (Fig. S4), and no clustering was observed when PCA analysis was applied to these spectra (Fig. 5C).

(A) Scatterplot of the first two principal component scores obtained after applying PCA to the UV-vis spectra of DMSO-disrupted R. mucilaginosa cell extract. Cells were grown at nine levels of naftifine concentrations (0 to 1.2 mg/L, indicated on the plot) in triplicate. The curved arrow indicates the position shift in the graph as the naftifine concentration increases. (B) The averaged UV-vis spectra used in the abovementioned PCA analysis at specific working concentrations. The arrows indicate spectral shifts as the naftifine concentration increases. (C) Scatterplot obtained identically to A, but with bifonazole used instead of naftifine. (D) Scatterplot of the first two principal component scores obtained after applying PCA to the UV-vis spectra of DMSO-disrupted R. mucilaginosa cell extract. Cells were grown in standard conditions (control, ctrl.), after repigmentation (in quintuplicate) and in hypoxic conditions (in triplicate).
To further investigate the influence of naftifine upon carotenoid synthesis, we performed experiments where the total carotenoids were monitored by absorbance at 512 nm after DMSO extraction from the cells and by OD600nm to evaluate the cell density (as a marker of cell survival). Contrary to our expectation, hypoxia and UV exposure alone led to depigmentation and had only a weak influence upon cell survival, while the addition of naftifine further suppressed the carotenoid content. Repigmentation of cells treated solely with naftifine (Ctrl. + naftifine) occurred after reinoculation in naftifine-free media (Fig. 6A). A significant profile change in the UV-Vis spectra of the control, repigmented and hypoxia-treated cells was observed by PCA analysis (Fig. 5D). Strikingly, further chromatographic analysis revealed that the relative ratio of the determined carotenoids and ergosterol was very different among the control, naftifine-treated and repigmented forms. The torulene and Cnd carotenoids were much lower in the repigmented forms than for the control batch, indicating an alteration of carotenogenesis after naftifine treatment, but β-carotene and ergosterol were much higher (Fig. 6B). This could be of particular importance for biotechnology involving carotenoid synthesis.

(A) OD600nm of the R. mucilaginosa cultures cultivated in the indicated conditions and the corresponding Abs at 512 nm after DMSO cell disruption (ANOVA test, p < 0.001 for both sets of histograms). The error bars indicate the standard deviations (n = 3). (B) Peak areas of the major components separated and identified in the HPLC chromatogram from Fig. 3A. Ergosterol corresponding values are divided by a factor of 4 to reach the same scale (ANOVA test, p < 0.001).
Following the chemical characterization, morphological and ultrastructural characteristics were evaluated using scanning and transmission electron microscopy. On the cell surface, a characteristic mucilage visible in scanning electron microscopy was observed (Fig. 7A). Ultrastructural characteristics of R. mucilaginosa cells include the electron-dense and lamellate cell wall, spherical to ovoid nucleus, plasmalemma, mitochondrion, endoplasmic reticulum, and lipid and glycogen accumulation in the cytoplasm. Numerous small lipid granules of uniform size were found in the cytoplasm of young cells, and larger lipid granules generated by fusion showed variable shapes in mature and senescent cells (Fig. 7B–D). Young R. mucilaginosa cells have a thin and electron-dense cell wall formed by elongation of the internal cell wall of the mother cell (Fig. 7B and C). The depigmented cells of R. mucilaginosa had a low-electron-density cell wall without visible mucilage or lamellate structure and with large lipid granules (Fig. 7 E and F). R. mucilaginosa control colonies were mucoid and pigmented because the cells possess a carotenoid biosynthetic pathway20, 25. Additionally, TEM revealed other ultrastructural characteristics as follows. The cells have lamellate and electron-dense cell walls and undergo enteroblastic budding similarly to basidiomycetous yeasts36, 37. We suspected that the number of layers of cell wall was correlated with and proportional to the number of buds generated by a cell. In addition to these features, the cells of R. mucilaginosa accumulate different storage materials in the main phases of growth, such as glycogen and lipids, similarly to R. glutinis 38.

Scanning electron micrograph (A) and transmission electron micrographs (B,C,D,E,F) of R. mucilaginosa showing ultrastructural components. (A) Visible mucilage on cell surface; (B) longitudinal section of bud and cell; (C) longitudinal section of bud and cell (zoomed in); (D) cross section of cell; (E) longitudinal section of two depigmented cells; (F) longitudinal section of a depigmented cell (detailed). Legend: b, bud; c, cell; CW, cell wall; ER, endoplasmic reticulum; G, glycogen; L, lipid; M, mitochondrion; m, mucilage; N, nucleus; P, plasmalemma; V, vacuole.
Our results complete the list regarding the antifungal effects of naftifine, which provides good activity against Candida and Aspergillus species39 and is an excellent candidate for the treatment of superficial dermatophytosis caused by Trichophyton rubrum, T. mentagrophytes, T. tonsurans, Epidermophyton floccosum and Microsporum canis 40. For R. mucilaginosa, the antifungal activity of naftifine occurs at higher concentrations (MIC = 0.05 mg/L for R. mucilaginosa) by preventing ergosterol synthesis41.
For the first time, we demonstrated that naftifine caused depigmentation of R. mucilaginosa colonies obtained on nutritive medium because the carotenoid biosynthetic pathway was affected. Our results are consistent with the results of other authors, who showed that naftifine has the ability to block the biosynthesis of carotenoids in Staphylococcus aureus by affecting the activity of the last enzyme in isoprenoid formation. Moreover, naftifine affected the budding of R. mucilaginosa cells, the synthesis of mucilage and the colour and lamellar structure of the cell wall. Bifonazole affected the biosynthetic pathway of ergosterol by inhibiting the demethylation of 4,4′,14-trimethylsterols or by inhibiting the microsomal HMG-CoA-reductase42. The carotenoid production was not affected in any way. Amphotericin B binds to ergosterol, determining the formation of pores in the cell membrane, and promotes the accumulation of ROS43. The MIC value determined against R. mucilaginosa was 1 µg/mL44.
Conclusions
Our research demonstrated that both naftifine (in Exoderil) and bifonazole (in Canespor) were effective antifungal agents for a R. mucilaginosa isolate causing onychomycosis, even though they acted differently. Of these fungicides, only naftifine affected the carotenoid synthesis pathway and produced depigmentation in R. mucilaginosa cells in a reversible fashion.
Furthermore, naftifine also reduced the relative carotenoid content after repigmentation. Future clinical studies are necessary to further investigate the implications of these findings.
Methods
Sample collection
Toenails affected by onychomycosis (Fig. 1A) were obtained from an 85-year-old female with chronic HBV hepatitis. After nail asepsis with 70% ethanol, the distal and lateral fragments of both toenail plates with subungual debris were removed with a sterile nail clipper.
The study was approved by the Ethics Committee of the Iuliu Hatieganu University of Medicine and Pharmacy, Cluj-Napoca (Romania) and an informed written consent was obtained from the patient before enrolling her in the study.
All experiments in this study were performed in accordance with relevant guidelines and regulations.
Fungal strain isolation and growth conditions
Small toenail fragments of 2–3 mm were disinfected in 20% ethanol for 1 minute and inoculated into SDA control media in Petri dishes45. Yeast growth was carried out using the standard method of triple culture (insemination of the inoculum in three points on the surface of SDA in Petri dishes) and incubated at 22 °C for 3 days. After incubation, the isolates were identified by morphological and cultural characteristics. All experiments were performed in triplicate.
Fungal colonies obtained on SDA control media were also grown in Yeast Extract-Peptone-Dextrose (YPD) medium containing 0.5% peptone, 0.3% yeast extract, 0.5% glucose and 30 mg/L chloramphenicol. The media were sterilized at 121 °C for 30 minutes before chloramphenicol addition. Cultures of 10 mL were grown for three days at 22 °C in 50-mL Erlenmeyer flasks on a rotary shaker at 100 rpm (GFL Orbital Shaker 3017, Gesellschaft für Labortechnik mbH, Burgwedel, Germany). In all experiments, the relative yeast growth was evaluated by optical densities at 600 nm (OD600nm). Inoculation was performed by adding 50 µL of R. mucilaginosa inoculum at 2.4 OD600nm (containing approximate 0.3 × 107 cells) in 10 mL culture media. Before running the experimental tests described in this study, preliminary studies involving OD600nm and Raman profile monitoring were performed to create a growth chart for our experimental conditions, as well as measuring pH changes during the growth of the yeast until maturation.
Fungal molecular confirmation
The DNA of the R. mucilaginosa isolate obtained on SDA control media was extracted using the Animal and Fungi DNA Preparation Kit® (Jena Bioscience) according to the manufacturers’ instructions. The ITS1 and ITS2 primer sets targeting the ITS (Internal Transcribed Spacer) region were used for identification46. For PCR amplification, the following mixture was used in a 25 µL final volume: 5 µL of 5X MangoTaq Colored Reaction Buffer (Bioline), 1.25 µL of 50 mM MgCl2 (Bioline), 0.5 µL of 10 mM dNTP (Bioline), 1.25 µL of each primer (Macrogen Sequencing Service, Korea), 0.25 µL of 5 U/µL MangoTaq (Bioline) and 2 µL of DNA. Negative control samples were used for each pair of primers. The amplification series consisted of 35 cycles of the following: 95 °C for 30 s, 56 °C for 30 s and 72 °C for 30 s. The fragment obtained after amplification was sequenced at Macrogen (Macrogen Sequencing Service, Korea).
Antifungal product effects
Two pharmaceutical antifungal products, Exoderil (containing 10 mg/mL of naftifine as active compound, Sandoz GmbH Kundl Austria) and Canespor (containing 10 mg/mL of bifonazole as active compound, KVP Pharma + Veterinär Produkte GmbH, Kiel, Germany), were tested against R. mucilaginosa. Minimum inhibitory concentrations (MIC50), the minimum concentrations of compound required to inhibit 50% of the population in terms of OD600nm values, for both antifungal agents was determined in liquid media and expressed in terms of the active compound concentration, followed by comparison with available data41. The tested concentrations were 0, 0.005, 0.01, 0.025, 0.05, 0.1, 0.2, 0.6, 1.2, 3.6, 10.8, 50, 100, 500, and 1000 mg/L for naftifine and 0, 0.015, 0.03, 0.06, 0.12, 1, 10, 50, 100, 500, and 1000 mg/L for bifonazole, respectively. Experiments were performed in duplicate.
The R. mucilaginosa strain isolated from the SDA control media was used as the inoculum for naftifine effects evaluation. The fungal depigmented colonies were obtained by using inoculum from control R. mucilaginosa isolate, exposed for 10 minutes to 1 mg/mL naftifine solution diluted with 20% ethanol, before application to nutritive medium, as inspired by practice recommendations of naftifine hydrochloride dosage and administration47. In another experimental variant, the R. mucilaginosa depigmented colonies, illustrated by photos, were obtained by including naftifine in small concentrations (0.1 mg/mL, 0.2 mg/mL) in SDA, before inoculation (Fig. 1C). The R. mucilaginosa repigmented colonies were obtained from fungal depigmented colonies by inoculation on SDA (Fig. 1D). All fungal colonies obtained on SDA in Petri dishes by method of triple cultures were incubated at 22 °C for 3 days, and all the experiments were performed in triplicate.
The concentration of 1.2 mg/L naftifine in YPD medium was chosen for subsequent investigations. An inoculum of 20 µL was used for 10 mL culture media. Two sets of experiments were carried out for R. mucilaginosa inoculated in YPD medium without naftifine and with naftifine. R. mucilaginosa previously treated with 1.2 mg/L naftifine was also used for inoculation in YPD medium without naftifine. For the first experiment, after inoculation the Erlenmeyer flasks were exposed to UV radiation for 40 minutes in a biological safety cabinet. For the second (hypoxic) experiment, flasks inoculated with R. mucilaginosa were sealed after argon was bubbled for 60 s in each flask. These tests were performed in quintuplicate. The results were reproduced in independent trials.
Carotenoid evaluation using UV-vis and resonant Raman spectroscopies
Cells from 2 mL of homogenous suspension were harvested and washed three times with 1 mL 0.9% NaCl solution; 100 µL of the final suspension in saline solution were used for Raman spectroscopy, while the other 900 µL were centrifuged and the saline solution was discarded. The pellet was resuspended in 1.8 mL of DMSO, immediately followed by vigorous shaking for complete cell disruption (transformation of the suspension into a clear solution) and carotenoid extraction. The clear solution was further centrifuged at 20000 g speed for cell debris deposition. The UV-vis spectra of the clear supernatant were measured between 200 and 800 nm using a Varian Cary 5000 UV-Vis-NIR Spectrophotometer (Agilent Technologies). Absorbance at 512 nm (maximum wavelength for these samples) was taken as a marker of the total carotenoid content.
Resonant Raman spectra were recorded using a confocal Renishaw inVia Reflex Raman Spectrometer at 5% of the 200 mW Cobolt Diode Pumped Solid State (DPSS) laser, emitting at 532 nm. The Raman back-scattered light was directed to a spectrometer equipped with 1800 lines/mm grating and a CCD detector. The spectral resolution was ~4 cm−1. The instrument was calibrated prior to each experiment using a silicon sample as an internal standard. Cells in saline buffer were applied to poly-L-lysine adhesive slides (Polysine™ Microscope Adhesion Slides, Erie Scientific via VWR) for efficient sample immobilization. The laser was focused on the sample using a 100x objective, and the spectra were recorded with 10-s exposure times and 2 accumulations. The experiments were done in triplicate.
Resonant Raman maps were obtained by raster-scanning the selected area with the 532-nm laser, focused on the sample using a 5x objective. The laser power was set to 10%. The exposure time for each spectrum was 2 s and the measurement step was 200 µm. The Raman signal contours map, as a function of the Raman intensity, is shown in Fig. 4C.
Chromatographic analysis
Both HPLC and HPTLC approaches were used for separating and evaluating the relative quantity of the carotenoids and ergosterol in R. mucilaginosa after DMSO cell disruption. An Agilent 1200 HPLC system (Waldbronn, Germany) equipped with an on-line vacuum degasser, quaternary pump, temperature-controlled sample tray, automatic injector, column thermostat compartment and DAD detector was employed, with a Nucleosil 100 C18 column (240 mm × 4.6 mm, 5 µm particle size) from Macherey-Nagel (Duren, Germany). The injection volume was 100 µL (0.2 µm filtered DMSO extract), the column temperature was set to 25 °C and the flow rate was 1.2 mL/minute. As a standard, β-carotene at a concentration of 1 mg/mL was used. An isocratic elution was employed using acetonitrile:methanol:isopropanol at 85:10:5, as previously described27. UV-Vis detection of compounds was accomplished using the DAD detector to measure the entire spectrum in the 190–700 nm range every 1 s, and the chromatograms were monitored at 254, 450, 490 and 512 nm. The chromatograms were exported and the graphs were developed in Excel and Origin 6, where the integration of the peaks was also performed. HPTLC was performed on HP silica gel plates (Silica gel 60, Merck, Darmstadt, Germany) using hexane-acetone-methanol (10:15:75 v/v/v) as the mobile phase.
Electron microscopy
R. mucilaginosa cells were examined using scanning electron microscopy (SEM) with a JEOL JSM 5510 LV electron microscope and transmission electron microscopy (TEM) with a JEOL JEM 1010 electron microscope (Japan Electron Optics Laboratory Co., Tokyo, Japan). The samples were fixed in 2.7% glutaraldehyde (in phosphate-buffered saline for 90 minutes). For SEM, the samples were critical-point dried in liquid CO2, mounted on sticky carbon tabs and sputter-coated with gold (10 nm). For TEM, the fixed and dried samples were infiltrated with resin (Epon 812), deposited onto colloidal-carbon-coated copper grids and negatively stained with lead citrate and uranyl acetate48. The chemicals needed for electron microscopy experiments were purchased as follows: glutaraldehyde, resin (Epon 812), lead citrate, uranyl acetate, bismuth subnitrate (Electron Microscopy Sciences, Fort Washington, USA); sticky carbon tabs, colloidal carbon coated grids (Agar Scientific, Cambridge, England). Details regarding the sample work protocol for electron microscopy were presented in other reports49.
Data analysis
Statistical analysis was performed using Statistica 7.0 for Windows (Stat-Soft, Inc., USA) and Excel. Principal component analysis (PCA) was applied to several data sets as indicated in the text using Statistica 7.0. T-tests were used to test the strength of association within the data. Where appropriate, using p < 0.05 as threshold for statistical significance, a statistical approach was formulated and the experimental data were also evaluated using one-way analysis of variance (ANOVA). The statistical parameters confirm the hypothesis that the differences between the results are either not significant (p > 0.05), significant (0.001 < p < 0.05) or highly significant (p < 0.001). The average of multiple measurements (duplicates, triplicates or more, depending on type of measurement) was used for plots, and the error bars are the standard error of the mean. For MIC determination, Origin 6 was used to fit the data and determine the MIC50.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Faergemann, J. & Baran, R. Epidemiology, clinical presentation and diagnosis of onychomycosis. Br J Dermatol 1–4 (2003).
Thomas, J. et al. Toenail onychomycosis: an important global disease burden. J Clin Pharm Ther 35, 497–519 (2010).
Westerberg, D. P. & Voyack, M. J. Onychomycosis: Current trends in diagnosis and treatment. Am Fam Physician 88, 762–70 (2013).
Azambuja, C. V. D. A., Pimmel, L. A., Klafke, G. B. & Xavier, M. O. Onychomycosis: clinical, mycological and in vitro susceptibility testing of isolates of Trichophyton rubrum. An Bras Dermatol 89(4), 581–586 (2014).
Cengiz, F. P. et al. Etiology of onychomycosis in patients, Turkey. J Am Podiatr Med Assoc, doi:https://doi.org/10.7547/16-139 (2017).
Velasquez-Agudelo, V. & Cardona-Arias, J. A. Meta-analysis of the utility of culture, biopsy, and direct KOH examination for the diagnosis of onychomycosis. BMC Infect Dis 17(1), 166, doi:https://doi.org/10.1186/s12879-017-2258-3 (2017).
Welsh, O. et al. Onychomycosis. Clin Dermatol 28, 151–9 (2010).
Martínez-Herrera, E. O., Arroyo-Camarena, S., Tejada-García, D. L., Porras-López, C. F. & Arenas, R. Onychomycosis due to opportunistic molds. An Bras Dermatol 90(3), 334–337 (2015).
da Cunha, M. M. L., dos Santos, L. P. B., Dornelas-Ribeiro, M., Vermelho, A. B. & Rozental, S. Identification, antifungal susceptibility and scanning electron microscopy of a keratinolytic strain of Rhodotorula mucilaginosa: a primary causative agent of onychomycosis. FEMS Immunol Med Microbiol 55, 396–403 (2009).
Zhou, J., Chen, M., Chen, H., Pan, W. & Liao, W. Rhodotorula minuta as onychomycosis agent in a Chinese patient: first report and literature review. Mycoses 57, 191–195 (2014).
Uludag Altun, H., Meral, T., Turk Aribas, E., Gorpelioglu, C. & Karabicak, N. A case of onychomycosis caused by Rhodotorula glutinis. Case Rep Dermatol Med 2014, 563261 (2014).
Miceli, M. H. et al. Emerging opportunistic yeast infections. Lancet Infect Dis 11, 142–151 (2011).
Wirth, F. & Goldani, L. Z. Epidemiology of Rhodotorula: An emerging pathogen. Interdiscip Perspect. Infect Dis 2012, 1–7 (2012).
Nunes, J. M., Bizerra, F. C., Ferreira, R. C. E. & Colombo, A. L. Molecular identification, antifungal susceptibility profile, and biofilm formation of clinical and environmental Rhodotorula species isolates. Antimicrob Agents Chemother 57, 382–389 (2013).
Nagahama, T., Hamamoto, M., Nakase, T., Takami, H. & Horikoshi, K. Distribution and identification of red yeasts in deep-sea environments around the northwest Pacific Ocean. Antonie Van Leeuwenhoek 80, 101–110 (2001).
Cordeiro, R. A. et al. Isolation of pathogenic yeasts in the air from hospital environments in the city of Fortaleza, northeast Brazil. Braz J Infect Dis 14, 30–34 (2010).
D D Almeida, G. M. et al. . Rhodotorula spp. isolated from blood cultures: clinical and microbiological aspects. Med Mycol 46, 547–556 (2008).
Savini, V. et al. Femoral prosthesis infection by Rhodotorula mucilaginosa. J Clin Microbiol 46, 3544–3545 (2008).
Gomez-Lopez, A., Mellado, E., Rodriguez-Tudela, J. L. & Cuenca-Estrella, M. Susceptibility profile of 29 clinical isolates of Rhodotorula spp. and literature review. J Antimicrob Chemother 55, 312–316 (2005).
Frengova, G. I. & Beshkova, D. M. Carotenoids from Rhodotorula and Phaffia: yeasts of biotechnological importance. J Ind Microbiol Biotechnol 36, 163–180 (2009).
Ellis, R. A. Principles and techniques of electron microscopy: biological applications. Cell 17(1), 235–236 (1979).
Hoog, G. S. de. Atlas of Clinical Fungi. (Centraalbureau voor Schimmelcultures, 2000).
Ellis, D., Davis, S., Alexiou, H., Handke, R., Bartley, R. Descriptions of Medical Fungi. 125-129 (Nexus Print Solutions, 2007).
Mata-Gómez, L., Montañez, J., Méndez-Zavala, A. & Aguilar, C. Biotechnological production of carotenoids by yeasts: an overview. Microb Cell Fact 13, 12 (2014).
Buzzini, P. et al. Carotenoid profiles of yeasts belonging to the genera Rhodotorula, Rhodosporidium, Sporobolomyces, and Sporidiobolus. Can J Microbiol 53, 1024–1031 (2007).
El-Banna, A. A., El-Razek, A. M. A. & El-Mahdy, A. R. Some factors affecting the production of carotenoids by Rhodotorula glutinis var. glutinis. Food Nutr Sci 3, 64–71 (2012).
Moliné, M. et al. Photoprotection by carotenoid pigments in the yeast Rhodotorula mucilaginosa: the role of torularhodin. Photochem Photobiol Sci 9, 1145–1151 (2010).
Moore, M. M., Breedveld, M. W. & Autor, A. P. The role of carotenoids in preventing oxidative damage in the pigmented yeast. Rhodotorula mucilaginosa. Arch Biochem Biophys 270, 419–31 (1989).
Goodwin, T. W. The Biochemistry of the Carotenoids 1–32 (Springer Netherlands, 1980).
Chen, F. et al. Small-molecule targeting of a diapophytoene desaturase inhibits S. aureus virulence. Nat Chem Biol 12, 174–179 (2016).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–402 (1997).
Moliné, M., Libkind, D. & van Broock, M. Production of torularhodin, torulene, and β-carotene by Rhodotorula yeasts. Method Mol Biol 898, 275–283 (2012).
Jehlička, J., Edwards, H. G. M. & Oren, A. Bacterioruberin and salinixanthin carotenoids of extremely halophilic Archaea and Bacteria: A Raman spectroscopic study. Spectrochim Acta Part A Mol Biomol Spectrosc 106, 99–103 (2013).
Geßner, R. et al. Identification of biotic and abiotic particles by using a combination of optical tweezers and in situ Raman Spectroscopy. Chem Phys Chem 5, 1159–1170 (2004).
Perrier, V., Dubreucq, E. & Galzy, P. Fatty acid and carotenoid composition of Rhodotorula strains. Arch Microbiol 164, 173–179 (1995).
Boekhout, T. & Kurtzman, C. P. Nonconventional Yeasts in Biotechnology 1–81 (Springer Berlin Heidelberg, 1996).
Kreger-Van Rij, N. J. W. & Veenhuis, M. A comparative study of the cell wall structure of Basidiomycetous and related yeasts. J Gen Microbiol 68, 87–95 (1971).
Mares, D. Ultrastructural and cytochemical study of Rhodotorula glutinis in the main growth phases. Mycopathologia 80, 179–188 (1982).
Gupta, A. K., Ryder, J. E. & Cooper, E. A. Naftifine: A Review. J Cutan Med Surg 12, 51–58 (2008).
Ghannoum, M. et al. In vitro antifungal activity of naftifine hydrochloride against dermatophytes. Antimicrob Agents Chemother 57, 4369–72 (2013).
Carrillo-Muñoz, A. J., Tur-Tur, C., Bornay-Llinares, F. J. & Arévalo, P. Comparative study of the in vitro antifungal activity of bifonazole, naftifine and sertaconazole against yeasts. J Chemother 11, 187–190 (1999).
Berg, D., Regel, E., Harenberg, H. E. & Plempel, M. Bifonazole and clotrimazole. Their mode of action and the possible reason for the fungicidal behaviour of bifonazole. Arzneimittelforschung 34, 139–46 (1984).
Mesa-Arango, A. C. et al. The production of reactive oxygen species is a universal action mechanism of amphotericin B against pathogenic yeasts and contributes to the fungicidal effect of this drug. Antimicrob Agents Chemother 58, 6627–6638 (2014).
Zaas, A. K. et al. Risk of fungemia due to Rhodotorula and antifungal susceptibility testing of Rhodotorula isolates. J Clin Microbiol 41, 5233–5 (2003).
Atlas, R. M. Handbook of Microbiological Media (CRC Press, 2010).
White, T. et al. Amplification and Direct Sequencing of Fungal Ribosomal RNA Genes for Phylogenetics. PCR Protocols: a Guide to Methods and Applications (Academic Press, 1990).
Naftifine Hydrochloride Monograph for Professionals https://www.drugs.com/monograph/naftifine-hydrochloride.html (accessed: 28th March 2017).
Hayat, M. A. Principles and Techniques of Electron Microscopy: Biological Applications. (Cambridge University Press, 2000).
Pârvu, M. et al. Chemical composition of celandine (Chelidonium majus L.) extract and its effects on Botrytis tulipae (Lib.) Lind fungus and the tulip. Not Bot Horti Agrobot Cluj-Napoca 41, 414–426 (2013).
Naewbanij, M., Seib, P. A., Burroughs, R., Seitz, L. M. & Chung, D. S. Determination of ergosterol using thin-layer chromatography and ultraviolet spectroscopy. Cereal Chem (1984).
Acknowledgements
This work was partly supported by CNCSIS/UEFISCDI [grant PN-II-RU-TE-2014-4-2555] and partly by TeMATIC-Art, project co-financed by FEDR through Competitiveness Operational Programme 2014–2020, Funding contract: 14/01.09.2016.
Author information
Authors and Affiliations
Contributions
A.C.M., M.P. and R.S.D. conceived and designed the experiments, A.C.M. performed chromatographic, UV-vis and statistical analysis and analysed the data, A.E.P., N.E.D. and N.L. performed the Raman measurements, performed the electron microscopy studies and analysed the data, O.R.C. and C.M. performed fungal culture growing and molecular confirmation, A.C.M. and M.P. wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Moț, A.C., Pârvu, M., Pârvu, A.E. et al. Reversible naftifine-induced carotenoid depigmentation in Rhodotorula mucilaginosa (A. Jörg.) F.C. Harrison causing onychomycosis. Sci Rep 7, 11125 (2017). https://doi.org/10.1038/s41598-017-11600-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-11600-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
