
Abstract
The Pacific whiteleg shrimp, Litopenaeus vannamei, is the most farmed aquaculture species worldwide with global production exceeding 3 million tonnes annually. Litopenaeus vannamei has been the focus of many selective breeding programs aiming to improve growth and disease resistance. However, these have been based primarily on phenotypic measurements and omit potential gains by integrating genetic selection into existing breeding programs. Such integration of genetic information has been hindered by the limited available genomic resources, background genetic parameters and knowledge on the genetic architecture of commercial traits for L. vannamei. This study describes the development of a comprehensive set of genomic gene-based resources including the identification and validation of 234,452 putative single nucleotide polymorphisms in-silico, of which 8,967 high value SNPs were incorporated into a commercially available Illumina Infinium ShrimpLD-24 v1.0 genotyping array. A framework genetic linkage map was constructed and combined with locus ordering by disequilibrium methodology to generate an integrated genetic map containing 4,817 SNPs, which spanned a total of 4552.5 cM and covered an estimated 98.12% of the genome. These gene-based genomic resources will not only be valuable for identifying regions underlying important L. vannamei traits, but also as a foundational resource in comparative and genome assembly activities.
Similar content being viewed by others
Introduction
Breeding programs for animal production species have traditionally been developed around phenotypic selection in conjunction with quantitative genetic theory. As with other realms of biology, animal production science is currently in the midst of a genomics revolution and there has been an increasing global focus on the development of genomic resources and subsequent identification of markers linked to genes of economic importance. Although still in its infancy as a production industry, aquaculture is perfectly situated to uptake recent advances in quantitative genetics and to integrate new genomic technologies into future breeding program designs1.
The globally important whiteleg shrimp or Pacific white shrimp, Litopenaeus vannamei, is an aquaculture species that would benefit substantially from the integration of genomic information into traditional breeding programs, particularly for disease resistance and other difficult to measure or low heritability traits. Unfortunately, even though several genetic linkage maps have been produced2, 3, comprehensive genomic information available for L. vannamei is still very limited and there is currently a poor understanding of fine scale genome structure and the genetic basis underlying complex commercially important traits. For example, current breeding programs for L. vannamei use traditional phenotypic selection to produce shrimp with improved growth and resistance to various viral pathogens like Taura syndrome virus (TSV)4, 5. While this traditional approach has been moderately successful in producing more productive shrimp strains, genetic progress using multi-trait phenotypic selection in L. vannamei has been significantly impeded by unfavourable genetic correlations between growth and disease resistance4, 6, as well as a poor correlated response in susceptibility to multiple diseases7,8,9. In light of these unfavourable genetic correlations between traits of interest in L. vannamei (i.e. growth and disease), breeding strategies would benefit from the integration of genetic markers tightly associated with trait variation (i.e. quantitative trait loci - QTL). The development of single nucleotide polymorphism (SNP) marker panels with the power to simultaneously identify genome-wide QTLs for complex and/or correlated traits would assist shrimp breeding strategies, as it would allow for the improved identification of selection candidates possessing advantageous genes. This would negate the current requirement for multiple selection lines and allow selection decisions for traits to be made directly on candidates, thereby increasing the accuracy of selection and resultant genetic gains. Despite recent increased research effort into L. vannamei genomics which have yielded SNP markers and moderate density linkage maps2, 3, 10,11,12,13,14, limited gene-based (Type-I) genomic resources are publically available, and production trait architecture and localisations are based on low density maps containing either AFLPs or microsatellites12, 14, 15. Therefore, there is still a need to develop comprehensive genome-wide SNP marker panels and dense genomic maps that allow the simultaneous detection of genome-wide QTLs for commercially important traits.
SNPs derived within expressed sequence tags (ESTs) which originate from gene coding and 3′-UTR regions are considered a valuable resource useful in linking genotype to phenotype. Anchoring EST-derived SNPs and their associated transcript sequences to a high density genomic map not only allows insights into genome structure and marker spacing across the genome, but also helps identify the biochemical pathways underlying traits of interest. This study aimed to extend on the current genomic resources available for L. vannamei by developing a gene-based commercial SNP array and genomic linkage map, demonstrate the placement of additional markers using novel locus ordering by linkage disequilibrium (LODE) methods16, 17, and describe the genome synteny between two commercially valuable penaeid species. The gene-based Type-I SNP marker panel and comparative genomic maps will be valuable resources for investigating genome-wide genetic trait associations, creating optimal marker sets for selective breeding and genomic prediction, understanding functional biology and genome evolution, and assisting in genome assembly.
Methods
Sequencing, assembly and annotation
To enable the identification and development of genome-wide Type I SNPs, total RNA was extracted from the tail muscle tissue of 30 L. vannamei individuals representing prominent domesticated industry lines (Global Gen, Indonesia), using TRIZOL® Reagent (Life Technologies). RNA from each individual were pooled together in equimolar amounts before being converted to double stranded cDNA using the Mint cDNA synthesis kit (Evrogen), and normalised using the Trimmer cDNA normalisation kit (Evrogen). Normalised cDNA was then sequenced using an Illumina GA-IIX genome analyser, which produced approximately 25 gigabases of 76 bp paired-end EST sequence data (~10× genome coverage). Illumina sequence adaptors and primers were screened and removed using the software Seqclean (https://sourceforge.net/projects/seqclean/). MOTHUR was used to remove sequences with an average quality score (Phred score) less than 15 (window size = 10 bp) and/or shorter than 50 bp in length18. The cleaned sequence data was assembled using Velvet V1.019 and OASES20. Assembly parameters consisted of no extra gap penalty with all other options at default or recommended settings. Transcript assemblies were conducted at kmer lengths of k39, k41, k43, k45, k47, k49, k51 and k53 before being clustered together at a 90% sequence identify threshold using the software CD-HIT21. Where multiple transcript sequences were identified, only the longest sequence was retained. Transcript redundancy removal was undertaken, since it is a requirement for SNP discovery.
Sequence annotation of Gene Ontology terms
Annotation of the assembled sequence database was achieved using a Blastx search algorithm22 and the NCBI non-redundant protein database conducted through the software package Blast2GO23. Where multiple annotations were returned, the one with the best bit score was retained. For each successfully annotated contig, gene ontology (GO) terms InterPro scan results were retrieved using Blast2GO.
SNP discovery and filtering
To ensure high-quality SNPs were produced, strict data integrity measures were implemented. Genome-wide SNPs were identified using stringent SNP discovery filtering within the software package SAMTOOLs24 and custom scripts. NOVACRAFT (Novocraft Technologies, Selangor, Malaysia) was used to align the cleaned sequence reads to the full sequence assembly. The SAMTOOLs pileup command was used to produce mapping qualities. The varFilter option in SAMTOOLs was employed to filter SNPs, keeping only the most informative [i.e. minimum minor allele frequency (MAF) of 0.25, a minimum read depth of 10 reads, a minimum of two minor allele reads, a minimum SNP mapping quality of 25, a minimum flanking sequence quality of 25]. Any SNP identified within 50 bp of a candidate SNP was excluded to ensure a conservative flanking region for probe design. In addition, multi-allelic SNPs and SNPs requiring type I Illumina Infinium Probes (A/T or C/G) were removed and sequence repeat elements were masked. The resultant SNPs with the highest MAF and read depth were prioritised and submitted for assay development analysis using Illumina’s Assay Design Tool (ADT). Any SNP that returned an ADT score of less than 0.7 was excluded from the array. To ensure no unintentional duplicate SNPs were included on the array, probes for each SNP were mapped to the initial assembly using NOVOCRAFT (Novocraft Technologies, Selangor, Malaysia) and only the probes that mapped uniquely were included in the array. Following this procedure, 8,967 SNPs (8,616 novel SNPs with the highest ADT score and 351 from the public domain including those mapped in Du et al.3 and Ciobanu et al.11) were incorporated into the Illumina Infinium ShrimpLD-24 v1.0 SNP genotyping array (Table 1 and Supplementary Table S1).
Infinium array genotyping
To validate the performance of the L. vannamei Illumina Infinium ShrimpLD-24 v1.0 beadchip, 2,004 samples were genotyped, including 1,134 female and 193 male broodstock that produced families, along with 677 nauplii DNA pools (pools of >300 nauplii larvae from an individual family). For some nauplii pools, one of the two parents was either unknown or not sampled. Consequently for these families, the full unknown parental genotype was reconstructed using methods described in Supplementary Methods and Peiris, et al.25. All families were raised indoors in a Nucleus Breeding Centre under biosecure conditions from founding individuals representing most of the prominent industry domesticated/selected lines. To ensure all genotypes calls were genuine and to identify aberrant SNPs and DNA samples, strict genotypic data integrity was undertaken in GenomeStudio V2011.1 following methods outlined in Jones, et al.26. Family groups were reconstructed using SNP genotypic data (as described below) to enable the assessment of Mendelian inheritance (MI) of alleles. Genotype reproducibility between batches and across arrays was tested using 52 replicate samples and 26 replicate SNPs.
Genomic DNA was extracted either from the 2,004 L. vannamei samples or pools using a modified CTAB protocol27. DNA was standardised to 50 ng/µl using PicoGreen dsDNA quantification (Invitrogen), while DNA quality was inspected by agarose gel electrophoresis. All array genotyping was undertaken at PathWest Medical Laboratories, Perth, Western Australia, following manufacturer instructions28. Genotypes were calculated within the genotyping module of Genome Studio V2011.1 (Illumina Inc.) using the GenTrain genotype clustering algorithm. A minimum GenCall (GC) score cut-off (quality metric for each genotype) of 0.15 was used in SNP genotype clustering. The proportion of loci that produced a genotype for a sample is the sample genotyping rate. The SNP conversion rate is defined as the number of SNPs that produced a genotype divided by the number of total SNPs included. SNP validation rates were calculated as the number of SNPs with a heterozygous call divided by the number of SNPs that produced a genotype. SNPs with a minor allele frequency of greater than 0.01 were considered polymorphic. Mendelian errors for each SNP were reported as in Mendelian agreement whereby;
All GenCall scores are reported as the 10th percentile of the GC scores (GC10 scores). All SNPs were investigated for conformation to expected Hardy-Weinberg Equilibrium (HWE) and Mendelian Inheritance (MI) patterns. All recorded pedigree information was validated in a number of subsequent iterations using the 1,800 highly reliable “first class” SNPs produced from the array and the parentage programs Cervus 3.029, 30 and COLONY31. Briefly, all individually genotyped females and male family relationships were confirmed using this integrated approach, whereby all maternal assignments were verified in COLONY (1,121), before being used to verify paternal assignments (750). Then using all validated parental relationships, COLONY was used to cluster pedigrees as an extra level of validation and to estimate unknown parents by inferring genotypes (N = 30). Any disagreements or pedigree alterations were resolved.
Linkage mapping families and map construction
After parental relationship validation and genotype reconstruction, a total of 631 progeny from 30 grandmaternal and 19 grandpaternal traced families were selected for linkage map construction (the number of progeny within a family ranged from 8 to 33; Supplementary Fig. S2 and Supplementary Table S3). The genotypic data of these individuals over all 6,379 high quality SNPs (as described below) was manually phased into hexadecimal encoding using custom scripts and linkage analysis was conducted in Carthagene V1.332, 33. Markers were segregated into linkage groups by the group function at a logarithm of odds (LOD) threshold of 10 and a distance threshold of 30 cM. Linkage groups were defined as groups of at least three markers ordered on a map at a LOD 3 threshold, and having agreement with independent linkage disequilibrium (LD) and LODE mapping assignment (as described below). The remaining 1,447 markers which did not have three markers ordered at a LOD 3 threshold and/or were not confirmed by LD and LODE analysis were designated as orphan markers. The defined linkage groups were subsequently constructed using a hierarchical approach whereby ordering was determined using consecutive thresholds of LOD3, LOD2 and the most likely marker position. For each consecutive threshold, maps were created using the buildfw function, followed by annealing, flips 6 and polish, until the best sex average map was produced. After all linkage groups were ordered, orphan markers were tested again using two-point to determine whether they could be inserted into any ordered linkage groups. In addition, the five distal markers from both ends of each linkage group were compared by two-point to identify if any linkage groups could be merged together. Sex specific maps were also produced by locking in the sex average marker order and re-calculating interval distances based on separate male and female informative recombination events. The Kosambi34 mapping function was used for all centi-Morgan (cM) calculations.
Sex- and family- specific recombination heterogeneity
To investigate sex-specific heterogeneity throughout independent linkage groups, the following goodness of fit heterogeneity test was utilised with one degree of freedom as described in Ott35 and Jones, et al.36;
where, \(Z({\hat{\theta }}_{m},{\hat{\theta }}_{f})\) is the joint sex-specific recombination rate and \(Z(\hat{\theta },\hat{\theta })\) represents the recombination rate when equal male and female recombination fractions are assumed. For each test, a false discovery rate (FDR) correction was applied to correct for multiple comparisons and minimise false positives37.
To detect any differences in sex-specific recombination rates, ratios of female-to-male map distances were calculated (R = X f /X m ) for each interval and linkage group as well as over the entire map. To ensure any observed sex-specific recombination was truly due to differences between the sexes, and not affected by variation in individual F1 parents, family specific heterogeneity was investigated for each F1 parent independently. LINKMFEX version 2.438 was used to calculate the recombination fraction, number of co-informative meiotic events (N) and the number of recombinations (r) for all mapped locus intervals for the maternal and paternal lines of each family separately. The Zmax score (LOD) was calculated for the mother and father in each family, and combined across all mothers and fathers respectively using methods outlined in Ott35. The following M-test was employed to investigate individual F1 recombination heterogeneity within each mapping family Ott35.
Here, \({Z}_{i}({\hat{\theta }}_{i})\) represents the LOD scores maximum likelihood estimation (MLE) for the ith F1 reference family for a pair of markers, with \(Z(\hat{\theta })\) being the total LOD score MLE of all ith reference families.
Segregation distortion
Segregation distortion was investigated to determine if there was any evidence of deviations from expected Mendelian Inheritance (MI) patterns. This was investigated using log-likelihood ratio tests for goodness of fit to Mendelian expectations on manually phased genotypic data across all markers from all dams and sires as described in Sokal and Rohlf 39 and Jones, et al.36.
The extent of linkage disequilibrium and integration of LODE-placed markers
Locus ordering by disequilibrium (LODE) is a novel methodology that allows the utilisation of additional linkage disequilibrium data to place unpositioned or orphan SNPs within genetic maps or scaffolds16, 17, 40, 41. The LODE procedure used in this study is an adaptation of the two step procedure described in Khatkar, et al.16. Firstly, SNPs are assigned to a chromosome or linkage group, then subsequently its position within this linkage group is estimated. Both of these steps rely on pair-wise estimates of linkage disequilibrium (LD). LD estimates (r 2 and D′) were computed among 6,379 SNPs and 1,963 individuals (631 individuals from mapping families, and 1,332 individuals representing prominent industry lines) using GOLD software42. The extent of LD among SNPs, within and across the linkage groups, was estimated using position of SNPs on the current linkage map. Placements of orphan SNPs using the LODE method were defined based on at least three pairs on a chromosome with r-squared 0.1 or more, but also looking at the maximum LD score.
Genome coverage
Genome coverage of the integrated linkage and LODE sex-average map was calculated using observed and expected genome lengths. The observed genome length (Goa) was calculated by adding the observed linkage group lengths. The expected genome length (Ge) was produced by multiplying the length (cM) of each linkage group by (m + 1)/(m − 1), where m is the number of loci in each linkage group43. The total expected genome length was then the sum of Ge from all linkage groups. Genome coverage (Coa), was calculated by dividing Goa by Ge44.
Comparative genome analysis
Syntenic relationships were explored for the integrated linkage and LODE map against three previously published maps, a L. vannamei SNP linkage map with 6,359 markers2, a L. vannamei SNP linkage map with 418 SNP markers3, and a Penaeus monodon linkage map with 3,959 SNPs45. Assignment of orthologous sequences were undertaken by reciprocal BLAST searches of contigs sequences from which SNPs were discovered in the present study against respective sequence databases available for the maps of Yu, et al.2, Du, et al.3 and Baranski, et al.45 (at an e-value threshold of >1e-5). Comparisons to Yu, et al.2 were undertaken using their contiguous sequences generated from their genome survey sequencing, bacterial artificial chromosomes (BACs) and marker sequences, whereas comparisons to Baranski, et al.45 were undertaken with the contig sequences associated with their mapped SNPs. The primary hit was retained in each case. In addition to sequence similarity search of the marker sequences published in Du, et al.3, 159 SNPs from a previously published low density SNP map3 were included on our genotyping array to allow the direct comparison of their linkage map to ours. Comparison of genome synteny in this case was undertaken by matching marker IDs of all SNPs from this current study that were directly genotyped and mapped with our integrated map. BLAST annotations to Daphnia pulex and Drosophila melanogaster for SNPs with common IDs were also carried across from Du, et al.3. Oxford Grids46 of the integrated map presented here versus Yu, et al.2, Du, et al.3 and Baranski, et al.45 were plotted using custom R scripts to confirm mapping position and illustrate genome synteny. An example linkage group (LG4) was drawn using ArkMap47 to illustrate genome conservation.
Data availability
The assembled contig sequences and mapped raw reads generated within the current study have been submitted to GenBank as a SRA database (Accession number: SRP094129). All SNPs included on the Illumina Infinium ShrimpLD-24 v1.0 array have been submitted to dbSNP on NCBI [Accession numbers: ss2137297825–ss2137306471 from the current study; rs159816077–rs159831399 mapped in Du et al.3; and rs142459135–rs142459627 developed in Ciobanu et al.11]. The Illumina Infinium ShrimpLD-24 v1.0 array is available from https://www.illumina.com/products/by-type/microarray-kits/infinium-shrimp-ld.html. All remaining data used and/or analysed during the current study are available from the corresponding author on reasonable request.
Results
Sequencing and assembly of transcripts
In total, over 25 Gb of sequence data (329 million raw EST sequences, 76 bp paired-end, ~10× genome coverage) was produced from an Illumina GA-IIx run. After clean-up and trimming, 19.7 Gb of sequence data was retained (average Phred score of 25.9). Assembly of the cleaned-up sequence data (including transcript redundancy removal) produced 76,963 contigs. The N50 of the assembly was 2,375 bp, the average contig length was 1,429 bp and median contig length was 955. Over 72% of the 76,963 contigs had a read depth coverage of greater than 50 reads (average read depth over all contigs was 2527.5 reads). The assembled contig sequences and mapped raw reads have been submitted to GenBank as a SRA database (Accession number: SRP094129). This is a significant genomic resource enabling the sequence data mining of 27,477 specific genes (see below) and in-silico detection of over 234,452 SNPs and 133,960 indels (Table 1).
Sequence annotation and gene ontology terms
Blastx searches against NCBI’s non-redundant protein database produced 30,317 hits from the 76,963 contigs. Of these sequences, 27,477 (24.7%) also had GO categories assigned, from which these genes were categorised into biological processes (21,333), molecular function (22,142) and cellular components (19,155) (Fig. 1 and Supplementary Table S4). Within the listed biological processes, most genes were involved in cellular and metabolic processes (32.7%). The most common molecular function designations were binding (43.6%) and catalytic activity (38.9%). Finally, cell (20.1%), cell part (20.0%) and organelle (15.2%) formed the most common GO terms within cellular component designations. A total of 12,957 unique gene hits were identified including Myosin and Myostatin/Growth Differentiation Factor-11, which are involved in muscle cell growth48, 49, as well as genes involved in immune response pathways such as apoptosis, MAPK signalling, toll-like receptor and antigen processing and presentation50.

Proportions of Gene Ontology (GO) annotations of the assembled 454 mantle tissue transcripts from Litopenaeus vannamei.
SNP discovery and development of commercial array
In-silico SNP discovery and filtering
From the assembled sequence dataset, 234,452 putative SNPs and 133,960 indels were identified in-silico before strict filtering parameters were applied. By filtering out all SNPs with a read depth less than 10 reads and a minor allele frequency (MAF) of less than 0.25, a total of 26,662 high-quality SNPs were identified. A further 2,445 multi-allelic SNPs, 4,565 SNPs requiring Type I Illumina Infinium probes and 1,054 highly repetitive SNP probes were removed before ADT analysis. Illumina’s ADT analysis calculates the effectiveness of the SNP probes on the array. A total of 1,142 SNPs did not return ADT values > 0.7 and 1,006 SNPs did not map to unique contigs and were removed. A further 7,003 SNPs were excluded due to being located within the flanking region of another SNP resulting in a final list of 9,447 SNPs. Of these, 8,967 SNPs (8,616 novel SNPs with the highest ADT score and 351 from the public domain including those developed in Ciobanu, et al.11 and mapped in Du, et al.3) were incorporated into the Illumina Infinium ShrimpLD-24 v1.0 SNP genotyping array enabling high throughput, cost effective and accurate genotyping (Table 1 and Supplementary Table S1). The average MAF and ADT score of these high-value SNPs was 37% and 0.95 respectively. All SNPs included on the Illumina Infinium ShrimpLD-24 v1.0 array have been submitted to dbSNP on NCBI (Accession numbers: ss2137297825 - ss2137306471 from the current study; rs159816077 - rs159831399 mapped in Du, et al.3; and rs142459135 - rs142459627 developed in Ciobanu, et al.11). The Illumina Infinium ShrimpLD-24 v1.0 array is available from https://www.illumina.com/products/by-type/microarray-kits/infinium-shrimp-ld.html.
Infinium array genotyping and validation
In total, 2,004 shrimp samples were genotyped, including 1,134 female and 193 male parents of families, along with 677 nauplii pools. From these samples, 70 individuals produced call rates of less than 90% and were subsequently removed from further analysis leaving 1,257 unique individuals to investigate SNP array performance. Analysis of the resulting genotypic data revealed that 6.01% of the SNPs did not amplify successfully (probe did not bind to the DNA) and 13.04% of the SNPs returned ambiguous clusters. From the resulting 7,259 SNPs, the SNP conversion rate was calculated to be 80.95%. Within the converted SNPs, 318 SNPs did not return heterozygous genotype calls and therefore were considered monomorphic. After the removal of the monomorphic SNPs, 6,941 remained resulting in a SNP validation rate of 95.62%. To estimate the proportion of informative or polymorphic SNPs, within this experimental population, a further 562 SNPs with deviations from HWE and MI errors were excluded, resulting in 6,379 SNPs (87.88%) with minimal errors (Table 2). Further stringent data integrity (i.e. excluding SNPs with a MAF < 0.01, SNP duplication, or low call rates) resulted in the exclusion of an additional 323 SNPs (Table 2). From the final dataset of 6,056 high quality SNPs, the SNP call rate was extremely high (98.92%) and the Mendelian inheritance concordance exceeded 99.9%. The average minor allele frequency of these high-value SNPs was 0.37. Summary statistics for all SNPs included on the array are included in Supplementary Table S1. A total of 52 replicate samples were included to evaluate final array genotyping performance. No major deviations between replicate samples were observed, resulting in sample concordance exceeding 99.9%. This provides strong support for highly reliable genotypic data across all validated SNPs.
Linkage map construction and LODE integration
A total of 708,209 phase known informative meiotic events were utilised to place and order SNPs across linkage groups. The average number of informative events per mapped locus was 147.02 (ranging from 4 to 444) compared to an average of 28.30 informative meiotic events for unmapped markers. A total of 4,370 SNPs were successfully mapped to their most likely position within the 44 linkage groups, which spans a total of 4,552.5 cM of the estimated sex-average 4,619.3 cM genome length, covering 98.12% of the L. vannamei genome. By utilising this linkage map in LODE analysis, an additional 447 markers were placed with high confidence. This integrated map (Build 1.2) contained a total of 4,817 SNPs which reduced the average marker interval across the genome to 0.97 cM, or 2.67 when all intervals of 0 cM were excluded (Fig. 2 and Table 3 and Supplementary Table S5). Linkage groups were ordered based on their total cM length.

Distribution of SNPs placed using linkage and LODE mapping methods across the 44 linkage groups.
Sex-specific and family-specific recombination heterogeneity
Sex-specific maps were also produced using the sex-average marker order to recalculate marker intervals based on 447,640 phase-known informative meiotic events for the female map and 260,569 phased known informative meiotic events for the male map. The total lengths of the female and male maps were 4,530.60 cM and 4,522.30 cM respectively (Table 3). Minimal differences in sex-specific recombination were observed throughout the linkage groups (Supplementary Fig. S6). However, LG23 and LG44 displayed slightly larger male maps and LG6, LG9, LG12, LG13 and LG24 displayed slightly larger female maps. Overall, the average female-to-male ratio was 1.02:1. The sex-specific log10 likelihood for each linkage group, averaged between the sexes ranged from −744.740 to −190.780 (average −516.333) and the total sex-specific log10 likelihood was −22,718.645. Cumulative cM distances of the sex average, female and male maps indicate that there is no pronounced pattern of sex-specific recombination throughout the 44 linkage groups (Supplementary Fig. S6).
Investigations into family specific heterogeneity also indicate minimal significant deviations throughout all maps after false discovery rate (FDR) corrections. Only one interval on the female map (66909_123–44494_691) and six intervals on the sex average map (75525_46–34150_668, 61146_423–57343_1404, 804_148–23736_451, 23736_451–24606_1440, 66909_123–44494_691 and 7007_119–48446_2196) returned significant recombination heterogeneity after FDR (χ 2 = 11.809–31.301, P = 0.00011–0.00059, df = 1–9).
Segregation distortion
A total of 540 significant segregation distortions were detected across all markers and families following FDR correction (Supplementary Table S7). The majority of these distortions (82.3%) were within families F1014, M1014 and F1002. As no significant family specific heterogeneity was detected for these distortions, they show no evidence of influencing calculations on mapping distances.
Extent of linkage disequilibrium
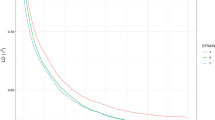
Linkage disequilibrium estimates declined gradually with increasing map intervals both throughout the genome and for each linkage group (Fig. 3 and Supplementary Table S8). This is accentuated by the relatively high mean r 2 values for SNP pairs less than 5 cM. Between the 4,817 adjacent SNP pairs, the mean r 2 estimates were 0.184 (with a median of 0.096).

Mean linkage disequilibrium (LD) estimates at different linkage map distances throughout the P. maxima genome for r 2 and D′.
Comparative genome analysis
Sequence similarity Blastn searches of assembled contig sequences to mapping sequence data published in Yu, et al.2 and Baranski, et al.45 returned 5,591 and 2,499 hits respectively. Of these hits, a total of 67 SNPs for Yu, et al.2 and 275 SNPs for Baranski, et al.45 were in common between our integrated and previously published maps (Supplementary Table S9). The small number of blast hit results to Yu, et al.2, is most likely due to the large number of contigs from which the SNPs originated in both studies (i.e. 7.4 million contigs from Yu, et al.2 and 76,963 from our transcriptome assembly), as well as the relatively low number of mapped SNPs (6,147 for Yu, et al.2 and 4,817 from our integrated map). Between these two studies, SNPs were also developed using different methodologies. Yu, et al.2 utilised genomic DNA sequencing and specific-locus amplified fragment sequencing (SLAF-seq), whereby the SNP discovery method utilised in the current study involved transcriptome sequencing, SNP identification and design which was optimal for developing Illumina custom array probes.
Of the 159 SNPs developed in Du, et al.3 and included on our genotyping array, 83 were assigned a map position in the integrated map (Supplementary Table S9). In addition, sequence similarity searches of our mapped contig sequences (from which the mapped SNPs were designed) to the marker sequences from Du, et al.3 returned 38 hits (evalue < 0.01, similarity >95%). Of these 38 marker sequence and contig matching pairs, 30 of the respective contigs were mapped adjacent to its pair confirming its placement and blast hit (average distance = 1.08 ± 2.1 cM).
The identified homologous sequences were used to identify homologous linkage groups across the independent maps (Figs 4, 5 and 6 and Supplementary Table S9). Comparing our integrated map to the previously published L. vannamei SNP map from Du, et al.3, 29 linkage groups were able to be matched. Marker order was highly conserved (Figs 4 and 7), although, six SNPs were placed on alternative linkage groups. Within this comparison, our integrated map indicates that LG11 and LG34 from Du, et al.3 may be able to be merged due to common SNPs on LG14 in the present map. In addition, the map produced in this study indicates that LG1 of Du, et al.3 may be a concatenation of two separate linkage groups. A total of 35 linkage groups from Yu, et al.2 were able to be matched to the linkage groups within the present integrated map (Fig. 5). Within the 67 common SNPs between these two maps, only two SNPs were placed on alternative linkage groups. However, homologous sequence matching indicates that LG7 and LG17, as well as LG26 and LG29 may be merged due to being mapped to LG35 and LG39 respectively in the present map. Comparisons to Baranski, et al.45 revealed 44 linkage groups matched by homologous sequences (Fig. 6). Marker synteny was highly conserved with only three markers being assigned to alternatively matched linkage groups (Figs 6 and 7). Both LG39 and LG43 from Baranski, et al.45 were matched to SNPs placed within LG2 reported here, which were placed with high significance (two-point LODs ranging from 10.8–39.6), indicating a potential merger.

Homologous linkage map relationships between the integrated linkage and LODE L. vannamei map and the L. vannamei linkage map produced in Du, et al.3. Each dot represents a homologous locus proportional to cM lengths (Kosambi).

Homologous linkage map relationships between the integrated linkage and LODE L. vannamei map and the L. vannamei linkage map produced in Yu, et al.2. Each dot represents a homologous locus proportional to cM lengths (Kosambi). Data from Yu, et al.2 was utilised under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).

Homologous linkage map relationships between the integrated linkage and LODE L. vannamei map and the P. monodon linkage map produced in Baranski, et al.45. Each dot represents a homologous locus proportional to cM lengths (Kosambi). Data from Baranski, et al.45 was utilised under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).

Demonstration of synteny analysis between LG4 of the integrated map, LG1 from Du, et al.3 and LG20 from Baranski, et al.45. Only matched markers are listed. Data from Baranski, et al.45 was utilised under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/).
Discussion
The rapid advancement of genetic and genomic technologies has made the generation of genome-wide genomic resources available for many non-model species. Genome-wide markers with known gene function and position are highly useful in comparative mapping studies, genome-wide association studies, and linking gene function to traits of interest. By using the high-throughput sequencing approach, this study provides over 234,452 putative in-silico SNPs and 26,662 filtered high-quality SNPs (MAF ≥ 0.25, read depth ≥ 10). A total of 8,967 high-utility SNPs were incorporated into a commercial array allowing cost-effective routine genotyping of validated content. In addition, 4,817 of these SNPs were placed within a moderate density integrated linkage and LODE map allowing insights into the genome structure of L. vannamei and comparisons to previously published genome maps in penaeids. These resources vastly improve the publically available genomic resources available for this important commercial species and have high-utility in studies aiming to identify genomic regions linked to traits of interest. Furthermore, the current integrated genetic map will help with forthcoming L. vannamei genome sequence assemblies, by providing robust gene-associated reference maps to anchor and orientate sequence data.
Highly reliable genotypes are integral to ensure the generation of integrated genetic maps and genome association studies are accurate. The success of Type I SNP assay development can be attributed to the quality of the EST sequence data used, sequence depth, in-silico MAF cutoff and SNP flanking region composition51, 52. By utilising a SNP mining approach within 25 Gb of assembled transcriptome-wide sequence data (~10× genome coverage), and applying strict SNP discovery filtering parameters (i.e. MAF of 0.25, read depth > 10, a minimum of two minor allele reads, a minimum flanking sequence quality of 25, and no observed variation in the flanking probe design region), we ensured that the SNPs reported within this study are of high utility and are dispersed throughout the genome. In addition, since all SNPs reported here were designed within large expressed contigs (N50 of 2,375 and average contig length of 1,429 bp) which are generally well conserved, they are not only known to be associated with functional genes, but will also be highly useful in ongoing comparative genomic analyses and genome sequence assemblies.
Standard measures of quantifying the success of genotyping arrays are the conversion (the proportion of SNPs producing genotypes) and validation (the proportion of SNPs that are polymorphic in a population) rates. Conversion and validation rates observed within this study (i.e. 80.95% and 95.62% respectively) were relatively high compared to Illumina genotyping arrays designed on other aquaculture species. Previous conversion and validation rate of Illumina panels from aquaculture species such as the silver-lipped pearl oyster (Pinctada maxima), the pacific oyster (Crassostrea gigas), European flat oyster (Ostrea edulis), rainbow trout (Oncorhynchus mykiss), Atlantic salmon (Salmo salar) and catfish (Ictalurus punctatus) range from 70.3–92.0% and 48.0–59.9% respectively26, 53,54,55,56,57. In comparison, the current L. vannamei array is very similar to the well-established Illumina livestock arrays such as the chicken, goat, bovine, porcine and the domestic horse which have conversation and validation rate that range from 88–97.5% and 78–99.1% respectively58,59,60,61,62. The current array was validated on a large number of samples distributed throughout the most prominent industry domesticated lines. This ensures that a high proportion of SNPs included on the commercial array will be polymorphic within the majority of farmed and wild populations of L. vannamei worldwide.
With the advent of genotype by sequencing (GBS) approaches for generating genotypic data, there has been a move away from solid state SNP genotyping arrays63. However, there are significant benefits of using solid state SNP arrays over GBS. For example, the laboratory techniques and procedures required to undertake genotyping as well as integral downstream bioinformatics analysis are much simpler and require less technical knowledge. As a result, genotyping arrays usually have a much quicker turnaround and are much more robust and less prone to errors64, 65. In addition, SNP genotyping arrays can be automated leading to higher reproducibility. Per sample, genotyping arrays are generally more expensive than GBS approaches, however, as long as the SNP arrays were designed with loci that are polymorphic within the populations for its intended use, there are many benefits that can be yielded over any GBS approach.
Assigning gene annotation and ontology terms to SNPs provide valuable insights into the functional biology and trait architecture particularly when coupled with location information within the genome. A total of 27,477 of the 76,963 contigs utilised in SNP discovery were annotated with one or more gene ontology terms. The proportion of annotated contigs reflects the still limited amount of annotated sequence data for decapods in the public domain. The major GO terms returned including cellular, metabolic and single-organism process in Biological Process; binding and catalytic activity in Molecular Function; and cell, organelle, membrane and macromolecular complexes in Cellular Components were all reflective of the proportions of GO terms returned within previous studies within penaeid transcriptome studies10, 66. In addition, the GO distribution patterns of protein coding genes and therefore gene compositions between the two shrimps was reported to be similar66.
Out of the 6,379 SNPs deemed suitable for linkage mapping, 4,370 were successfully placed via linkage analysis. An additional 447 SNPs were placed when integrating LODE methodologies resulting in a total of 4,817 SNPs mapped. The power of placing SNPs on a linkage map comes from the number of informative meiosis events within the reference mapping families. The average number of informative meiotic event across all mapped SNPs was 147.0, compared to 28.3, for all unmapped SNPs. This significant drop in the number of informative events is a major contributor to the ability to firstly assign a SNP to a linkage group, and finally order the markers unambiguously. In addition to the placement of markers, the number of informative meiotic events adds power to teasing apart the LD blocks or binning of markers placed at the same location on the linkage map. There were a number of clusters of co-localised markers within the map presented in this study (i.e. there were 4,817 markers places within 1,752 unique locations, indicating that on average, there were 2.75 markers co-localised throughout the genome). An increase in either the number of individuals per family, or the number of families should result in higher power to refine the position of these co-localised markers. Nevertheless, considering there were only minimal evidence of sex-specific recombination, family specific recombination and segregation distortion, the assignment of the SNP markers throughout the linkage mapping procedure is considered to be highly accurate.
To assign additional orphaned markers to the linkage map that were not assigned a position within linkage analysis, a locus ordering by disequilibrium (LODE) mapping procedure was implemented as described in Khatkar, et al.16. LODE methodologies rely on the linkage disequilibrium (LD) information from unrelated samples within a population and do not require specific resource populations or mapping families. A reliable estimate of r 2 (statistical measure of LD) can be successfully obtained from a minimum samples size of 75 unrelated individuals40. Within this study, a total of 1,963 individuals (from prominent domesticated industry lines) were included in LODE analysis which allowed the generation of accurate estimates of LD. In doing so, the LODE method was able to harness additional samples and linkage disequilibrium information (outside of linkage mapping families) that was useful to place 447 additional orphan SNPs.
The estimation of LD does have some assumptions and limitations. The precision of determining locations of orphan SNPs by this method depends on the extent of linkage disequilibrium and density of the framework map. Furthermore, the extent of linkage disequilibrium, among other factors, depends on population structure. Therefore the number of LODE placed SNPs within this study may be increased by utilising larger random mating wild populations (to stabilise genetic drift).
Using the observed length of the integrated linkage and LODE map, the expected genome length of L. vannamei was calculated to be 4,619.3 cM (sex average) and the estimated genome coverage is 98.12%. This is comparable to the sex-average linkage map from Yu, et al.2, which incorporated 4,817 loci and had a total genome map length of 4,341.39 cM and genome coverage of 98.39%. Previous linkage maps to this contained fewer loci and as such are less accurate and smaller in size (i.e. 3677.65–4025.5 cM; refs 12,13,14).
To date, no complete shrimp genomes have been published due to their large genome size (ranging from 2.17 Gb to 2.64 Gb) and high levels of duplication2, 66. In addition, previous comparative mapping in penaeid shrimps has been very limited, although, some comparative mapping and divergence times have been recently conducted in decapod shrimps within the Pancrustacea clade using transcriptome data66. By comparing homologous loci between L. vannamei and P. monodon, this study reports one of the first genomic comparisons for gene order and synteny within penaeids. Homology was investigated in two L. vannamei maps2, 3 and one P. monodon linkage map45.
Within the homologous loci between the L. vannamei linkage map published in Du, et al.3 and the integrated map within this study, the gene order is well conserved. In total, only six out of the 83 homologous loci were assigned to alternative linkage groups). Positional two-point LOD thresholds of five of the six SNPs calculated from the current map ranged from 4.5 to 27.1 (informative meiotic events ranging from 98 to 230) indicating high statistical support for their current assignments. Only one marker, LV1007 had a low two-point LOD placement threshold of 0 resultant from 70 informative meiotic events. With a higher density of markers and number of informative meiotic events in the current integrated map, the placement of the large majority of SNPs is expected to be more accurate. Similarly, only two out of the 67 common SNPs were assigned to alternative linkage groups when comparing the map presented in this study to the L. vannamei map published in Yu, et al.2. These two SNPs had high positional two-point LOD thresholds at 14.4 and 19.3, indicating strong support for their placement in the current map.
The total length of the female and male maps for P. monodon map were 4,060 cM and 2,917 cM respectively. Within P. monodon, the female map was 28% larger than the male map indicating greater recombination frequency in female over males. Even though large differences in recombination rate between sexes have been reported in other penaeids12, 13, 45, 67,68,69, it was not observed within the L. vannamei map reported in this study (female and male map length of 4,530.6 cM and 4,522.3 cM respectively). Sex-specific recombination is highly variable throughout existing reported penaeid maps, which may come down to the number of markers mapped to the respective sex maps, whereby various numbers of markers mapped for respective sexes could be influencing the recombination rates observed. The incorporation of many more markers, families and offspring (and therefore more total informative meiosis events) within the integrated map is expected to produce a more accurate estimation of sex-specific recombination rate than the existing maps.
P. monodon and L. vannamei share an identical chromosome number of 2n = 4413, 67. A comparison of the 44 LGs from our integrated map to the P. monodon map produced in Baranski, et al.45 returned substantial macrosynteny throughout the 275 homologous loci identified despite their estimated divergence of 95 million years ago66. Only three SNPs were assigned to alternative linkage groups, whereby the two-point LOD SNP placements for the three SNPs placed on different linkage groups ranged from 6.6 to 22.3 (with informative meiotic events ranging from 79 to 226). There is minor evidence of interchromosomal rearrangements and marker shuffling between the species [i.e. within linkage group pairs LG6 (L. vannamei) and LG14 (P. monodon); LG40 (L. vannamei) and LG22 (P. monodon); LG34 (L. vannamei) and LG41 (P. monodon); and LG15 (L. vannamei) and LG44 (P. monodon)], however, comparisons between a higher density of markers is required before inferences of chromosomal rearrangements can be made reliably. Overall, the robustness of the marker orders in all maps is demonstrated by the high correlations between marker orders across most linkage groups calculated from independent analysis. The sporadic marker disagreements may be due to either genotyping errors, or differences in the mapping algorithms used during map construction1.
Penaeid genomics has come a long way in the last 15 years. Many species now have significant genomic resources which will enable more advanced methods of breeding such as marker assisted and genomic selection70. These novel techniques may help increase disease resistance to specific emerging diseases which is a major priority for current shrimp breeding programs. It is predicted from simulated genetic advancement using genomic information in selection programs for survival and disease resistance was up to 2.6 times as effective than that of phenotypic sib-selection alone70. Furthermore, considering vaccination is not an option and management interventions to curve disease are usually unfeasible, several shrimp breeding programs have already been implemented in a number of countries to improve disease resistance as reviewed in Neira71, Rye72, and Castillo-Juárez, et al.70. With the continuing development of genomic resources in penaeids, incorporation of genomic information into breeding programs is a viable option promising to increase the accuracy of selection and therefore response compared to conventional selection70.
Developing a large set of type I genome-wide molecular markers and genomic maps for L. vannamei is a fundamental step towards further understanding the genomic structure and genetic contribution to commercially important traits. The development and validation of a large EST-derived SNP database and commercial genotyping array as described within this study will expedite the development and incorporation of genomic information into advanced selective breeding programs by enabling researchers to cost effectively genotype a large number of individuals within breeding programs. In addition, this study provides an integrated linkage and LODE map for L. vannamei, which revealed high macrosynteny between L. vannamei and P. monodon with only a small number of occurrences of inter chromosomal rearrangements. Combined, these data provide an important resource for genetic association studies, comparative genomics, and assisting in genome assemblies across several related shrimp taxa.
References
Yue, G. H. Recent advances of genome mapping and marker-assisted selection in aquaculture. Fish Fish. 15, 376–396 (2014).
Yu, Y. et al. Genome survey and high-density genetic map construction provide genomic and genetic resources for the Pacific White Shrimp, Litopenaeus vannamei. Sci. Rep. 5, 15612 (2015).
Du, Z. Q. et al. A gene-based SNP linkage map for pacific white shrimp. Litopenaeus vannamei. Anim. Genet. 41, 286–294 (2010).
Argue, B. J., Arce, S. M., Lotz, J. M. & Moss, S. M. Selective breeding of Pacific white shrimp (Litopenaeus vannamei) for growth and resistance to Taura Syndrome Virus. Aquaculture 204, 447–460 (2002).
Moss, S. M., Moss, D. R., Arce, S. M., Lightner, D. V. & Lotz, J. M. The role of selective breeding and biosecurity in the prevention of disease in penaeid shrimp aquaculture. J. Invertebr. Pathol. 110, 247–250 (2012).
Gitterle, T. et al. Response to multi-trait selection for harvest weight, overall survival, and resistance to white spot syndrome virus (WSSV) in Penaeus (Litopenaeus) vannamei. Aquaculture 272(Supplement 1), S262 (2007).
Moss, D. R., Arce, S. M., Otoshi, C. A., Doyle, R. W. & Moss, S. M. Effects of inbreeding on survival and growth of Pacific white shrimp Penaeus (Litopenaeus) vannamei. Aquaculture 272(Supplement 1), S30–S37 (2007).
Moss, S. M., Doyle, R. W. & Lightner, D. V. Breeding shrimp for disease resistance: challenges and opportunities for improvement. Diseases of Asian Aquaculture V. Fish Health Section. American Fisheries Society, Manila, Philippines, 379–393 (2005).
Wyban, J. A. Breeding for fast growth and virus resistance. Global Aquaculture Advocate 3, 32–33 (2000).
Yu, Y. et al. SNP discovery in the transcriptome of white Pacific shrimp Litopenaeus vannamei by next generation sequencing. PLoS One 9, e87218 (2014).
Ciobanu, D. C. et al. A major SNP resource for dissection of phenotypic and genetic variation in Pacific white shrimp (Litopenaeus vannamei). Anim. Genet. 41, 39–47 (2010).
Zhang, L. et al. A genetic linkage map of Pacific white shrimp (Litopenaeus vannamei): sex-linked microsatellite markers and high recombination rates. Genetica 131, 37–49 (2007).
Pérez, F., Erazo, C., Zhinaula, M., Volckaert, F. & Calderón, J. A sex-specific linkage map of the white shrimp Penaeus (Litopenaeus) vannamei based on AFLP markers. Aquaculture 242, 105–118 (2004).
Alcivar-Warren, A. et al. Shrimpmap: a low-density, microsatellite-based linkage map of the Pacific whiteleg shrimp, Litopenaeus vannamei: identification of sex-linked markers in linkage group 4. J. Shellfish Res. 26, 1259–1277 (2007).
Andriantahina, F., Liu, X. & Huang, H. Genetic map construction and quantitative trait locus (QTL) detection of growth-related traits in Litopenaeus vannamei for selective breeding applications. PLoS One 8, e75206 (2013).
Khatkar, M. S. et al. Assignment of chromosomal locations for unassigned SNPs/scaffolds based on pair-wise linkage disequilibrium estimates. BMC Bioinformatics 11, 171 (2010).
Khatkar, M. et al. In 9th World Congress on Genetics Applied to Livestock Production (German Society for Animal Science, Germany, 2010).
Schloss, P. D. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537 (2009).
Zerbino, D. R. & Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829 (2008).
Schulz, M. H., Zerbino, D. R., Vingron, M. & Birney, E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092 (2012).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Peiris, B. L., Ralph, J., Lamont, S. J. & Dekkers, J. C. M. Predicting allele frequencies in DNA pools using high density SNP genotyping data. Anim. Genet. 42, 113–116 (2010).
Jones, D. B., Jerry, D. R., Forêt, S., Konovalov, D. A. & Zenger, K. R. Genome-wide SNP validation and mantle tissue transcriptome analysis in the silver-lipped pearl oyster, Pinctada maxima. Mar. Biotechnol. 15, 647–658 (2013).
Sambrook, J. & Russell, D. W. Molecular Cloning: a Laboratory Manual. (Cold spring harbor laboratory press, 2001).
Steemers, F. J. & Gunderson, K. L. Whole genome genotyping technologies on the BeadArray™ platform. J. Biotechnol. 2, 41–49 (2007).
Kalinowski, S. T., Taper, M. L. & Marshall, T. C. Revising how the computer program cervus accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 16, 1099–1106 (2007).
Kalinowski, S. T., Taper, M. L. & Marshall, T. C. Corrigendum. Mol. Ecol. 19, 1512–1512 (2010).
Jones, O. R. & Wang, J. COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol. Ecol. Resour. 10, 551–555 (2010).
Schiex, T. & Gaspin, C. CARTHAGENE: constructing and joining maximum likelihood genetic maps. Proc. Int. Conf. Intell. Syst. Mol. Biol. 5, 258–267 (1997).
de Givry, S., Bouchez, M., Chabrier, P., Milan, D. & Schiex, T. CarthaGène: multipopulation integrated genetic and radiation hybrid mapping. Bioinformatics 21, 1703–1704 (2005).
Kosambi, D. The estimation of map distances from recombination values. Ann. Eugen. 12, 172–175 (1944).
Ott, J. Analysis of Human Genetic Linkage (Third Edition) (Johns Hopkins University Press, 1999).
Jones, D. B., Jerry, D., Khatkar, M., Raadsma, H. & Zenger, K. A high-density SNP genetic linkage map for the silver-lipped pearl oyster, Pinctada maxima: a valuable resource for gene localisation and marker-assisted selection. BMC Genomics 14, 810 (2013).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 57, 289–300 (1995).
Danzmann, R. (2006).
Sokal, R. R. & Rohlf, F. J. Biometry: the Principles and Practise of Statistics in Biological Research. 4th Edition (W. H., Freeman and Co., 2012).
Khatkar, M. et al. Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genomics 9, 187 (2008).
Miller, S. P., Hayes, B. J. & Goddard, M. E. 21–14 (Instituto Prociência, Minas Gerais, 2006).
Abecasis, G. R. & Cookson, W. O. C. GOLD—Graphical overview of linkage disequilibrium. Bioinformatics 16, 182–183 (2000).
Cartwright, D. A., Troggio, M., Velasco, R. & Gutin, A. Genetic mapping in the presence of genotyping errors. Genetics 176, 2521–2527 (2007).
Liao, M. et al. Development of silver carp (Hypophthalmichthys molitrix) and bighead carp (Aristichthys nobilis) genetic maps using microsatellite and AFLP markers and a pseudo‐testcross strategy. Anim. Genet. 38, 364–370 (2007).
Baranski, M. et al. The development of a high density linkage map for black tiger shrimp (Penaeus monodon) based on cSNPs. PLoS One 9, e85413 (2014).
Edwards, J. H. The Oxford Grid. Ann. Hum. Genet. 55, 17–31 (1991).
Paterson, T. & Law, A. ArkMAP: integrating genomic maps across species and data sources. BMC Bioinformatics 14, 1–10 (2013).
Gao, Y. et al. Whole transcriptome analysis provides insights into molecular mechanisms for molting in Litopenaeus vannamei. PLoS One 10, e0144350 (2015).
Qian, Z. et al. cDNA cloning and expression analysis of myostatin/GDF11 in shrimp, Litopenaeus vannamei. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 165, 30–39 (2013).
Chen, X. et al. Transcriptome analysis of Litopenaeus vannamei in response to white spot syndrome virus infection. PLOS One 8, e73218 (2013).
Wang, S. et al. Quality assessment parameters for EST-derived SNPs from catfish. BMC Genomics 9, 450 (2008).
Andreassen, R., Lunner, S. & Hoyheim, B. Targeted SNP discovery in Atlantic salmon (Salmo salar) genes using a 3′UTR-primed SNP detection approach. BMC Genomics 11, 706 (2010).
Lapègue, S. et al. Development of SNP-genotyping arrays in two shellfish species. Mol. Ecol. Resour. 14, 820–830 (2014).
Liu, S. et al. Development of the catfish 250K SNP array for genome-wide association studies. BMC Res. Notes. 7, 135–135 (2014).
Sanchez, C. et al. Single nucleotide polymorphism discovery in rainbow trout by deep sequencing of a reduced representation library. BMC Genomics 10, 559 (2009).
Lien, S. et al. A dense SNP-based linkage map for Atlantic salmon (Salmo salar) reveals extended chromosome homeologies and striking differences in sex-specific recombination patterns. BMC Genomics 12, 615 (2011).
Dominik, S. et al. Evaluation of an Atlantic salmon SNP chip as a genomic tool for the application in a Tasmanian Atlantic salmon (Salmo salar) breeding population. Aquaculture 308(Supplement 1), S56–S61 (2010).
Tosser-Klopp, G. et al. Design and characterization of a 52K SNP chip for goats. PLOS One 9, e86227 (2014).
Groenen, M. A. et al. The development and characterization of a 60K SNP chip for chicken. BMC Genomics 12, 274 (2011).
Matukumalli, L. K. et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS One 4, e5350 (2009).
Ramos, A. M. et al. Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS One 4, e6524 (2009).
McCue, M. E. et al. A high density SNP array for the domestic horse and extant Perissodactyla: Utility for association mapping, genetic diversity, and phylogeny studies. PLoS Genet. 8, e1002451 (2012).
Robledo, D., Palaiokostas, C., Bargelloni, L., Martínez, P. & Houston, R. Applications of genotyping by sequencing in aquaculture breeding and genetics. Reviews in Aquaculture, doi:10.1111/raq.12193 (2017).
Hong, H. et al. Technical reproducibility of genotyping SNP Arrays used in genome-wide association studies. PLOS One 7, e44483 (2012).
Wall, J. D. et al. Estimating genotype error rates from high-coverage next-generation sequence data. Genome Res. 24, 1734–1739 (2014).
Yuan, J. et al. Comparative genomics analysis of decapod shrimps in the Pancrustacea clade. Biochem. Syst. Ecol. 64, 111–121 (2016).
You, E. M. et al. Construction of integrated genetic linkage maps of the tiger shrimp (Penaeus monodon) using microsatellite and AFLP markers. Anim. Genet. 41, 365–376 (2010).
Maneeruttanarungroj, C. et al. Development of polymorphic expressed sequence tag-derived microsatellites for the extension of the genetic linkage map of the black tiger shrimp (Penaeus monodon). Anim. Genet. 37, 363–368 (2006).
Staelens, J. et al. High-density linkage maps and sex-linked markers for the black tiger shrimp (Penaeus monodon). Genetics 179, 917–925 (2008).
Castillo-Juárez, H., Campos-Montes, G. R., Caballero-Zamora, A. & Montaldo, H. H. Genetic improvement of Pacific white shrimp [Penaeus (Litopenaeus) vannamei]: perspectives for genomic selection. Front. Genet. 6, 93 (2015).
Neira, R. Breeding in aquaculture species: genetic improvement programs in developing countries. In Proceedings of the 9th World Congress on Genetics Applied to Livestock Production. Vol. 8. (2010).
Rye, M. Current status and prospects for the application of genetic improvement in aquaculture species. In Proceedings of the 9th Biennial Symposium of the Brazilian Society of Animal Breeding (2012).
Acknowledgements
Illumina GA-IIx sequencing was conducted at AGRF QLD Australia, and Illumina array genotyping was conducted at PathWest medical laboratories in Perth, Western Australia. Global Gen Ltd. provided the experimental animals and funding for this study. The commercial array was developed in collaboration with Cindy Lawler from Illumina Inc. Giana Bastos Gomes and Tansyn Noble helped with the genetic validation of pedigree relationships. Hoc Tan Dao helped with the reconstruction of parental genotypes from allele frequencies of pooled families.
Author information
Authors and Affiliations
Contributions
D.R.J., K.R.Z., H.W.R. and H.v.d.S. conceptualised, developed and oversaw the project. J.P. and H.v.d.S. managed the rearing and tissue sampling of shrimp families over the course of two years. K.R.Z. prepared the RNA and cDNA for transcriptome sequencing. K.R.Z. and S.F. cleaned and assembled all transcriptome sequence data. K.R.Z. and D.B.J. mined the transcriptome data and developed a list of SNPs for inclusion within the custom Illumina genotyping array. D.B.J. performed the laboratory analysis associated with DNA preparation and QA/QC for sequencing and genotyping, the filtering of SNP genotype data and general data integrity, the annotation of the transcriptome, the generation of the linkage map and integration of LODE-placed markers as well as all associated statistical tests and comparative mapping analysis, and was the major contributor in writing the manuscript. M.S.K. and H.W.R. performed the LD and LODE analysis and gave advice on map integration. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jones, D.B., Jerry, D.R., Khatkar, M.S. et al. A comparative integrated gene-based linkage and locus ordering by linkage disequilibrium map for the Pacific white shrimp, Litopenaeus vannamei . Sci Rep 7, 10360 (2017). https://doi.org/10.1038/s41598-017-10515-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10515-7
This article is cited by
-
Genome-wide QTL and eQTL mapping reveal genes associated with growth rate trait of the Pacific white shrimp (Litopenaeus vannamei)
BMC Genomics (2024)
-
Development and validation of a RAD-Seq target-capture based genotyping assay for routine application in advanced black tiger shrimp (Penaeus monodon) breeding programs
BMC Genomics (2020)
-
Genomic selection for white spot syndrome virus resistance in whiteleg shrimp boosts survival under an experimental challenge test
Scientific Reports (2020)
-
De novo assembly, characterization, functional annotation and expression patterns of the black tiger shrimp (Penaeus monodon) transcriptome
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
