
Abstract
Phthalate, an environmental toxin, has been considered as an endocrine-disrupting chemical. Growing evidence has demonstrated links between endocrine-disrupting chemicals, tissue development, and reproductive physiology, but the mechanisms of gene expression regulation by environmental factors that affect cell differentiation are unclear. Herein, we investigated the effects of butyl benzyl phthalate (BBP) on human endometrial mesenchymal stem/stromal cell (EN-MSC) differentiation and identified a novel signaling pathway. Differentiation of endometrial mesenchymal stem/stromal cells decreased after administration of BBP. We analyzed BBP regulation of gene expression in EN-MSC using cDNA microarrays and Ingenuity Pathway Analysis software to identify affected target genes and their biological functions. PITX2 emerged as a common gene hit from separate screens targeting skeletal and muscular disorders, cell morphology, and tissue development. BBP decreased transcription of PITX2 and elevated expression of the microRNA miR-137, the predicted upstream negative regulator of PITX2. These data indicated that BBP affects PITX2 expression through miR-137 targeting of the 3′ untranslated region of PITX2 mRNA. PITX2 down-regulation also decreased MyoD transcript levels in EN-MSC. Our results demonstrate that BBP decreases EN-MSC myogenic differentiation through up-regulation of miR-137, contribute to our understanding of EN-MSC differentiation, and underline the hazardous potential of environmental hormones.
Similar content being viewed by others

Introduction
Human mesenchymal stem/stromal cells (hMSCs) are adult stem cells that maintain tissue homeostasis by serving as a source of renewable progenitor cells to repair injured tissues and replace cells in routine cellular turnover throughout adult life1, 2; they may be isolated from a variety of tissues. Human mesenchymal stem cells (MSCs) have been isolated from a variety of tissues, including bone marrow, blood, adiopose, endometrium and other adult tissues. Among the diverse origins, we used MSCs derived form endometrium tissues. The human endometrium is a highly regenerative tissue that undergoes menstrual cycles involving growth, differentiation, and shedding during a woman’s reproductive life. The differentiation ability of the endometrium is based on endometrial stem cells3,4,5. Therefore endometrial adult stem cell populations are thought to be responsible for this remarkable regenerative capacity3, 4. Endometrial mesenchymal stem/stromal cells (EN-MSCs) are multi-potent stem cells that may be isolated and induced in vitro to differentiate into a variety of cell lineages that include adipocytes, osteocytes, chondrocytes, and myocytes5. EN-MSC differentiation is controlled by regulatory genes that induce progenitor cell differentiation into a specific lineage; in addition, environmental factors, such as phthalates, may influence gene expression during cell differentiation6. However, how environmental factors affect cell differentiation through gene expression regulation is unclear.
The pollutant butyl benzyl phthalate (BBP) is ubiquitously present in the environment. BBP is widely used as a plasticizer in the polyvinyl chloride industry and is commonly found in a variety of products such as automotive trim, food packaging, medical products and children’s toys7. BBP is an external plasticizer, i.e., used in resin softening without chemical binding to the final product. Therefore, BBP tends to migrate slowly out of discarded plastics and disperse into aqueous environments8, 9; hence, BBP may enter the food chain10. In addition, phthalates have been classified as endocrine-disrupting chemicals (EDCs) and may interfere with the endocrine system to produce adverse developmental, reproductive, neurological, and immunological effects11,12,13. In previously study, Upson K et al. finding that urinary concentration of the BBP metabolite MBzP (mono-n-benzyl phthalate) may be associated with increased risk of endometriosis14. Reddy et al. has demonstrated the relationship between exposure to polyethylenes such as BBP and the occurrence of endometriosis in infertile women15.
MicroRNAs (miRNAs) are small, endogenous non-coding RNAs that regulate gene expression by forming imperfect base pairing to sequences in the 3′ untranslated regions (UTRs) of their target mRNAs, thereby triggering translational repression. miRNAs influence a variety of biological processes including development, tissue morphogenesis, cell growth, and maintenance of tissue identity16. Emerging evidence indicates that miRNAs have a critical role in the self-renewal and differentiation of MSCs17.
In addition to the contribution of genetic background, increasing evidence indicates that EDCs may affect MSC differentiation18, 19. Therefore, the aim of the present study was to evaluate the effect of BBP on the differentiation of EN-MSCs and to investigate the relationship between BBP and epigenetic-modulated gene expression.
Results
EN-MSCs differentiate into adipogenic, osteogenic, chondrogenic, and myogenic lineages
To investigate the differentiation potential of EN-MSCs, we cultured cells from endometrium under conditions that favored adipogenic, osteogenic, chondrogenic, or myogenic differentiation. Cytochemical and immunofluorescence staining and quantitative real-time PCR (qPCR) were performed to determine the capacity for EN-MSCs to undergo various lineage differentiations after induction. Adipogenic, osteogenic, or chondrogenic differentiation of EN-MSCs was identified by Oil Red O, Alizarin Red S, or Alcian Blue staining of the respective markers (Supplementary Fig. S1A and B). Subsequent qPCR analysis revealed increased expression of the respective adipogenic-, osteogenic-, and chondrogenic-specific markers FABP4, Runx2, and collagen II (Supplementary Fig. S1C).
Myogenic differentiation, as assayed by immunoreactivity for MyoD (last two panels; Supplementary Fig. S1A) and qPCR analysis of transcript levels of myogenic markers, was also increased (last one panel; Supplementary Fig. S1B). These data suggested that the endometrium contains cells that have MSC properties associated with multiple lineage differentiation.
BBP decreases EN-MSC adipogenic and myogenic differentiation
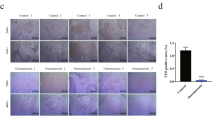
To assess how BBP influences EN-MSC differentiation, cells were treated with 1 μM BBP during the induction period corresponding to multiple lineage differentiation. After EN-MSC differentiation induction and/or BBP treatment, adipogenic, osteogenic, chondrogenic, and myogenic differentiation was analyzed by cytochemical and immunofluorescence staining. In the absence of BBP treatment, EN-MSC differentiation after induction into the four lineages of interest was not affected; in contrast, in the presence of the BBP, EN-MSC differentiation was affected (Fig. 1A and Supplementary Fig. S2A and B). Further, we assessed whether BBP could influence the expression of marker genes during differentiation. BBP decreased the expression of the adipogenic marker FABP4, PPARγ2 and myogenic marker MyoD, Myf5 in each of the non-differentiated and differentiated condition (Fig. 1B and Supplementary Fig. S2C). These data revealed that BBP affected EN-MSC differentiation. We next examined the phenotype of BBP affected the myogenic differentiation of EN-MSC, we performed the RNA extraction and PCR to detect the level of endometrial MSC markers sushi Domain Containing 2 (SUSD2). The data showed that the level of SUSD2 markers was decreases in BBP treated EN-MSC, suggesting BBP affected EN-MSC differentiation through loss of the EN-MSC phenotype (Fig. 1C).

Effect of BBP on EN-MSC differentiation. (A) EN-MSCs were cultured in differentiation medium for 2 weeks and treated with or without 1 μM BBP every day. Staining and magnification were carried out as in (A). Differentiation is apparent in control differentiation samples, whereas there is signal reduction in the BBP-treated differentiation samples. (B) Gene expression analysis of myogenic markers in differentiated EN-MSCs by real-time PCR analysis. Expression was analyzed with qPCR, using 18S as an internal control. The BBP treatment protocol was as in (A). (C) The RNA extraction and PCR to detect the level of endometrial MSC markers SUSD2. The data shown represent the mean ± SD of three experiments with three different batches of cells. *P < 0.05.
cDNA microarray and signaling pathways
Next, we investigated how phthalate affected EN-MSC differentiation using whole-genome cDNA microarrays to examine BBP regulation of gene expression. For these experiments, we added 1 μM BBP to a culture of EN-MSCs for 24 h prior to the isolation of total RNA and subsequent cDNA synthesis. We focused the down-regulated gene after BBP-treatment in MSCs.
Table 1 lists the top 15 genes that were down-regulated after BBP treatment, which underscores the remarkable potential of this compound to alter EN-MSC differentiation. Analysis of cDNA microarray data revealed how individual genes interacted and coordinated to affect regulation of biological functions and signaling pathways. The top biofunctions associated with the gene expression profiles of BBP-treated MSCs were identified using Ingenuity Pathway Analysis and are listed in Supplementary Table S1: diseases and disorders, molecular and cellular functions, and physiological system development and function.
Finally, we identified the major biofunction categories associated with cellular differentiation. Table 2 lists the genes associated with skeletal and muscular disorders, cell morphology, and tissue development. Biofunctions of the candidate genes responsible for BBP-induced alterations of EN-MSC differentiation were determined.
Gene expression in BBP-treated MSCs
The gene was found to have overlapping contributions among the BBP deregulated genes whose functions were associated with skeletal and muscular disorders, cell morphology, and tissue development and were excluded which are irrelevant to MSCs from intersection of all three categories. We found the three genes, namely, PITX2 (fold change = 0.596, P = 0.00007), SRC (fold change = 0.532, P = 0.000082), and TLR2 (fold change = 0.645, P = 0.00078) (Fig. 2A). First, we examined gene expression levels of these three genes in EN-MSCs after 1 μM BBP treatment for 24 h. The data revealed that expression of the three genes decreased in BBP-treated MSCs (Fig. 2B). These data suggested that PITX2, SRC, and TLR2 play a vital role in mediating the effects of BBP on EN-MSC differentiation. In previous studies, PITX2, a homeodomain transcription factor, is essential for normal development and differentiated of tissue20, 21. Recent studies have reported that SRC plays a role in signal pathways involved in cell proliferation, growth, survival osteoclast and intestinal epithelial cell differentiation22, 23. In addition, TLR2 play a central role in the innate immune system and is associated with B cell differentiation24. We then investigated whether miRNAs that target SRC, PITX2, and/or TLR2 mRNAs mediate the effects of BBP on EN-MSC differentiation. We targeted PITX2, which has been reported to be associated with myogenesis25,26,27,28. We attempted to identify miRNAs that serve as upstream regulators of EN-MSC differentiation, i.e., miRNAs that might be affected by BBP; specifically that were affected by BBP in EN-MSC differentiation, and specifically we used the prediction software miRanda (http://www.microrna.org/) to select three candidate miRNA regulators of PITX2: miR-137, miR-141 and miR-200a. BBP treatment increased the level of miR-137 in EN-MSCs, whereas the levels of miR-141 and miR-200a were not affected (Fig. 2C). Next, we examined whether PITX2 expression was affected by miR-137. Overexpression of precursor-miR-137 in EN-MSCs reduced the PITX2 transcript level (Fig. 2D). These data suggested that the BBP-induced effects on the level of PITX2 transcript are mediated through miR-137.

mRNA levels of the three identified genes. (A) Venn diagram with the number of genes differentially expressed between biofunctions in three individual categories: skeletal and muscular disorders, cell morphology, and tissue development. The shaded area shows intersection of all three categories and denotes three shared genes that emerged: PITX2, SRC, and TLR2. (B) qRT-PCR analysis of mRNA levels of the three identified genes; all genes showed significant reduction in expression after BBP treatment as compared with controls. (C) The level of miR-137 was increased in BBP-treated EN-MSCs compared with control cells. U6 was detected as an internal control. (D) Validation of miR-137 target. Precursor-miR-137 effectively decreased the transcript level of PITX2. The data shown represent the mean ± SD of three experiments with three different batches of cells. *P < 0.05.
miR-137 down-regulates PITX2 by targeting the 3′UTR
To test whether miR-137 targets PITX2, the 3′UTR of PITX2, which contains a miR-137 binding site, was cloned into the pGL-2 control vector to create a luciferase reporter system (Fig. 3A). Co-transfection was performed with pre-miR-137 (precursor control) and pGL2-PITX2 3′UTR (mutant version of pGL2-PITX2 3′UTR). Cotransfection was performed with pre-miR-137 (precursor control) and either wild-type pGL2-PITX2 3′UTR or a mutant derivative. The luciferase reporter showed that miR-137 inhibited the PITX2 wild-type reporter but did not affect the PITX2 mutant reporter (Fig. 3B).

PITX2 mRNA is a direct target of miR-137. (A) Sequences of target sites for miR-137 in the wild-type and mutant (Mut) versions of 3′-UTR of PITX2 mRNA. (B) Cells were co-transfected with precursor-miR-137 or precursor control and the pGL2 vector containing wild-type (Wt) or mutant version of the putative PITX2 3′UTR miR-137 binding site. Luciferase activity was normalized to the control. (C) Western blot analysis was used to detect the expression of MyoD and PITX2 in response to miR-137 alteration. (D) EN-MSCs were cultured in differentiation medium for 2 weeks and transected with miR-137. Gene expression analysis of myogenic markers in differentiated EN-MSCs by real-time PCR analysis. Expression was analyzed with qPCR, using 18S as an internal control. The data shown represent the mean ± SD of three experiments with three different batches of cells. *P < 0.05.
Further, we performed western blotting to confirm whether miR-137 affects the protein level of PITX2 and MyoD. Over-expression of miR-137 decreased the level of PITX2 and MyoD, whereas knock-down of mir-137 increase the levels of PITX2 and MyoD (Fig. 3C). These results indicated that, in our experimental system, PITX2 was indeed a direct target of miR-137.
Taken together, these results showed that BBP reduced PITX2 expression in EN-MSC differentiation (Fig. 2B) via increased expression of miR-137 (Fig. 2C), its upstream negative regulator. Therefore, we investigated whether myogenesis was affected by miR-137 in EN-MSCs. Ectopic miR-137 expression decreased the expression of the myogenic marker MyoD, PITX2 in differentiated condition (Fig. 3D), which supports the hypothesis that BBP exerts its effect on EN-MSC myogenic differentiation through the action of miR-137.
miR-137 affects myogenesis through PITX2
To understand the extent to which miR-137 affects EN-MSC differentiation through PITX2 directly, we used a short hairpin RNA (shRNA) to knockdown PITX2 expression (Fig. 4A). When EN-MSC was transfected with PITX2-shRNA-1 or PITX2-shRNA-2, PITX2 and MyoD expression were down-regulated (Fig. 4B and E). PITX2 overexpression (Fig. 4C) in the cells increased PITX2 and MyoD expression (Fig. 4D and E). These data confirmed that PITX2 expression level had a significant effect on EN-MSC myogenic differentiation.

Knockdown and overexpression of PITX2 affect MyoD expression. (A) EN-MSCs transfected with PITX2 shRNA-1, PITX2 shRNA-2 or scrambled shRNA (a negative control for PITX2 shRNA). (B) qPCR analysis of mRNA levels of MyoD. Expression levels were normalized to 18S rRNA levels. (C) EN-MSCs were transfected with PITX2 or control vector. qPCR analysis of mRNA levels of PITX2. (D) mRNA levels of MyoD were analyzed with qPCR. Expression levels were normalized to 18S rRNA levels. (E) Western blotting indicated that PITX2 and MyoD level were positively correlated. The data shown represent the mean ± SD of three experiments with three different batches of cells. *P < 0.05.
Discussion
Phthalates are omnipresent toxins in the environment, and they have been classified as EDCs that can interfere with elimination of natural hormones that are responsible for homeostasis and essential for growth and development29. In previously study, Upson K et al. finding that urinary concentration of the BBP metabolite MBzP (mono-n-benzyl phthalate) may be associated with increased risk of endometriosis14. Reddy et al. has demonstrated the relationship between exposure to polyethylenes such as BBP and the occurrence of endometriosis in infertile women15. These speculate the correlation between the exposures of BBP and endometrial diseases. In addition, the effects of exposure to BBP and its main metabolite MnBP (mono-n-butyl phthalate) and MBzP may have same effect in animal model30, 31. The pattern of malformations produced by MnBP was similar to that produced by BBP31. Previous study also reported that BBP, MnBP and MBzP caused embryolethality and malformations in mice30.
MSCs play an important role in tissue homeostasis, serving as a source of renewable progenitor cells to replace or repair tissue cells throughout adult life32, 33. In the present study, we found that BBP decreased EN-MSC differentiation. We identified one such target gene PITX2, which is a homeobox transcriptional factor that regulates muscle development21, 34. Normally, PITX2 and MyoD transcription levels increase during myogenic differentiation21; thus, the observed reduction in PITX2 and MyoD transcripts offers further evidence that BBP alters transcriptional regulation during stem cell differentiation in endometrial tissues. In addition, PITX2 is essential for development of multiple organs, including the lung, heart and pituitary gland35. Therefore, environmental hormones might affect tissue development through PITX2.
In the present study, we used microarray analysis to identify genes whose expression levels were altered by BBP. Our results found that TLR2 and SRC were dysregulated in response to BBP treatment. This finding is consistent with previous studies, we found TLR2 gene related to immune24, and SRC which has been described to be related with epithelial cell differentiation23. A previous study has investigated the alteration in the gene expression following phthalate treatment in which exposure to this compound caused a dys-regulation in the expression of many genes, including apoptosis-, cell proliferation-, and immune response- related genes36, 37.
TLR2 plays a key role in immune system and is found in immune cell, such macrophages, B cell and mast cells24. MSC display unique suppressive properties on T-cell immunity, since TLR expressed on human MSC enhanced the immunosuppressive phenotype of MSC38, 39. Immunosuppressive properties of MSC most probably depend on environmental factors40. Interestingly, functional role of phthalate-elicited differential gene expression is associated with immune system. It may be informative to investigate the potential mechanism related to BBP effects on immune system.
In the present study, we investigated how phthalate affected EN-MSC differentiation using whole-genome cDNA microarrays to examine BBP regulation of gene expression. Although the top 15 down-regulated genes were not found to have overlapping contributions among the BBP deregulated genes whose functions were associated with skeletal and muscular disorders, cell morphology, and tissue development. However, some studies have reported that these genes, TDGF141, ALX142, LGI343, ADH444, VSX145, ZIC346, 47, are relevant to stem cell differentiation or tissue development. As expected, phthalate exposure might affect cell differentiation or development.
Recently, several studies showed that exposure to various environmental or growth factors regulates the expression of certain specific miRNAs48, 49. Although several studies have demonstrated that miR-137 functions in neurogenesis or adipogenesis in stem cells50, 51, ours is the first study to demonstrate that BBP administration modulates miR-137 level and to identify PITX2 as a novel miR-137 downstream target during myogenic differentiation.
In conclusion, we characterized the roles of miR-137 in myogenic hMSC differentiation and elucidated the mechanisms of BBP action in this process. These findings contribute to our understanding of hMSC differentiation and underscore the hazardous potential of environmental hormones.
Methods
Cell line
EN-MSCs were isolated and collected from 3 different endometrium biopsies after hysterectomy for non-endometrial benign pathological condition, such as uterine prolapse. These women had not taken exogenous hormones for three months prior to surgery. This study was approved by the Institutional Review Board of Kaohsiung Medical University, and informed consent was obtained from each patient (KMUH-IRB-20140031). All experiments were performed in accordance with relevant guidelines and regulations. Written informed consent was obtained from each participant. EN-MSCs were isolated and purified as described5. Briefly, endometrial tissue was minced with sterile scissors and subjected to enzymatic digestion with 1 mg/ml type II collagenase for 60 to 90 minutes. After digestion, these digested tissues were filtered by wire sieves with serial different pores (100 μm, 70 μm and 40 μm diameter pores) to remove epithelial cells. These endometrial stromal cells were collected. For EN-MSC isolation, endometrial stromal cells (passage 5) were seeded in triplicate at clonal density; 200 cells per 100 mm Petri dish. After incubation of 21 days, large colonies were isolated and separated into single suspended cells by trypsinization. These cells were diluted and seeded in a 96-well plate, density at one cell per well. After incubation of 14 days, proliferated cells (which were from one single cell) were trypsinized and cultured in a 100 mm Petri dish. These early-passage EN-MSCs were used in the following experiments. EN-MSCs were characterized using MSC phenotypes and differentiation induction (i.e., adipogenesis, osteogenesis, and chondrogenesis,) and by gene expression, i.e., POU5F1 (previously known as OCT-4), CD29, CD44, CD49f, CD90, CD105, CD146, CD140b, and SUSD2 by flow cytometry5 (Supplementary Material). EN-MSCs were cultured in modified MCDB 153 medium (Keratinocyte-SFM, Gibco-Life Technologies, Carlsbad, CA) and Dulbecco’s Modified Eagle’s Medium: Nutrient Mixture F12 (Gibco-Life Technologies) (1:2, v/v) supplemented with 10% fetal bovine serum (Gibco-Life Technologies), 2 mM N-acetyl-l-cysteine (A8199, Sigma-Aldrich, St. Louis, MO) and 0.2 mM l-ascorbic acid 2-phosphate (Asc 2P; A8960, Sigma-Aldrich), and incubated at 37 °C in a humidified atmosphere with 5% CO2.
Differentiation experiments
EN-MSCs were seeded at 5 × 104 cells per well in a 6-well plate; differentiation conditions were applied the following day. Adipogenic differentiation of EN-MSCs was induced by treatment with 500 μM of 3-isobutyl-1-methylxanthine (I7018, Sigma-Aldrich), 1 μM dexamethasone (D8893, Sigma-Aldrich), 1 μM indomethacin (I8280, Sigma-Aldrich), and 10 μg/mL insulin (I1882, Sigma-Aldrich) (IDI-I medium) for 2 d, and followed by insulin treatment for 1 d. After 4 cycles of treatment over 12 d, the cells were fixed with 4% paraformaldehyde and stained with 0.2% Oil Red O for 30 min (O0625, Sigma-Aldrich)52. For osteogenic differentiation, 10 nM dexamethasone (D8893, Sigma-Aldrich), 50 μM Asc-2P, and 10 mM β-glycerophosphate disodium (G9891, Sigma-Aldrich), also commonly known as DAG medium, were added to the growth medium for 2 weeks. Medium changes and treatments were renewed once every 3 d52. At 14 d after the initiation of differentiation, the cells were stained with 2% Alizarin Red S (A5533, Sigma-Aldrich) to assay for osteocytes53. For myogenic differentiation, 5 μM 5-azacytidine (A2385, Sigma-Aldrich) was added to the growth medium for 24 h, after which the myogenic induction medium was replaced with normal growth medium54. The medium was changed every 3 d for the remainder of the culture. On day 14 after induction of myogenic differentiation, cells were fixed in 4% paraformaldehyde, stained for MyoD (ab64159, Abcam), and then examined for the presence of myocytes by immunofluorescence. After reaching 90% confluence, the cells were harvested and reseed in 15 ml tube at 2.5 × 105 cells/tube. Chondrogenic differentiation of EN-MSCs was induced by treatment with 10 ng/mL TGF-β1 (T1654, Sigma-Aldrich), 50 μM Asc-2P, and 6.25 μg/mL insulin (TAI medium) in the 24 well plates. Medium was changed every 3 d52, 55, 56. After 14 d, the micromass was fixed in 4% paraformaldehyde and then examined for chondrocytes by staining with, 1% Alcian Blue 8-Gx, pH 1.0 (A5268, Sigma-Aldrich).
Chemicals and Reagents
BBP (98%) was purchased from Sigma-Aldrich and diluted with ethanol to a concentration 1000-fold higher than the final concentration that was used in cell culture.
quantitative real-time PCR analysis
qPCR was used to assess gene and miRNA expression. RNAs were extracted from EN-MSCs using TRI Reagent (Sigma-Aldrich). Reverse transcription was carried out with 1.5 μg of RNA using the Deoxy + HiSpec RT kit (Yeastern, Taipei, Taiwan) and TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems, Foster City, CA). The expression of various transcripts and mature miRNAs was assessed by real-time PCR with Power SYBR Green PCR Master Mix (Applied Biosystems) and TaqMan MicroRNA Assay using an ABI 7900 Real-Time PCR system (Applied Biosystems). The primer sets used in this study are listed in Supplemental Table S4. Changes in gene expression were calculated relative to 18S RNA using the 2−ΔΔCt method. MiRNAs expression were normalized to endogenous small nuclear U6B RNA using the 2−ΔΔCt method.
Immunofluorescence
After treatment, EN-MSCs were rinsed several times with PBS and fixed in 4% paraformaldehyde for 5 min, permeabilized with 0.5% Triton X-100 in PBS for 5 min. The fixed cells were probed with Rabbit-MyoD antibody (1:1000, ab64159, Abcam) and secondary antibody followed by Alexa Fluor 568–conjugated goat anti-rabbit IgG (1:500, A11011, Gibco-Life Technologies) for 45 min. Nuclei were counterstained with 4,6-diamidino-2-phenylindole (DAPI, 1 μg/ml, Roche). Images were obtained using a fluorescence microscope (Nikon Eclipse TE 300, Tokyo, Japan).
cDNA microarray and data analysis
RNA was extracted from EN-MSCs using TRI Reagent. RNA integrity number >7.0 were used to synthesize the first strand cDNA via reverse transcription using an Illumina Total Pre RNA Amplification Kit (Ambion, Austin, TX, USA). Amplified cRNA samples were hybridized with streptavidin-Cy3 and scanned on the Illumina Beadstation GX. To determine differentially expressed genes, microarray data (n = 2 in each group) were analyzed using the gene expression module in Illumina Beadstudio software, version 3.3.7. Intensity data were normalized using the Beadstudio cubic spline algorithm and calculated with Beadstudio software according to the manufacturer’s protocols. The gene expression fold change of the stimulated cells was calculated as the average signal value relative to the average signal value for the control cells. Genes were selected based on a p-value cut-off (after adjustment) of p < 0.05 to control the false discovery rate (FDR)57, 58. A significant down-regulation was defined as a foldchange ≥1.5. We applied the Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, California) tool for analysis of canonical pathways and participating networks using the cDNA microarray data.
Ingenuity Pathway Analysis
The molecular functions of the unique gene analysis of the BBP-induced genes were performed using Ingenuity Pathway Analysis (IPA) software (IPA, Ingenuity Systems, Redwood City, California). Genes from the data set that met the cutoff of and were associated with biological functions and/or diseases in the Ingenuity Pathways Knowledge Base were included in the analysis.
Transfection
Transfection of each of miRNAs, shRNA, plasmid DNA, and reporter vectors was performed using TransIT-LT1 Transfection Reagent (Mirus Bio, Madison, WI). The following plasmids were used: Precursors of miR-137 and anti-miR-137 plasmids were purchased from System Biosciences. PITX2 plasmid DNA was from the Bioresource Collection and Research Center, Hsinchu, Taiwan. The shRNAs included shRNA-PITX2#1 (TRCN0000020481), shRNA-PITX2#2 (TRCN0000235583), and scrambled control shRNA (TRCN0000040032) (National RNAi Core Facility at the Institute of Molecular Biology, Academia Sinica, Taipei, Taiwan). Cells were harvested 2 d after transfection.
Luciferase assay
Cells were seeded onto 48-well culture plates and co-transfected with 200 ng of vector pGL2-PITX2–3′UTR or pGL2 that contained a mutant version of the PITX2 3′UTR, 200 ng pre-miR-137 or a precursor control, 30 ng luciferase reporter, and 5 ng Renilla luciferase reporter. Luciferase activity was measured by the Dual-Luciferase Reporter Assay system (Promega, Madison, WI). Firefly luciferase activity was normalized to Renilla luciferase activity for each sample. The luciferase signal was read with a TD-20/20 luminometer (Turner Biosystems, Sunnyvale, CA).
Western Blot
The proteins were extracted with RIPA lysis buffer (Millipore, Billerica, MA, USA) containing several protease and phosphatase inhibitors (GBiosciences, St Louis, MO, USA). The protein content was determined by a Bio-Rad Protein Assay system (Bio-Rad, Hercules, CA, USA). Equal amounts of protein were separated by 10% SDS-PAGE and transferred to PVDF membranes (Millipore, Bedford, MA, USA). Then the membrane was incubated with primary antibodies: anti-MyoD (ab126726, 1:1000, abcam), anti-PITX2 (ab55599, 1:1000, abcam), and anti-actin (1:5000; Sigma-Aldrich). The secondary antibodies used were goat-anti-mouse or anti-rabbit IgG conjugated to HRP (Santa Cruz Biotechnology), and the ECL reagents (Millipore) were used for immunodetection
Statistical Analysis
Statistical analyses were performed using One-way ANOVA followed by Tukey’s HSD test for comparing differences between multiple groups and Student’s t-test for comparing differences between two groups. Data represented the mean ± standard deviation. P values < 0.05 were considered statistically significant.
References
Lv, F. J., Tuan, R. S., Cheung, K. M. & Leung, V. Y. Concise review: the surface markers and identity of human mesenchymal stem cells. Stem cells 32, 1408–1419 (2014).
Phinney, D. G. & Sensebe, L. Mesenchymal stromal cells: misconceptions and evolving concepts. Cytotherapy 15, 140–145 (2013).
Gargett, C. E. Uterine stem cells: what is the evidence? Human reproduction update 13, 87–101 (2007).
Gargett, C. E., Nguyen, H. P. & Ye, L. Endometrial regeneration and endometrial stem/progenitor cells. Reviews in endocrine & metabolic disorders 13, 235–251 (2012).
Kao, A. P. et al. Comparative study of human eutopic and ectopic endometrial mesenchymal stem cells and the development of an in vivo endometriotic invasion model. Fertility and sterility 95, 1308–1315 e1301 (2011).
Chen, S. S., Hung, H. T., Chen, T. J., Hung, H. S. & Wang, D. C. Di-(2-ethylhexyl)-phthalate reduces MyoD and myogenin expression and inhibits myogenic differentiation in C2C12 cells. The Journal of toxicological sciences 38, 783–791 (2013).
Heudorf, U., Mersch-Sundermann, V. & Angerer, J. Phthalates: toxicology and exposure. International journal of hygiene and environmental health 210, 623–634 (2007).
Liu, X., Shi, J., Bo, T., Li, H. & Crittenden, J. C. Occurrence and risk assessment of selected phthalates in drinking water from waterworks in China. Environmental science and pollution research international 22, 10690–10698 (2015).
Dominguez-Morueco, N., Gonzalez-Alonso, S. & Valcarcel, Y. Phthalate occurrence in rivers and tap water from central Spain. The Science of the total environment 500–501, 139–146 (2014).
National Toxicology, P. NTP-CERHR Monograph on the Potential Human Reproductive and Developmental Effects of Butyl Benzyl Phthalate (BBP). Ntp Cerhr Mon, i–III90 (2003).
Thor Larsen, S., My Lund, R., Damgard Nielsen, G., Thygesen, P. & Melchior Poulsen, O. Di-(2-ethylhexyl) phthalate possesses an adjuvant effect in a subcutaneous injection model with BALB/c mice. Toxicology letters 125, 11–18 (2001).
Grande, S. W., Andrade, A. J., Talsness, C. E., Grote, K. & Chahoud, I. A dose-response study following in utero and lactational exposure to di(2-ethylhexyl)phthalate: effects on female rat reproductive development. Toxicological sciences: an official journal of the Society of Toxicology 91, 247–254 (2006).
Ma, M. et al. Exposure of prepubertal female rats to inhaled di(2-ethylhexyl)phthalate affects the onset of puberty and postpubertal reproductive functions. Toxicological sciences: an official journal of the Society of Toxicology 93, 164–171 (2006).
Upson, K. et al. Phthalates and risk of endometriosis. Environmental research 126, 91–97 (2013).
Reddy, B. S., Rozati, R., Reddy, B. V. & Raman, N. V. Association of phthalate esters with endometriosis in Indian women. BJOG: an international journal of obstetrics and gynaecology 113, 515–520 (2006).
Zhao, Y. & Srivastava, D. A developmental view of microRNA function. Trends in biochemical sciences 32, 189–197 (2007).
Tome, M. et al. miR-335 orchestrates cell proliferation, migration and differentiation in human mesenchymal stem cells. Cell death and differentiation 18, 985–995 (2011).
Strong, A. L. et al. Effects of the endocrine-disrupting chemical DDT on self-renewal and differentiation of human mesenchymal stem cells. Environmental health perspectives 123, 42–48 (2015).
Wadia, P. R. et al. Low-dose BPA exposure alters the mesenchymal and epithelial transcriptomes of the mouse fetal mammary gland. PloS one 8, e63902 (2013).
Shang, Y., Yoshida, T., Amendt, B. A., Martin, J. F. & Owens, G. K. Pitx2 is functionally important in the early stages of vascular smooth muscle cell differentiation. The Journal of cell biology 181, 461–473 (2008).
L’Honore, A., Ouimette, J. F., Lavertu-Jolin, M. & Drouin, J. Pitx2 defines alternate pathways acting through MyoD during limb and somitic myogenesis. Development 137, 3847–3856 (2010).
GuezGuez, A. et al. 3BP2 adapter protein is required for receptor activator of NFkappaB ligand (RANKL)-induced osteoclast differentiation of RAW264.7 cells. The Journal of biological chemistry 285, 20952–20963 (2010).
Seltana, A., Guezguez, A., Lepage, M., Basora, N. & Beaulieu, J. F. Src family kinase inhibitor PP2 accelerates differentiation in human intestinal epithelial cells. Biochemical and biophysical research communications 430, 1195–1200 (2013).
Ganley-Leal, L. M., Liu, X. & Wetzler, L. M. Toll-like receptor 2-mediated human B cell differentiation. Clinical immunology 120, 272–284 (2006).
Diehl, A. G. et al. Extraocular muscle morphogenesis and gene expression are regulated by Pitx2 gene dose. Investigative ophthalmology & visual science 47, 1785–1793 (2006).
Dong, F. et al. Pitx2 promotes development of splanchnic mesoderm-derived branchiomeric muscle. Development 133, 4891–4899 (2006).
L’Honore, A. et al. Sequential expression and redundancy of Pitx2 and Pitx3 genes during muscle development. Developmental biology 307, 421–433 (2007).
Shih, H. P., Gross, M. K. & Kioussi, C. Expression pattern of the homeodomain transcription factor Pitx2 during muscle development. Gene expression patterns: GEP 7, 441–451 (2007).
Markey, C. M., Rubin, B. S., Soto, A. M. & Sonnenschein, C. Endocrine disruptors: from Wingspread to environmental developmental biology. The Journal of steroid biochemistry and molecular biology 83, 235–244 (2002).
Saillenfait, A. M., Sabate, J. P. & Gallissot, F. Comparative embryotoxicities of butyl benzyl phthalate, mono-n-butyl phthalate and mono-benzyl phthalate in mice and rats: in vivo and in vitro observations. Reproductive toxicology 17, 575–583 (2003).
Ema, M., Kurosaka, R., Amano, H. & Ogawa, Y. Developmental toxicity evaluation of mono-n-butyl phthalate in rats. Toxicology letters 78, 101–106 (1995).
Li, L. & Xie, T. Stem cell niche: structure and function. Annual review of cell and developmental biology 21, 605–631 (2005).
Javazon, E. H., Beggs, K. J. & Flake, A. W. Mesenchymal stem cells: paradoxes of passaging. Experimental hematology 32, 414–425 (2004).
Zacharias, A. L., Lewandoski, M., Rudnicki, M. A. & Gage, P. J. Pitx2 is an upstream activator of extraocular myogenesis and survival. Developmental biology 349, 395–405 (2011).
Gage, P. J., Suh, H. & Camper, S. A. Dosage requirement of Pitx2 for development of multiple organs. Development 126, 4643–4651 (1999).
Moral, R. et al. In utero exposure to butyl benzyl phthalate induces modifications in the morphology and the gene expression profile of the mammary gland: an experimental study in rats. Environmental health: a global access science source 10, 5 (2011).
Hong, C. C., Shimomura-Shimizu, M., Muroi, M. & Tanamoto, K. Effect of endocrine disrupting chemicals on lipopolysaccharide-induced tumor necrosis factor-alpha and nitric oxide production by mouse macrophages. Biological & pharmaceutical bulletin 27, 1136–1139 (2004).
Hwa Cho, H., Bae, Y. C. & Jung, J. S. Role of toll-like receptors on human adipose-derived stromal cells. Stem cells 24, 2744–2752 (2006).
Tomchuck, S. L. et al. Toll-like receptors on human mesenchymal stem cells drive their migration and immunomodulating responses. Stem cells 26, 99–107 (2008).
Pevsner-Fischer, M. et al. Toll-like receptors and their ligands control mesenchymal stem cell functions. Blood 109, 1422–1432 (2007).
Bianco, C. et al. Role of Cripto-1 in stem cell maintenance and malignant progression. The American journal of pathology 177, 532–540 (2010).
Ettensohn, C. A., Illies, M. R., Oliveri, P. & De Jong, D. L. Alx1, a member of the Cart1/Alx3/Alx4 subfamily of Paired-class homeodomain proteins, is an essential component of the gene network controlling skeletogenic fate specification in the sea urchin embryo. Development 130, 2917–2928 (2003).
Park, W. J. et al. Leucine-rich glioma inactivated 3 induces neurite outgrowth through Akt and focal adhesion kinase. Neurochemical research 35, 789–796 (2010).
Gao, W. et al. Ethanol negatively regulates hepatic differentiation of hESC by inhibition of the MAPK/ERK signaling pathway in vitro. PloS one 9, e112698 (2014).
Chow, R. L. et al. Control of late off-center cone bipolar cell differentiation and visual signaling by the homeobox gene Vsx1. Proceedings of the National Academy of Sciences of the United States of America 101, 1754–1759 (2004).
Lim, L. S. et al. Zic3 is required for maintenance of pluripotency in embryonic stem cells. Molecular biology of the cell 18, 1348–1358 (2007).
Zhu, L. et al. Identification of a novel role of ZIC3 in regulating cardiac development. Human molecular genetics 16, 1649–1660 (2007).
Hou, L., Wang, D. & Baccarelli, A. Environmental chemicals and microRNAs. Mutation research 714, 105–112 (2011).
Hsu, C. Y. et al. Synthetic Steroid Hormones Regulated Cell Proliferation Through MicroRNA-34a-5p in Human Ovarian Endometrioma. Biology of reproduction 94, 60 (2016).
Sun, G. et al. miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nature communications 2, 529 (2011).
Shin, K. K., Kim, Y. S., Kim, J. Y., Bae, Y. C. & Jung, J. S. miR-137 controls proliferation and differentiation of human adipose tissue stromal cells. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology 33, 758–768 (2014).
Lin, T. M., Tsai, J. L., Lin, S. D., Lai, C. S. & Chang, C. C. Accelerated growth and prolonged lifespan of adipose tissue-derived human mesenchymal stem cells in a medium using reduced calcium and antioxidants. Stem cells and development 14, 92–102 (2005).
Smink, J. J. et al. Transcription factor C/EBPbeta isoform ratio regulates osteoclastogenesis through MafB. The EMBO journal 28, 1769–1781 (2009).
Wakitani, S., Saito, T. & Caplan, A. I. Myogenic cells derived from rat bone marrow mesenchymal stem cells exposed to 5-azacytidine. Muscle & nerve 18, 1417–1426 (1995).
Pittenger, M. F. et al. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 (1999).
Denker, A. E., Nicoll, S. B. & Tuan, R. S. Formation of cartilage-like spheroids by micromass cultures of murine C3H10T1/2 cells upon treatment with transforming growth factor-beta 1. Differentiation; research in biological diversity 59, 25–34 (1995).
Reiner, A., Yekutieli, D. & Benjamini, Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 19, 368–375 (2003).
Benjamini, Y. & Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc B 57 (1995).
Acknowledgements
This study was supported by the Kaohsiung Medical University Hospital Research Fund (Grant No. KMUH104-4M31, KMUH103-10V07, KMUH-105-M532 and KMUH105-5R31), Kaohsiung Medical University (KMU-TP105A06, KMU-TP105G02 and KMU-TP105E19), and Ministry of Science and Technology, Taiwan (Grant No. MOST 105-2314-B-037 -052 -MY3).
Author information
Authors and Affiliations
Contributions
H.S.C., C.Y.H., T.H.H., and E.M.T. conceived the work. T.H.H., C.Y.H., and Y.C.C. performed most of the experiments. H.Y.C., C.Y.L., H.S.C., C.Y.H., T.H.H., and E.M.T. interpreted the experimental data. H.S.C. and C.Y.H. wrote the manuscript, and E.M.T. edited the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chen, HS., Hsu, CY., Chang, YC. et al. Benzyl butyl phthalate decreases myogenic differentiation of endometrial mesenchymal stem/stromal cells through miR-137-mediated regulation of PITX2. Sci Rep 7, 186 (2017). https://doi.org/10.1038/s41598-017-00286-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-00286-6
This article is cited by
-
PM2.5-bound phthalates and phthalate substitutes in a megacity of southern China: spatioseasonal variations, source apportionment, and risk assessment
Environmental Science and Pollution Research (2022)
-
Epimutational effects of electronic cigarettes
Environmental Science and Pollution Research (2021)
-
Mitochondrial noncoding RNA-regulatory network in cardiovascular disease
Basic Research in Cardiology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
