
Abstract
N6-methyladenosine (m6A) modification is widespread in messenger RNAs and increasing evidence suggests the crucial roles of m6A in cell differentiation and tissue development. However, whether m6A modulates the osteogenic differentiation of mesenchymal stem cells (MSCs) has not been fully elucidated. Here we show that conditional knockout of the demethylase Alkbh5 in bone marrow MSCs strengthened bone mass in mice. Loss- and gain-of-function studies demonstrated that ALKBH5 negatively regulates the osteogenic differentiation of MSCs in vitro. At a mechanistic level, meRIP-seq and RNA-seq in MSCs following knockdown of ALKBH5 revealed changes in transcripts of PRMT6 containing consensus m6A motifs required for demethylation by ALKBH5. Furthermore, we found that ALKBH5 accelerates the degradation rate of PRMT6 mRNA in an m6A-dependent manner, and that the ALKBH5-PRMT6 axis regulates the osteogenesis of MSCs, mainly through activation of the PI3K/AKT pathway. Thus, our work reveals a different facet of the novel ALKBH5-PRMT6 axis that modulates the osteogenic differentiation of MSCs, which can serve as a target to improve the clinical use of MSCs.
Similar content being viewed by others


Introduction
Bone is an active organ that undergoes persistent metabolism1. Maintaining the body’s bone mass balance depends on the steady state of bone tissue, which is regulated by osteoclasts and osteoblasts2. Mesenchymal stem cells (MSCs) are pluripotent stem cells that exist in connective tissues and are an important source of osteoblasts in the body3. Due to their functional properties, MSCs are considered to be promising cell types for various applications and are used most frequently in regenerative medicine, especially for bone repair4,5,6. However, the regulatory mechanism controlling the osteogenic differentiation of MSCs remains largely unknown and hampers the further application of MSC-based cell therapies7. Understanding the molecular mechanism that regulates the osteogenic differentiation of MSCs will therefore allow us to further accelerate the differentiation of MSCs to osteoblasts, shorten the treatment time, and improve the safety and effectiveness of the clinical treatment, which is of great significance for the clinical application of treatments.
As a genetic medium in the process of transcription and translation, RNA is thought to be only an intermediate product that transfers genetic information from DNA to protein. In recent years, research has found that there are also abundant epigenetic modifications on RNA, playing a critical role in modulating biological processes. N6-methyladenosine (m6A) is the most common RNA methylation modification in humans8,9. Existing research shows that under the control of methyltransferases and demethylases, m6A can change dynamically in different biological processes; in addition, m6A-binding proteins recognize m6A and are responsible for regulating the metabolism of RNA10. Studies have shown that m6A can regulate cell differentiation and participate in various pathophysiological processes. After downregulation of fat mass and obesity-associated protein (FTO) in adipose precursor cells, the adipogenic differentiation ability of the cells is weakened11; methyltransferase 14 (METTL14) participates in the self-renewal and proliferation of embryonic neural stem cells by regulating histone modifications12. Due to the strong correlation between m6A and cell differentiation, we wondered whether m6A is also involved in regulating the osteogenic differentiation of MSCs, which has not been fully elucidated.
The current study only found two demethylases, alkB homolog 5 (ALKBH5) and FTO. A previous study found that FTO plays a role in bone mass13. However, whether ALKBH5 can regulate the osteogenic differentiation of MSCs and its role are unclear. In our study, we demonstrated that conditional knockout of Alkbh5 in bone marrow MSCs strengthened bone mass in mice. Mechanistically, we revealed that the m6A demethylase ALKBH5 plays a negative role in regulating the osteogenic capacity of MSCs by increasing the mRNA decay rate of protein arginine methyltransferase 6 (PRMT6) and discovered the phosphatidylinositol 3-kinase (PI3K)/AKT pathway as a crucial downstream target of the ALKBH5-PRMT6 axis. Together, our research unveils a different facet of the novel ALKBH5-PRMT6 axis that modulates the osteogenic differentiation of MSCs, which suggests a potential strategy for improving the efficiency of MSC-based tissue engineering for bone regeneration.
Results
Conditional knockout of Alkbh5 expression in mouse MSCs strengthened bone mass
To study the role of Alkbh5 in the osteogenic differentiation of bone marrow MSCs, we first constructed Prx1-Cre; Alkbh5fl/fl mice (Supplementary Fig. 1A, B). Compared with the Alkbh5fl/fl control littermates, Prx1-Cre; Alkbh5fl/fl mice showed no significant difference in size and weight (Fig. 1A, B). Western blotting detection confirmed that Prx1-Cre; Alkbh5fl/fl mouse bone tissue completely lacks Alkbh5 expression, but no change in the expression of the Alkbh5 protein was observed in adipose tissue and muscle tissue (Fig. 1C).

A Growth curves for Prx1-Cre; Alkbh5fl/fl and control mice over 24 wk (n = 8). B Representative photograph of a Prx1-Cre; Alkbh5fl/fl mouse and an Alkbh5fl/fl control littermate at 24 wk of age. C Western blotting to detect the expression of Alkbh5 in mouse femur, fat, and muscle. n = 8 in each group and only the results for three mice are displayed. D Representative computer renderings of bone structure in the femurs from Prx1-Cre; Alkbh5fl/fl and Alkbh5fl/fl mice at 24 wk (male, n = 8). E Quantitative μCT analyses of the distal end of femurs at 24 wk (male, n = 8). F Representative H&E staining of femoral sections from Prx1-Cre; Alkbh5fl/fl and Alkbh5fl/fl male mice. Scale bar, 100 µm (male, n = 8). G Representative Bglap immunohistochemical staining of femoral sections from Prx1-Cre; Alkbh5fl/fl and Alkbh5fl/fl male mice. Scale bars: left, 100 µm; right, 500 µm. All data are presented as the means ± SDs. **p < 0.01.
Micro-computed tomography (µCT) was used to scan the femurs of the mice and the results showed that Prx1-Cre; Alkbh5fl/fl mice had increased bone mass compared with their Alkbh5fl/fl control littermates (Fig. 1D). Analysis of the trabecular bone of the distal femur metaphysis revealed that compared with those in Alkbh5fl/fl mice, the bone mineral density and bone volume/tissue volume ratio (BV/TV) in Prx1-Cre; Alkbh5fl/fl mice increased by 32% and 29%, respectively. Furthermore, Alkbh5 deletion also increased the trabecular number (Tb. N), trabecular thickness (Tb. Th), and cortical thickness but decreased the trabecular separation (Fig. 1E). The hematoxylin and eosin (H&E) staining results were consistent with the µCT analysis results. Prx1-Cre; Alkbh5fl/fl mice had more trabecular bones than their Alkbh5fl/fl littermates (Fig. 1F). Immunohistochemical staining showed that the expression of bone gamma carboxyglutamate protein (Bglap) in bone tissue of Prx1-Cre; Alkbh5fl/fl mice increased compared with that in their control littermates (Fig. 1G). Overall, the results indicate that conditional knockout of Alkbh5 in mouse MSCs strengthened bone mass in mice.
The overall level of m6A is upregulated during the osteogenic differentiation of MSCs
We next considered whether ALKBH5 also plays an important role in the osteogenic differentiation of human MSCs. By selecting different induction time points for alizarin red S (ARS) staining, alkaline phosphatase (ALP) staining, and ALP activity detection, we confirmed the osteogenic differentiation ability of isolated and cultured MSCs in vitro (Fig. 2A, B), which was consistent with previous tests14. Next, we detected m6A methylation by an m6A mRNA dot blot and an m6A RNA methylation assay. As a result, the overall level of m6A gradually increased during osteogenic differentiation and peaked on the 14th day (Fig. 2C, D). Furthermore, the western blotting results showed that the expression of ALKBH5 gradually decreased (Fig. 2E, F). Correlation analysis showed that the expression level of ALKBH5 was negatively correlated with ALP activity (Fig. 2G). The above results suggest that the overall level of m6A during the osteogenic differentiation of MSCs was increased by the downregulation of the demethylating enzyme ALKBH5.

MSCs were cultured in osteogenic medium for up to 14 days. A ARS staining (top) and ALP staining (bottom) during the osteogenic differentiation of MSCs. Scale bar, 100 µm. B ALP activity was determined as units per gram of protein per 15 min and ARS staining was quantified as the absorbance at 562 nm (n = 3 independent experiments with three different MSC lines). C m6A dot blot detection of the overall m6A modification level at different osteogenic differentiation time points (n = 6). D An m6A RNA methylation assay was used to detect the overall m6A modification level at different time points (n = 6). E, F Protein level of ALKBH5 during osteogenic differentiation of MSCs (n = 9). G Correlation analysis of the expression level of ALKBH5 and ALP activity (n = 15). All data are presented as the means ± SDs. *p < 0.05.
ALKBH5 negatively regulates the osteogenic differentiation capacity of MSCs
Next, we detected the regulatory effect of ALKBH5 on osteogenic differentiation of MSCs. Quantitative PCR (qPCR) and western blotting verified that siALKBH5 had good knockout efficiency (Fig. 3G). The results showed that after interfering with ALKBH5 expression, ALP staining and activity increased. ARS staining found that the siALKBH5 group had a significantly greater number of calcium nodules than the control group (Fig. 3A, B). The immunofluorescence of collagen I showed increased expression in the siALKBH5 group compared to the control group (Fig. 3C). After overexpressing WT-ALKBH5, ALP staining and activity decreased. The ARS staining results were consistent with the ALP staining results (Fig. 3D, E). Studies have shown that a mutant ALKBH5 protein with the iron ligand residue H204 substituted to Ala completely lost its demethylation activity15. To determine whether the ALKBH5-mediated regulation of the osteogenic differentiation of MSCs is dependent on m6A modification, we also constructed the ALKBH5 catalytically active mutant Mut-ALKBH5 (H204A). The results showed that compared with the control group, there was no significant change in ALP staining, ALP activity, or ARS staining in the Mut-ALKBH5 group (Fig. 3D, E). The immunofluorescence of collagen I was generally consistent with the ARS staining results (Fig. 3F).

ALKBH5 was knocked down and overexpressed using siRNA and lentivirus, respectively. A ALP staining and ARS staining in the ALKBH5 knockdown or control group. Scale bar, 100 µm. B ALP activity and ARS staining quantification in the ALKBH5 knockdown or control group (n = 3 independent experiments with three different MSC lines). C The immunofluorescence of collagen I in the ALKBH5 knockdown or control group. Scale bar, 100 µm (n = 3 independent experiments with three different MSC lines). D ALP staining and ARS staining after using wild-type lentivirus (WT-ALKBH5) and mutant lentivirus (Mut-ALKBH5) to overexpress ALKBH5. Scale bar, 100 µm. E ALP activity and ARS staining quantification in the ALKBH5 overexpression or control group (n = 3 independent experiments with three different MSC lines). F The immunofluorescence of collagen I in the ALKBH5 overexpression or control group. Scale bar, 100 µm (n = 3 independent experiments with three different MSC lines). G Protein levels of the osteogenesis-associated markers Runx2 and SP7 were determined by western blot analysis (n = 3 independent experiments with three different MSC lines). All data are presented as the means ± SDs. *p < 0.05.
In addition, we determined the expression levels of the osteogenic markers RUNX family transcription factor 2 (RUNX2) and Sp7 transcription factor (SP7). The results showed that knocking down ALKBH5 increased the expression of RUNX2 and SP7, whereas overexpressing WT-ALKBH5 inhibited the expression of RUNX2 and SP7. However, overexpressing Mut-ALKBH5 did not change the expression of these two marker proteins (Fig. 3G). In summary, our data revealed that the demethylase ALKBH5 has a negative regulatory effect on the osteogenesis of MSCs through its m6A demethylation active site.
Whole-transcriptome m6A-seq and RNA-seq detection of ALKBH5 downstream regulatory genes
As ALKBH5 regulated the osteogenic differentiation of MSCs through m6A demethylation activity, we performed m6A sequencing (m6A-seq) on the control group and siALKBH5 group with two independent biological replicates. Consistent with previous m6A-seq results16,17, the canonical RRACH motif was highly enriched within m6A sites in both the control and small interfering RNA (siRNA) groups (Fig. 4A). These m6A modifications were predominantly located in the coding sequence and the 3′-untranslated region (Fig. 4B). m6A-seq analysis identified 43,872 and 44,701 m6A peaks from 14,472 to 14,637 m6A-modified genes in the control and siALKBH5 groups, respectively (Supplementary Fig. 2A). We further analyzed the total m6A peak density of mRNAs according to the m6A-seq results. The analysis showed that m6A peaks were abundant in the vicinity of the start and stop codons, especially at the position of the stop codon (Fig. 4C).

A Specific motif sequences of the siControl and siALKBH5 groups (two biological repeats per group). B Proportion of m6A peak distribution in the 5′-UTR, start codon, CDS, stop codon or 3′-UTR across the entire set of mRNA transcripts. C Density distribution of m6A peaks across mRNA transcripts. D Heatmap of genes with significantly different expression between the siControl and siALKBH5 groups. siCon-1 and siCon-2 are biological repeats; E GO enrichment analysis of differentially expressed genes. F–H IGV software analysis of the m6A peaks of SUZ12, SUV39H1, and PRMT6.
Next, we used RNA sequencing (RNA-seq) to reveal gene expression changes resulting from decreased ALKBH5 expression in MSCs. RNA-seq results showed that after knocking down ALKBH5 expression, there were a total of 790 genes with expression changes, including 377 upregulated genes and 413 downregulated genes (Fig. 4D and Supplementary Fig. 2B). Gene Ontology (GO) enrichment analysis of these changed genes indicated that a handful of the genes were associated with cell differentiation, histone methyltransferase activity, and activation of mitogen-activated protein kinase activity (Fig. 4E), and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses also revealed that genes altered by ALKBH5 were clustered in signaling pathways regulating the pluripotency of stem cells, Extracellular matrix (ECM)–receptor interactions, and the PI3K/Akt signaling pathway (Supplementary Fig. 2C), further indicating that ALKBH5 plays a role in the regulation of osteogenic differentiation of MSCs. By comparing the peak calling of related genes in the histone methyltransferase activity pathway through Integrative Genomics Viewer (IGV) analysis, it was found that SUV39H1 and SUZ12 did not have specific m6A peaks (Fig. 4F, G), whereas the PRMT6 was modified by m6A and the m6A peak in the siALKBH5 group was higher than that in the control group (Fig. 4H). Therefore, we speculated that PRMT6 may be the downstream target of ALKBH5 and selected this protein for further analysis.
ALKBH5 downregulates the expression of PRMT6 through m6A modification
To determine whether PRMT6 is directly regulated by ALKBH5, we found through methylated RNA immunoprecipitation qPCR (meRIP-qPCR) experiments that compared with the IgG group, m6A-specific antibodies can significantly enrich PRMT6, which is consistent with the m6A-seq results; after knocking down ALKBH5 expression, the enrichment of m6A methylation in PRMT6 was higher than that in the siControl group (Fig. 5A). Furthermore, the results of RNA immunoprecipitation qPCR (RIP-qPCR) showed that compared with the IgG group, the anti-ALKBH5 antibody significantly enriched PRMT6 (Fig. 5B). We found by qPCR and western blotting that knocking down ALKBH5 increased the expression of PRMT6 (Fig. 5C–E). The correlation analysis showed that there was a negative correlation between PRMT6 and ALKBH5 expression (Fig. 5F). The results confirmed that the methylation level of PRMT6 was regulated by ALKBH5.

A meRIP-qPCR analysis of PRMT6 mRNA in the siControl and siALKBH5 groups (n = 6). B RIP-qPCR detected whether ALKBH5 and PRMT6 were directly bound (n = 6). C qPCR results of PRMT6 after interfering with ALKBH5 expression (n = 9). D, E After interfering with ALKBH5 expression, western blotting was used to detect and quantify PRMT6 (n = 9). F Correlation analysis of PRMT6 and ALKBH5 during osteogenic differentiation of MSCs (n = 15). G Nuclear and cytoplasmic fractionation assays following qPCR in the siControl and siALKBH5 groups (n = 6). actin β (ACTB) and U6 small nuclear 1 (U6) were used as the positive controls. H After diminishing ALKBH5 expression, qPCR was used to detect the distribution of PRMT6 in the cytoplasm and nucleus (n = 6). I The RNA lifetime for PRMT6 in MSCs transfected with control siRNA or siRNA targeting ALKBH5 was determined by monitoring transcript abundance after adding actinomycin D (n = 9). All data are presented as the means ± SDs. **P < 0.01; ns, not statistically significant.
Next, we explored the specific regulatory mechanism of ALKBH5-mediated downregulation of PRMT6. qPCR results showed successful separation of cytoplasmic and nuclear mRNA (Fig. 5G). However, comparison of the expression of PRMT6 in the cytoplasm and nucleus showed that there was no difference between the control group and siALKBH5 group (Fig. 5H). Subsequently, we detected the mRNA decay rate of PRMT6 after knocking down ALKBH5 expression. The results showed that the degradation rate of PRMT6 was significantly slowed in the siALKBH5 group (Fig. 5I), indicating that ALKBH5 regulates the expression of PRMT6 mainly by affecting the mRNA degradation rate.
ALKBH5 regulates the osteogenic differentiation ability of MSCs mainly through PRMT6
As the functions of PRMT6 in the osteogenic differentiation of MSCs remain unclear, we first examined the expression pattern of PRMT6 during the osteogenic differentiation of MSCs. The results demonstrated that RPMT6 expression was upregulated after induction to the osteogenic lineage (Supplementary Fig. 3A). The correlation analysis showed that there was a positive correlation between PRMT6 and RUNX2 or SP7 mRNA expression (Supplementary Fig. 3B). ALP staining and activity were markedly suppressed upon PRMT6 knockdown. ARS staining showed that calcium nodules were significantly reduced after PRMT6 knockdown (Fig. 6A, B). Western blotting results showed that the expression of RUNX2 and SP7 decreased in the siPRMT6 group (Fig. 6C, D). In contrast, the ALP assay values and ARS staining were both increased after overexpressing PRMT6 (Fig. 6E, F). Western blotting demonstrated that PRMT6 overexpression increased the expression of RUNX2 and SP7 (Fig. 6G, H). Our data indicate that PRMT6 positively regulates the osteogenic differentiation of MSCs.

A ALP staining and ARS staining after simultaneous interference with ALKBH5 and PRMT6 expression. Scale bar, 100 µm. B ALP activity and ARS staining quantification after simultaneous interference with ALKBH5 and PRMT6 expression (n = 3 independent experiments with three different MSC lines). C, D Protein levels of the osteogenesis-associated markers RUNX2 and SP7 were determined by western blot analysis (n = 3 independent experiments with three different MSC lines). E ALP staining and ARS staining after overexpression of ALKBH5 and PRMT6. Scale bar, 100 µm. F ALP activity and ARS staining quantification after overexpression of ALKBH5 and PRMT6 (n = 3 independent experiments with three different MSC lines). G, H Western blot detection of RUNX2 and SP7 expression after overexpression of ALKBH5 and PRMT6 (n = 3 independent experiments with three different MSC lines). All data are presented as the means ± SDs. **p < 0.01.
Further studies demonstrated that simultaneous knockdown of ALKBH5 and PRMT6 decreased ARS and ALP assay values to the levels observed in the siControl group compared with the siALKBH5 group (Fig. 6A, B). In addition, simultaneous knockdown of ALKBH5 and PRMT6 reduced the expression of RUNX2 and SP7 to the level in the control group (Fig. 6C, D). In contrast, simultaneous overexpression of ALKBH5 and PRMT6 rescued the ARS and ALP results compared with those in the OE-ALKBH5 group (Fig. 6E, F). Western blotting results showed that overexpression of ALKBH5 and PRMT6 simultaneously increased the expression of RUNX2 and SP7 compared with that in the OE-ALKBH5 group (Fig. 6G, H). These results suggest that PRMT6 is a critical downstream target of ALKBH5-mediated inhibition of osteogenic differentiation of MSCs.
The ALKBH5-PRMT6 axis controls the activation of the AKT signaling pathway to modulate the osteogenesis of MSCs
Previous studies demonstrated that PRMT6 can regulate the activation of PI3K/AKT signaling pathway18,19 and the PI3K/AKT pathway plays an important role in regulating the osteogenesis of MSCs20. In addition, KEGG pathway enrichment analyses found that the gene-enriched pathway contains the PI3K/AKT pathway (Supplementary Fig. 1C). Therefore, we speculated that the ALKBH5-PRMT6 axis regulates osteogenesis of MSCs through the PI3K/AKT pathway. Western blotting results showed that the activation level of the AKT signaling pathway was increased after ALKBH5 knockdown but decreased in the WT-ALKBH5 group (Fig. 7A). In addition, the phosphorylation level of AKT was decreased in the siPRMT6 group (Fig. 7B). Further studies demonstrated that simultaneous knockdown of ALKBH5 and PRMT6 decreased the activation level of AKT to those observed in the siControl group compared with the siALKBH5 group, whereas simultaneous overexpression of ALKBH5 and PRMT6 rescued the phosphorylation level of AKT compared with that in the OE-ALKBH5 group (Fig. 7C, D).

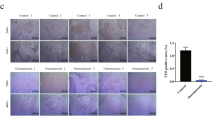
A Western blot detection of AKT phosphorylation level after interference with ALKBH5 expression. B Western blot detection of AKT phosphorylation level after knocking down PRMT6 expression. C, D Western blot detection of AKT phosphorylation level after simultaneous knockdown of ALKBH5 and PRMT6, and simultaneous overexpression of ALKBH5 and PRMT6. E, F ALP assay and ARS assay after adding LY294002 (PI3K/AKT pathway inhibitor), while knocking down ALKBH5. Scale bar, 100 µm. G ALP assay and ARS assay after adding SC79 (PI3K/AKT pathway activator), while overexpressing ALKBH5. Scale bar, 100 µm. H ALP assay and ARS assay after adding SC79, while knocking down PRMT6. Scale bar, 100 µm. All data are presented as the means ± SDs. **p < 0.01 (n = 3 independent experiments with three different MSC lines).
Subsequently, we evaluated the role of the PI3K/AKT pathway in the osteogenic differentiation of MSCs regulated by the ALKBH5-PRMT6 axis. Adding LY294002 (PI3K/AKT pathway inhibitor) while knocking down ALKBH5 decreased ARS and ALP assays to the level observed in the siControl group compared with siALKBH5 group (Fig. 7E, F), whereas adding SC79 (PI3K/AKT pathway activator) while overexpressing ALKBH5 rescued ARS and ALP assays compared with that in the OE-ALKBH5 group (Fig. 7G). Western blotting results demonstrated that the expression of RUNX2 and SP7 decreased in the siALKBH5 + LY294002 group compared with the siALKBH5 group (Supplementary Fig. 4A) but increased in the WT-ALKBH5 + SC79 group compared with the WT-ALKBH5 group (Supplementary Fig. 4B). Furthermore, ARS and ALP assays were increased to the level observed in the siControl group compared with the siPRMT6 group after adding SC79, while knocking down PRMT6 (Fig. 7H). Western blotting results showed that the expression of RUNX2 and SP7 in the siPRMT6 + SC79 group was rescued to the level of the siControl group (Supplementary Fig. 4C).

ALKBH5 negatively regulates the osteogenic differentiation of MSCs by increasing the mRNA decay rate of PRMT6, so as to inhibit the activation of PI3K/AKT pathway in bone development.
Discussion
In this study, we demonstrated that conditional knockout of Alkbh5 in bone marrow MSCs strengthened bone mass in mice. The overall level of m6A modification increased during the osteogenic differentiation of MSCs and the expression of the demethylase ALKBH5 gradually decreased, which negatively regulated the osteogenic differentiation of MSCs. Further research found that ALKBH5 accelerates the degradation rate of PRMT6 in an m6A-dependent manner, and that PRMT6 influences the activity of the PI3K/AKT signaling pathway to regulate the osteogenic differentiation of MSCs in vitro (Fig. 8).
The discovery of the m6A modification expands our understanding of epigenetic regulation in the life sciences. To date, a number of studies have shown that m6A modification participates in a variety of biological functions and plays an important role in regulating cell differentiation21. Interfering with the expression of METTL3 in pluripotent embryonic stem cells affects the differentiation of embryonic stem cells and makes them stay in a naive state22; imbalance of m6A modification regulation impairs the differentiation of fat precursor cells11 and hematopoietic stem cells23. In our study, we found that the overall level of m6A modification during osteogenesis of MSCs increased, demonstrating that m6A modification has different levels of activity before and after osteogenic differentiation. Subsequently, correlation analysis showed that the expression level of ALKBH5 was negatively correlated with ALP activity. Through functional studies, our research demonstrated that the demethylase ALKBH5 negatively regulates the osteogenic differentiation of MSCs through m6A modification, which has not been directly reported before.
To uncover the downstream target of ALKBH5, we performed m6A-seq and RNA-seq. By performing GO enrichment analysis, we found that a handful of genes were associated with histone methyltransferase activity. Previous studies have found that m6A modification can play a regulatory role through the histone pathway12 and histone modification plays an important role in regulating the osteogenic differentiation of MSCs24,25. Therefore, we wondered whether ALKBH5 can regulate the osteogenic differentiation of MSCs through the histone pathway. Then, we found that PRMT6 was a critical downstream target of ALKBH5. PRMT6 can catalyze the asymmetric dimethylation of histone H3 (Arg2 and Arg42), histone H2A (Arg29), and nonhistone proteins, and regulate cell signal transduction, transcriptional regulation, RNA splicing, and DNA repair26,27. At a mechanistic level, we demonstrated that ALKBH5 regulates the expression of PRMT6 mainly by affecting the degradation rate of PRMT6 mRNA but not by regulating its nucleocytoplasmic transport. We also found for the first time that PRMT6 promotes the osteogenic differentiation of MSCs. Furthermore, we uncovered that the PI3K/AKT pathway is an important downstream signaling pathway of the ALKBH5-PRMT6 axis. The PI3K/AKT signaling pathway plays not only an important role in regulating MSC osteogenic differentiation but also a key regulatory pathway for cell proliferation, growth, and survival28. Recent studies have indicated that m6A methylation regulates the activity of the PI3K/AKT pathway in endometrial cancers29 and acute myeloid leukemia30. Our research unexpectedly revealed that the demethylase ALKBH5 also influences the activity of the PI3K/AKT pathway in MSCs to regulate osteogenic differentiation. However, the specific genes regulated by the ALKBH5-PRMT6 axis in the PI3K/AKT pathway need to be further explored.
A primary study found that ALKBH5 is highly expressed in mouse testes and participates in the sperm development of mice15. A subsequent study found that ALKBH5 is essential for maintaining mouse brain development under hypobaric hypoxia31. In our research, we found that knocking out ALKBH5 in mouse MSCs had no significant effect on the growth of mice, but the bone mass and osteogenic activity of osteoblasts in Prx1-Cre; Alkbh5fl/fl mice increased compared with that in their control littermates. Recent studies have reported that the conditional knockout mice Prx1-Cre;Mettl3 fl/fl and Lepr-Cre;Mettl3 fl/fl have lower bone mass than their control littermates, proving that METTL3 has a positive effect on regulating the osteogenic differentiation of MSCs32. In addition, two other studies have confirmed the above results33,34. By generating Prx1-Cre; Alkbh5fl/fl mice, our data show that changing the m6A modification by regulating the expression of the methyltransferase METTL3 or demethylase ALKBH5 can lead to the regulation of MSC differentiation. Our research interestingly expands the understanding of the m6A modification-mediated regulation of the osteogenic differentiation of MSCs from a different aspect.
Under the influence of aging or pathological factors, the balance of osteoblasts and osteoclasts is often disrupted, resulting in a decrease in bone mass. Therefore, how to promote the conversion of bone tissue to osteoblasts and reverse the negative balance is of great significance for patients with low bone mass. Due to the critical role of m6A in modulating pathophysiological processes, some researchers are devoted to exploring small-molecule compounds related to methylases. Selberg et al.35 identified small-molecule ligands that increase m6A levels in RNA. In terms of inhibitors, the study also found several small-molecule inhibitors of the m6A demethylase FTO. Huang et al.36 developed a small-molecule compound, FB23-2, which can inhibit FTO, and found that FB23-2 significantly inhibits the progression of acute myeloid leukemia. Peng et al.37 identified entacapone, a U.S. Food and Drug Administration-approved drug that can serve as FTO inhibitors to treat metabolic disorders such as obesity and diabetes. A previous study also demonstrated that the nonsteroidal anti-inflammatory drug meclofenamic acid acts as a highly selective inhibitor of FTO38. Based on the studies reported above, we believe that the development of small-molecule compounds for methylase will have excellent application prospects in clinical treatment. Our research demonstrated that the demethylating enzyme ALKBH5 inhibits the osteogenic differentiation ability of MSCs. In future research, we can develop small-molecule inhibitors against ALKBH5, which will help improve the clinical effect of MSC transplantation in the treatment of low bone mass-related diseases.
In summary, we uncover the critical role of the novel ALKBH5-PRMT6 axis in modulating the osteogenic differentiation of MSCs. Our research expands the understanding of the role of m6A modification in regulating MSCs osteogenic differentiation, which is expected to provide new strategies for the treatment of patients with low bone mass by using MSCs. However, additional research still needs to be performed. To further confirm the therapeutic effect of ALKBH5, it is necessary to use pretreated MSCs to repair bone defects or osteoporosis and we are currently preparing to apply this theory to preclinical practice.
Materials and methods
Generation of conditional Alkbh5-knockout mice
Alkbh5fl/+ mice with C57BL/6 background were generated by Cyagen (Suzhou, China) using CRISPR/Cas-mediated genome engineering. Briefly, the Alkbh5 gene (NCBI Reference Sequence: NM_172943, Ensembl: ENSMUSG00000042650) is located on mouse chromosome 11. Exon 1 was selected as the conditional knockout region. To engineer the targeting vector, homologous arms and the Conditional knockout (CKO) region were generated by PCR using the BAC clone RP23-329 M3 as the template. Cas9, guide RNA, and the targeting vector were coinjected into fertilized eggs for CKO mouse production. The pups were genotyped by PCR followed by sequencing analysis.
Prx1-Cre transgenic mice were purchased from The Jackson Laboratory. We crossed Prx1-Cre mice with Alkbh5fl/+ mice to obtain Prx1-Cre; Alkbh5fl/+ mice as heterozygous conditional Alkbh5-knockout mice. By mating Prx1-Cre; Alkbh5 fl/+ male mice with Alkbh5fl/fl female mice, we obtained Prx1-Cre; Alkbh5fl/fl mice as homozygous conditional Alkbh5-knockout mice. The genotype of the transgenic mice was identified by PCR analyses of genomic DNA extracted from mouse tails. Primers for floxed Alkbh5-knockout allele genotyping were as follows: forward (5′-CAGGTTTGAAGTGGCCATAGTAGC-3′) and reverse (5′-GAGGCCAAGACAGGAGAATCAGAC-3′). Primers for Cre transgene genotyping were as follows: forward (5′-GCTCTGATGTTGGCAAAGGGGT-3′) and reverse (5′-AACATCTTCAGGTTCTGCGGG-3′). All mice were bred and maintained under specific pathogen-free conditions. All procedures involving animals were approved by the Animal Use and Care Committee of the Eighth Affiliated Hospital of Sun Yat-sen University.
MSC isolation and expansion
This study was approved by the Ethics Committee of Sun Yat-sen Memorial Hospital of Sun Yat-sen University, Guangzhou, China. Recruited healthy volunteers aged 20–30 years were fully informed of the relevant precautions and potential risks of bone marrow extraction, and signed the informed consent form. Bone marrow was collected from the posterior superior iliac spine of the volunteers and the MSCs were separated by density gradient centrifugation. The MSCs were resuspended in Dulbecco’s modified Eagle’s medium (DMEM, 1000 mg/L glucose, Gibco) containing 10% fetal bovine serum (FBS, Hangzhou Sijiqing Biological Engineering Material Company, Limited) and placed in a cell incubator containing 5% CO2 at 37 °C. After 48 h, the medium was changed to remove nonadherent cells. After 3 days, the medium was changed once. When the cell density reached 80–90%, the cells were digested with trypsin containing 0.53 mM EDTA and re-seeded in new flasks at passage 1. Cells from passage 3 to passage 5 were used for experiments.
Osteogenic differentiation induction
MSCs were seeded in a 12-well plate at a density of 0.6 × 105/well. After 12 h, when the cells adhered to the wells, the culture medium of the MSCs was changed to osteogenic differentiation medium consisting of DMEM (1000 mg/L glucose) with 10% FBS, 100 IU/mL penicillin, 100 IU/mL streptomycin, 0.1 μM dexamethasone, 10 mM β-glycerol phosphate, and 50 μM ascorbic acid (Sigma-Aldrich). The medium was changed every 3 days and continued to be induced to the desired time point.
ARS staining and quantification
The original medium was discarded from the well plate, 500 μl of 4% paraformaldehyde was added to each well and the plate was placed at room temperature for 30 min to fix the cells. After paraformaldehyde fixation, the plate was rinsed twice with phosphate-buffered saline (PBS), 500 μl of 1% ARS (pH 4.3) was added, and staining was performed for 20 min. Then, PBS was added to rinse the plate and the central field of view of the well plate was selected under the microscope to be photographed. For ARS quantification, 10% cetylpyridinium chloride monohydrate (Sigma-Aldrich) was used to destain the cells for 1 h at room temperature. Then, 200 μl of the liquid was transferred to a 96-well plate and the spectrophotometric absorbance was measured at 562 nm.
ALP staining and activity assay
ALP staining was performed according to the instructions of the BCIP/NBT Alkaline Phosphatase Color Development Kit (Beyotime Institute of Biotechnology). Briefly, MSCs were fixed in 4% paraformaldehyde for 15 min and then stained with mixture solution at 37 °C in the dark for 15 min. The central field of view of the well plate was selected to be photographed under the microscope.
For the ALP activity assay, an ALP assay kit (Nanjing Jiancheng Bioengineering Institute, China) was used. Then, 80 μl of radioimmunoprecipitation assay (RIPA) buffer was added to lyse cells in a well plate, followed by centrifugation at 14,000 r.p.m. for 10 min. A 96-well plate was prepared and 50 μl of A/1 solution and 50 μl of B/2 solution were added. Then, 30 μl of deionized water was added to the blank well, 30 μl of control solution was added to the standard well, and 30 μl of sample supernatant was added to the sample well. Then, the 96-well plate was placed in a 37 °C incubator for 15 min. After adding stop solution, the absorbance was measured at 405 nm. In addition, the protein concentration of the sample was determined by the Bicinchoninic acid (BCA) method, and finally, the ALP activity was calculated based on the measured ALP absorbance (optical density, OD) value and protein concentration; the unit of ALP activity was unit/g pro/15 min.
Western blotting
RIPA solution containing phosphatase inhibitor and protease inhibitor was added to lyse the cells. After measuring the protein concentration by the BCA method, equal amounts of each sample were diluted in 5× sodium dodecyl sulfate (SDS) loading buffer and denatured by boiling. Protein lysates were separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Millipore). The membranes were blocked in 5% nonfat dry milk dissolved in TBST (150 mM NaCl, 50 mM Tris-HCl pH 7.5, and 0.05% Tween-20) at room temperature for 1 h and then incubated overnight with primary antibodies against ALKBH5 (1 : 1000, ab195377, Abcam), total AKT (1/5000, ab179463, Abcam), p-AKT (1 : 1000, ab38449, Abcam), SP7 (1 : 1000, ab209484, Abcam), RUNX2 (1 : 1000, ab23981, Abcam), PRMT6 (1 : 1000, 14641 S, Cell Signaling Technology), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1 : 3000, AF0006, Beyotime) overnight at 4 °C. The protein signals were detected using chemiluminescent reagents (Millipore) according to the manufacturer’s instructions.
Quantitative reverse-transcriptase PCR
Total RNA from MSCs was extracted using TRIzol (Invitrogen) reagent. The RNA concentration was measured with a NanoDrop 2000 (Thermo Fisher Scientific). cDNA was transcribed by using PrimeScript RT Master Mix (Takara). Quantitative reverse-transcriptase PCR was performed using SYBR Green Premix Ex Taq (TaKaRa) in a Light-Cycler® 480 PCR System (Roche). Relative gene expression was normalized to GAPDH expression using the 2−ΔΔCt method. The primers are listed in Supplementary Table 1.
Cells transfection and chemical inhibition
The siRNAs were designed and synthesized by GenePharma (Shanghai, China). Three interference sequences were designed for the target gene. The interference efficiency was verified by qPCR and western blotting methods. Two sequences with a knockdown efficiency of >70% were selected for further experiments. Details of the sequences are shown in Supplementary Table 2. Transfection was carried out according to the instructions. After transfection, osteogenic induction medium was added to induce the induction of MSCs into osteoblasts. On the seventh day of osteoinduction, siRNA was transfected again for a second knockdown. The overexpression lentivirus was constructed by OBiO Technology (Shanghai, China). Mut-ALKBH5 contains a mutation of histidine to alanine at position 204. MSCs were incubated with the lentiviruses for 24 h at a multiplicity of infection of 30.
LY294002 and SC79 were purchased from Calbiochem and dissolved into dimethyl sulfoxide. MSCs were treated with 10 μM LY294002 and 4 µg/ml SC79 after siRNA or lentivirus transfection.
m6A dot blot
Total RNA was extracted from the cells using TRIzol and the RNA concentration was determined by a NanoDrop 2000 (Thermo Fisher Scientific). The RNA concentration of different samples was adjusted to 250 and 500 ng/µl. Two microliters of RNA was spotted onto the nylon membrane (Sigma-Aldrich, GERPN1210B), followed by 1500 J ultraviolet crosslinking twice. One of the membranes was dyed in methylene blue solution for 30 min. After 30 min, the membrane was rinsed twice with ultrapure water and photographed. The other membrane was blocked with 5% nonfat dry milk dissolved in PBST (PBS with 0.1% Tween-20) for 1 h, followed by incubation with m6A antibody (1 : 1000, 202003, Sysy) for 14–16 h. Then, PBST was added to wash the membrane for 5 min, which was repeated three times. An horseradish peroxidase-conjugated AffiniPure goat anti-mouse IgG dilution (1 : 3000, BA1050, BOSTER) was added and the membrane was incubated at room temperature for 1 h. Then, PBST solution was added again to rinse the membrane three times. The protein signals were detected using chemiluminescent reagents (Millipore) according to the manufacturer’s instructions.
m6A RNA methylation assay
This experiment was performed using the m6A RNA Methylation Assay Kit (ab185912, Abcam) according to the manual. In short, TRIzol was used to extract total RNA from the cells, the concentration was adjusted to 100 ng/μl after determining the RNA concentration, and 2 μl of RNA, 2 μl of negative control, and 2 μl of positive control were added to the well plate. Then, 100 μl of Developer Solution was added and the plate was incubated at room temperature for 6 min in the dark. Then, 100 μl of Stop Solution was added to stop the enzyme-linked reaction. The absorbance value was measured in a microplate reader (450 nm). The overall m6A methylation level was calculated according to the absorbance value and the formula used was as follows:
where Sample OD is the OD value of sample well, NC OD is the OD value of negative control well, PC OD is the OD value of positive control well, S is total mass of RNA added to sample well, and P is the total mass of the positive control added to the positive control well.
m6A-seq and RNA-seq
After using siRNA to interfere with the expression of ALKBH5 in MSCs, osteogenic differentiation was induced for 3 days. Total RNA was extracted using TRIzol reagent (Invitrogen). Approximately 50 µg of total RNA was subjected to isolation of poly(A) mRNA with poly-T oligo-attached magnetic beads (Invitrogen). After fragmentation, RNA was incubated with an m6A-specific antibody (No. 202003, Synaptic Systems, Germany) for immunoprecipitation. Then, eluted m6A-containing fragments and untreated input control fragments were converted to the final cDNA library in accordance with strand-specific library preparation by the dUTP method. Paired-end 2 × 150 bp sequencing was performed on an Illumina NovaSeq™ 6000 platform at LC-BIO Biotech, Ltd (Hangzhou, China).
meRIP-qPCR and RIP-qPCR
The Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore) was used for the RIP assay. Briefly, 1 × 107 cells were collected by RIP lysis buffer and then incubated with RIP buffer containing magnetic beads conjugated to anti-m6A (202003, sysy) and anti-ALKBH5 (ABE547, Merck Millipore) antibodies. Purified rabbit IgG was used as a negative control. Then, immunoprecipitated RNAs were isolated and purified for qPCR analysis to detect the presence of the target mRNA.
Nuclear and cytoplasmic fractionation
This experiment was performed according to the instructions of the PARIS™ Kit (AM1921, Thermo Fisher). The cells were collected after digestion with trypsin. Cell fractionation buffer was added to the cells and the cells were then centrifuged. Then, Cell Disruption Buffer was added to lyse the nuclei. Then, the mixture was transferred to a filter column, preheated elution solution was added to dissolve the RNA after centrifugation, and the RNA concentration was measured. After reverse transcription to cDNA, qPCR was used to detect the expression of the target gene.
RNA decay assay
After MSCs were transfected with siRNA, they were added to osteogenic induction medium and cultured for 3 days. Subsequently, 5 μg/ml actinomycin D was added. Total RNA at 0, 1, 2, and 4 h was extracted using TRIzol reagent (Invitrogen) and the relative expression was detected by qPCR according to a previously described method39.
µCT and histomorphometric analyses
We selected 6-month-old male mice for bone mass analysis. µCT was used to analyze the bone structure of the femur. The collected bone tissues were fixed in 4% polyoxymethylene for 2 days and then stored in 70% ethanol at 4 °C before being processed. To analyze the trabecular bone, images were acquired at an effective pixel size of 9.56 μm, a voltage of 80 kV, a current of 500 μA, and an exposure time of 1500 ms in each of the 360 rotational steps. The BV/TV, bone surface area/BV, Tb. Th, Tb. N, and trabecular spacing were analyzed according to the guidelines40. Two-dimensional and three-dimensional bone structure image slices were reconstructed.
H&E staining and immunohistochemical staining
The dissected femurs were fixed in 4% polyoxymethylene for 2 days and decalcified in 10% EDTA for 2 weeks before sectioning (5 μm). For H&E staining, the experiment was performed according to the manufacturer’s instructions (Beijing Solarbio Science & Technology Co, Ltd). For immunohistochemical staining, slides were treated with trypsin solution at 37 °C for 30 min for antigen retrieval and then incubated with rabbit anti-osteocalcin (1 : 500, Abcam, catalog number ab93876).
Immunofluorescence staining
MSCs were seeded on sterile glass coverslips and osteogenic differentiation was induced for 10 days. Cells were fixed in 4% paraformaldehyde for 30 min and then 0.1% Triton X-100 was added for 15 min at room temperature. Normal goat serum (5%) was used to block cells for 30 min. A primary antibody against collagen I (1 : 500, Abcam, ab34710) was added and the cells were incubated overnight at 4 °C. Then, anti-rabbit IgG (1 :5 00, Cell Signaling Technology, 4413) was added and incubated for 1 h at room temperature. 4′,6-Diamidino-2-phenylindole (DAPI) was used to counterstain the nuclei. Thereafter, the samples were viewed under a laser scanning confocal microscope at wavelengths of 555 nm (red, collagen I) and 405 nm (blue, DAPI).
Statistical analysis
The experiments in this study were independently repeated at least three times. SPSS 18.0 software (SPSS, Chicago, IL, USA) was used to analyze the experimental data and the results are expressed as the mean ± SD. Student’s t-test and one-way analysis of variance followed by the Bonferroni test was performed for statistical analyses. The bone mass of mice in the CKO and wild-type (WT) groups was compared using an unpaired two-tailed Student’s t-test. P < 0.05 was considered statistically significant; *P < 0.05, **P < 0.01, and ns represents no statistical significance.
Data availability
All Seq data have been deposited into Sequence Read Archive database with the identifier PRJNA727434.
References
Kartsogiannis, V. & Ng, K. W. Cell lines and primary cell cultures in the study of bone cell biology. Mol. Cell Endocrinol. 228, 79–102 (2004).
Florencio-Silva, R., Sasso, G. R. D. S., Sasso-Cerri, E., Simões, M. J. & Cerri, P. S. Biology of bone tissue: structure, function, and factors that influence bone cells. Biomed. Res. Int. 2015, 1–17 (2015).
Ding, D., Shyu, W. & Lin, S. Mesenchymal stem cells. Cell Transplant. 20, 5–14 (2011).
Marolt Presen, D., Traweger, A., Gimona, M. & Redl, H. Mesenchymal stromal cell-based bone regeneration therapies: from cell transplantation and tissue engineering to therapeutic secretomes and extracellular vesicles. Front. Bioeng. Biotechnol. 7, 352 (2019).
Guan, M. et al. Directing mesenchymal stem cells to bone to augment bone formation and increase bone mass. Nat. Med. 18, 456–462 (2012).
Iaquinta, M. R. et al. Adult stem cells for bone regeneration and repair. Front. Cell Dev. Biol. 7, 268 (2019).
Trounson, A. & McDonald, C. Stem cell therapies in clinical trials: progress and challenges. Cell Stem Cell 17, 11–22 (2015).
Gilbert, W. V., Bell, T. A. & Schaening, C. Messenger RNA modifications: form, distribution, and function. Science 352, 1408–1412 (2016).
Batista, P. J. The RNA modification N 6 -methyladenosine and its implications in human disease. Genomics Proteomics Bioinformatics 15, 154–163 (2017).
Meyer, K. D. & Jaffrey, S. R. The dynamic epitranscriptome: N-6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 15, 313–326 (2014).
Zhao, X. et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 24, 1403–1419 (2014).
Wang, Y. et al. N(6)-methyladenosine RNA modification regulates embryonic neural stem cell self-renewal through histone modifications. Nat. Neurosci. 21, 195–206 (2018).
Zhang, Q. et al. The RNA demethylase FTO is required for maintenance of bone mass and functions to protect osteoblasts from genotoxic damage. Proc. Natl Acad. Sci. USA 116, 17980–17989 (2019).
Li, J. et al. TRAF4 positively regulates the osteogenic differentiation of mesenchymal stem cells by acting as an E3 ubiquitin ligase to degrade Smurf2. Cell Death Differ. 26, 2652–2666 (2019).
Zheng, G. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013).
Dominissini, D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).
Meyer, K. D. et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646 (2012).
Jiang, N. et al. PRMT6 promotes endometrial cancer via AKT/mTOR signaling and indicates poor prognosis. Int. J. Biochem. Cell Biol. 120, 105681 (2020).
Li, T., Fanning, K. V., Nyunoya, T., Chen, Y. & Zou, C. Cigarette smoke extract induces airway epithelial cell death via repressing PRMT6/AKT signaling. Aging (Albany, NY) 12, 24301–24317 (2020).
Liu, X. et al. Lifelong accumulation of bone in mice lacking Pten in osteoblasts. Proc. Natl Acad. Sci. USA 104, 2259–2264 (2007).
Shi, H., Wei, J. & He, C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 (2019).
Geula, S. et al. m(6)A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 347, 1002–1006 (2015).
Zhang, C. et al. m(6)A modulates haematopoietic stem and progenitor cell specification. Nature 549, 273 (2017).
Deng, P., Chen, Q., Hong, C. & Wang, C. Histone methyltransferases and demethylases: regulators in balancing osteogenic and adipogenic differentiation of mesenchymal stem cells. Int. J. Oral Sci. 7, 197–204 (2015).
Yi, S. et al. Bone remodeling: histone modifications as fate determinants of bone cell differentiation. Int. J. Mol. Sci. 20, 3147 (2019).
Guccione, E. & Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 20, 642–657 (2019).
Peng, C. & Wong, C. C. L. The story of protein arginine methylation: characterization, regulation, and function. Expert. Rev. Proteomics 14, 157–170 (2017).
Kandel, E. S. & Hay, N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp. Cell. Res. 253, 210–229 (1999).
Liu, J. et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 20, 1074–1083 (2018).
Vu, L. P. et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 23, 1369–1376 (2017).
Ma, C. et al. RNA m6A methylation participates in regulation of postnatal development of the mouse cerebellum. Genome Biol. 19, 1–18 (2018).
Wu, Y. et al. Mettl3-mediated m6A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat. Commun. 9, 4772 (2018).
Yan, G. et al. m6A methylation of precursor-miR-320/RUNX2 controls osteogenic potential of bone marrow-derived mesenchymal stem cells. Mol. Ther. Nucleic Acids 19, 421–436 (2020).
Tian, C., Huang, Y., Li, Q., Feng, Z. & Xu, Q. Mettl3 regulates osteogenic differentiation and alternative splicing of Vegfa in bone marrow mesenchymal stem cells. Int. J. Mol. Sci. 20, 551 (2019).
Selberg, S. et al. Discovery of small molecules that activate RNA methylation through cooperative binding to the METTL3-14-WTAP complex active site. Cell Rep. 26, 3762–3771 (2019).
Huang, Y. et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell 35, 677 (2019).
Peng, S. et al. Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Sci. Transl. Med. 11, u7116 (2019).
Huang, Y. et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 43, 373–384 (2015).
Huang, H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295 (2018).
Bouxsein, M. L. et al. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res. 25, 1468–1486 (2010).
Acknowledgements
We thank American Journal Experts for providing English language editing of the manuscript. We are also grateful for the assistance given by the Experimental Animal Center of the East Campus of Sun Yat-sen University.
Funding
This work was supported by the National Natural Science Foundation of China [81971518 and 81871750], Key-Area Research and Development Program of Guangdong Province [2019B020236001], Shenzhen Key Laboratory of Ankylosing Spondylitis [ZDSYS20190902092851024], and Health Welfare Fund Project of Futian District [FTWS2019020].
Author information
Authors and Affiliations
Contributions
Z.L. performed most of the experiments and wrote the manuscript. P.W. and J.L. designed the experiments, analyzed the data, and generated the figures. Z.X., S.C., and M.L. isolated and cultured human bone marrow mesenchymal stem cells. W.L., G.Y., G.Z., and M.M. performed the animal experiments in mice. S.W. and W.Y. analyzed the m6A-seq results. Y.W. and H.S. conceived this study, supervised it, and wrote and edited the manuscript.
Corresponding authors
Ethics declarations
Ethics statement
This study was approved by the Ethics Committee of The Eighth Affiliated Hospital and Sun Yat-sen Memorial Hospital, Sun Yat-sen University. All of the health donors were given and accepted informed consent form prior to their enrollment.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Y. Shi
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Z., Wang, P., Li, J. et al. The N6-methyladenosine demethylase ALKBH5 negatively regulates the osteogenic differentiation of mesenchymal stem cells through PRMT6. Cell Death Dis 12, 578 (2021). https://doi.org/10.1038/s41419-021-03869-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-021-03869-4
This article is cited by
-
Analysis of m6A regulators related immune characteristics in ankylosing spondylitis by integrated bioinformatics and computational strategies
Scientific Reports (2024)
-
Epitranscriptomic modifications in mesenchymal stem cell differentiation: advances, mechanistic insights, and beyond
Cell Death & Differentiation (2024)
-
FOXO1 regulates osteogenic differentiation of periodontal ligament stem cells through the METTL3 signaling pathway
Journal of Orthopaedic Surgery and Research (2023)
-
Hypoxia-cleavable and specific targeted nanomedicine delivers epigenetic drugs for enhanced treatment of breast cancer and bone metastasis
Journal of Nanobiotechnology (2023)
-
ALKBH5 facilitates CYP1B1 mRNA degradation via m6A demethylation to alleviate MSC senescence and osteoarthritis progression
Experimental & Molecular Medicine (2023)
