
Abstract
The Alectoris Chukar (chukar) is the most geographically widespread partridge species in the world, demonstrating exceptional adaptability to diverse ecological environments. However, the scarcity of genetic resources for chukar has hindered research into its adaptive evolution and molecular breeding. In this study, we have sequenced and assembled a high-quality, phased chukar genome that consists of 31 pairs of relatively complete diploid chromosomes. Our BUSCO analysis reported a high completeness score of 96.8% and 96.5%, with respect to universal single-copy orthologs and a low duplication rate (0.3% and 0.5%) for two assemblies. Through resequencing and population genomic analyses of six subspecies, we have curated invaluable genotype data that underscores the adaptive evolution of chukar in response to both arid and high-altitude environments. These data will significantly contribute to research on how chukars adaptively evolve to cope with desertification and alpine climates.
Similar content being viewed by others

Background & Summary
Alectoris Chukar (chukar) commonly known as “chukar”, is a member of the Galliformes order and the Phasianidae family, hailing from the stony semi-desert regions of Asia, Western Europe, and the Middle East. This species has been introduced to numerous other countries such as the United States, Canada, England, and New Zealand, primarily for stocking on game farms or releasing for hunting purposes1,2. In recent years, there has been an uptick in the use of chukars for meat production under controlled husbandry conditions. Owing to their rapid growth, high productivity, and superior meat quality, chukars are ideally suited for commercial production3,4,5,6. The domestication of these partridges coupled with selection for growth traits enhances their potential as a prime source of high-quality protein for human consumption.
The wild chukar, a polytypic species with 22 subspecies scattered globally, exhibits a broad spectrum of environmental adaptations. Among these are the six subspecies of pubescens, potanini, pallida, falki, dzungarica, and pallescens, with pubescens and pallida exclusively found in China. These subspecies have evolved to survive in their specific habitats, which span a wide range of temperatures and altitudes. For instance, while falki is adapted to a drier environment, pallescens thrives in the highlands of Tibet (altitude >4000 m).
This environmental adaptation capacity and genetic diversity among the chukar subspecies underline the significance of comprehensive genomic research in this species. Whole-genome sequence assembly has proven to be a fundamental tool for extensive genomics initiatives, including evolutionary studies and efficient breeding strategies. Over the years, critical poultry species like chickens, turkeys, and ducks have substantially reaped the benefits of these genomic resources7,8,9,10. Notably, the chicken reference genome has undergone several refinements, making it one of the superior vertebrate genomes available and establishing it as a model for avian research9. However, the chukar partridge’s genomic advancement is currently stymied due to the absence of a reference genome. The introduction of phased-genome assembly, renowned for its precision in resolving complex genomic variations11, could be instrumental in breaking this impasse, thus transforming the genetic improvement of chukar breeding, spurring evolutionary studies, and securing genetic resource conservation.
In this study, we employ a de novo assembly strategy to present the first continuous, accurate, phased-resolved genome for the chukar. Utilizing this genome, we resequenced and analyzed five wild subspecies and one domestic population of chukar. Our research provides valuable resources for investigating adaptive evolution, breeding, and conservation genomics of crucial ecological species.
Method
Ethics statement
The collection and handling of the samples in this study were carried out in accordance with approved guidelines and regulations from both Lanzhou University and Shanghai Jiao Tong University.
Sample collections
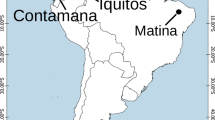
We sequenced the genome of a female domesticated chukar collected from Tianming Alectoris chukar Farm (Guangzhou, Guangdong, China), which was primarily used for genome assembly. The transcriptomes of two adult female domesticated chukars, which were collected from Tianming Alectoris Chukar Farm (including the one used for genome assembly), were sequenced for annotation of coding genes in the genome. In addition, we included the genomes resequenced from a total of 58 chukars for genotype identification. These samples include 14 pubescens, 12 potanini, 9 pallida, 14 falki, 2 pallescens, and 7 domestic chukars (refer to Table 1 for more details). Pubescens, potanini, pallida, and falki muscle samples were obtained from 10 distinct locales including Akesai(AK), Changji(CJ), Dongdashan(DD), Helanshan(HL), Jingtai(JT), Kuerle(KE), Quzi (QZ), Subei (SB), Tongchuan (TC) and Wudu (WD) during 2002–2008, representing the majority of chukar’s geographical area in China and reflecting various geographic, topographic, and climatic conditions (Fig. 1, Table 1). To avoid sampling near relatives, each bird within a location was collected from a different portion of the colony. These 49 samples were donated by Lanzhou University. Three and four blood samples were collected at random from domestic (DOM) chukars in Tianming farm (Jieyang, China) and Qinxiangyuan farm (Jieyang, China), respectively. To avoid the selection of relatives, the pedigrees of these DOM chukar were investigated (Table 1). Two muscle samples of pallescens sampled in Tibet (TB) province were received from the animal branch of the southwest China germplasm bank of wildlife (Yunnan, China) (Table 1).

Sampling distribution map of chukar subspecies. Pubescens (orange), potanini (red), pallida (green), falki (blue), pallescens (brown), and domestic (pink) samples were obtained from 12 distinct locales including Akesai(AK), Changji(CJ), Dongdashan(DD), Helanshan(HL), Jingtai(JT), Kuerle(KE), Quzi (QZ), Subei (SB), Tongchuan (TC),Wudu (WD), Tibet (TB), and Jieyang(DOM). The horizontal axis represents longitude, and the vertical axis represents latitude.
De novo sequencing and assembly of the chukar phased genome
The DNA samples of the female domesticated chukar blood were extracted and sequenced using PacBio single-molecule real-time (SMRT) sequencing and Illumina paired-end sequencing technology. We carried out SMRT DNA sequencing of ∼20 kb inserts using the PacBio Sequel II platform (Personal Biotechnology Co., Ltd. Shanghai, China). Next, 400 bp paired-end libraries (refer to Illumina TruSeq DNA Sample Preparation Guide) constructed from the same genomic DNA were sequenced on the Illumina HiSeq platform (Personal Biotechnology Co., Ltd. Shanghai, China). The DNA sample of the chukar muscle was extracted and sequenced using Hi-C technology in Beijing Nuohezhiyuan Technology Service Co, Ltd. We filtered and trimmed the Illumina and Hi-C reads to remove adapters and low-quality bases using the standard settings in SOAPnuke v1.5.012.
After low-quality and adaptor reads were filtered, we obtained ~136.59 Gb long sequencing data that were used to assemble the genome (Table S1a). De novo assembly followed the PacBio string graph assembler process, using FALCON v2.1.4, FALCON-Unzip v0.4.0, and FALCON-phase v0.2.013 to generate long-range phased haplotypes. FALCON computes an initial assembly by correcting errors in raw reads and subsequently assembling them using a string graph formulated from read overlaps. Following this, FALCON-Unzip identified read haplotypes based on the phasing information derived from detected heterozygous positions. These phased reads are then deployed to assemble both haplotigs and primary contigs. As a result of this assembly process, there were 363 and 1,711 contigs for the primary contigs and haplotigs respectively (Table 2). The cumulative lengths of these two haploid assemblies were 1.03 Gb and 0.85 Gb, with their Contig N50 lengths reaching approximately 29.3 Mb and 1.1 Mb each. Subsequently, FALCON-Phase inputs the partially phased long-read assembly from FALCON-Unzip, and extends the phasing on the contigs using ~182 Gb filtered Hi-C data from the same sample. This achieves a phased contig-level genome assembly of chukar, resulting in the production of both Hap1 Contigs and Hap2 Contigs with a total length of 1.03 Gb and contig N50 length of 1.06 Mb (Table 2, Table S1a). Furthermore, Jellyfish v2.1.4 (https://github.com/gmarcais/Jellyfish) was used in conjunction with GenomeScope v1.0.014 to calculate genome size and heterozygosity in the chukar genome a k-mer frequency of 18. The genome size estimated (~1.0 Gb) was consistent with the genome assembled (Fig. 4a).
After the contig-level assemblies were generated, they were respectively polished using Pilon v1.2215, with the help of ~124.12 Gb high-quality Illumina data. Scaffolding was performed on these polished sequences using ALLHiC v0.9.816, with the support of ~182 Gb Hi-C sequencing data. In our next steps, Ragag Scaffold v1.1.017 was employed for a reference genome-assisted methodology to construct more comprehensive chukar haplotype genomes and ascertain chromosome IDs of scaffolds. The chicken chromosome-assembled genome (GRCg6a)18 served as the reference due to its close evolutionary relationship with chukar and shared chromosome number (n = 78)19. This stage merely oriented and ordered draft assembly sequences into longer sequences without modifying the input query sequence. Finally, we generated two phased pseudo-haplotype genomes of chukar (Hap1 Scaffolds and Hap2 Scaffolds) with 31 pairs of chromosomes (Fig. 2b and Table S2). The N50 length for both Hap1 Scaffolds and Hap2 Scaffolds was significantly enhanced, reaching 93.6 Mb and 93.5 Mb respectively.

Diagram of genome assembly and phylogenetic relationship of Alectoris chukar (chukar). (a) Photograph of the chukar. (b) Synteny map of primary and associated assembly.
We evaluated the completeness of two haplotype genomes via BUSCO v5.1.220 benchmarking using the aves_odb10 dataset. BUSCO reported 96.8% and 96.5% complete universal single-copy orthologs and a low rate of duplication (0.3% and 0.5%) for the Hap1 scaffolds and Hap2 scaffolds, respectively (Fig. 3). This indicated that the genome quality of Hap1 scaffolds and Hap2 scaffolds was better than those of other recently published bird genome assembly21,22,23,24. Because of the higher completeness and quality of the scaffolded Hap1 assembly, it was selected as the chukar reference genome for downstream analysis. The pairwise genome alignments of Hap1 and Hap2 were performed using MUMer v4.0.025. The collinearity analysis of the Hap1 scaffolds and Hap2 scaffolds is shown in Fig. 2b.

Histogram of BUSCO assessment of the chukar haplotype genome.

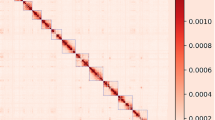
(a) The distribution of the k-mer frequency of the chukar genome. (b) Genome-wide Hi-C heatmap of Alectoris chukar, where both the horizontal and vertical axes represent genomic loci along chromosomes. The chromosomes are arranged sequentially from top to bottom (on the vertical axis) and from left to right (on the horizontal axis), displaying chromosomes Chr1 through Chr28, followed by Chr30, Chr31, Chr33, ChrZ, and ChrW. (c)The pairwise genome alignments of the chukar genome and the chicken genome are shown, displaying chromosomes with a length greater than 1 Mb.
Repeat sequence and gene annotation
To identify genomic repeats, we used the RepeatMasker v4.1.2 (http://www.repeatmasker.org/) to scan the chukar genome sequence of primary assembly. RMBlast v2.11.0 (https://www.repeatmasker.org/rmblast/) was used as the alignment engine. The Dfam 3.026 and Repbase27 were used and we specified the species library as ‘chicken’ for the chukar. The overall GC content of the chukar genome was estimated to be 41.81%, which is similar to that of the other reference bird species. Interspersed repeats accounted for approximately 9.8% of the whole genome, spanning 101.47 Mb, and consisted of approximately 90.78 Mb retroelements and 10.27 Mb DNA transposons. Approximately 7.19% of the sequences were identified as long interspersed nuclear elements (LINEs), which were thus the largest component, whereas 1.52% of the sequences were identified as long terminal repeats (LTRs). The chicken repeat 1 group was the most abundant, occupying 99.9% of the identified LINEs. The overall level of repetitive content in the chukar (Table 3) was similar to that in the common pheasant21 and chicken18 and greater than those of the turkey7 as well as most sequenced birds28, which may be attributed to the advantage of the long-read sequencing technology.
Gene annotation was performed using a combined strategy of ab initio predictions, homologue prediction, and transcriptome evidence. RNA from the liver, spleen, muscle, thymus gland, bursa of fabricius, and kidney of these two chukars was isolated, library prepared, and sequenced using Illumina technology in Beijing Nuohezhiyuan Technology Service Co, Ltd. Equal amounts of RNA from each of these six tissues were mixed for single-molecule long-read RNA sequencing (Iso-Seq) to obtain full-length transcriptomic data. Illumina reads were filtered by Trimmomatic v0.3929 and then combined to input to Trinity v2.14.0 (https://github.com/trinityrnaseq/trinityrnaseq) for transcript assembly. Iso-Seq reads were processed using SMRT tools v5.1.0 and ISO-SEQ v3.1 software packages. The pipeline includes five main steps to obtain high-quality sequences: (1) generating circular consensus (CCS) reads, (2) demultiplexing and primer removal and classifying full-length CCS reads, (3) clustering full-length non-chimeric (FLNC) sequences, and, finally, (4) polishing FLNC sequences (Table S1d). We use PASA v2.5.2 (https://github.com/PASApipeline/PASApipeline) to align assembled transcripts obtained from Illumina reads and Iso-Seq reads to the chukar genome sequences and then TransDecoder v5.50 (https://github.com/TransDecoder/TransDecoder) from the Trinity v2.14.0 to identify the likely open reading frame within the transcripts. The ab initio gene prediction was performed using Augustus based on chicken models30. For homology-based annotation, GeMoMa v1.931 was employed using gene annotation information from chicken. Finally, all the results were integrated using the EVidenceModeler pipeline v1.1.132. Alternative splicing analysis of the transcripts uses SUPPA v2.333, which generates seven different alternative splicing types, including skipped exon (SE), alternative 5′/3′ splice sites (A5/A3), mutually exclusive exons (MX), retained intron (RI) and alternative first/last exons (AF/AL). The final gene models comprised 20,082 transcripts, spanning a 302 Mb genomic region. These genes were annotated by using the UniProt/SwissProt protein database and validated 17,997 protein products. In addition, 109,477 splicing events were identified corresponding to all genes. Among seven type splicing events, the largest number of splicing events in chukar was alternative first exons (28,722, 26%), followed by Retained Intron (25865, 24%) and alternative 5′ splice site (17235, 16%) (Table S2b).
Population-based resequencing and variation calling
Genomic resequencing was performed for each individual on MGI-SEQ. 2000 platform. Raw reads were subjected to SOAPnuke v1.5.012 processing to remove sequencing adapters and low-quality reads. Following whole-genome resequencing of domestic chukar and five wild chukar subspecies, a total of 1,071.8 Gb of the clean base was obtained, with an average depth of 18× (Table S1b). High-quality reads were aligned to the chukar genome that we assembled above using the Burrows−Wheeler Aligner v0.5.934. Variants were called using the GATK tool suite v4.2.6.135. Briefly, potential PCR duplicates were marked using MarkDuplicates option. The HaplotypeCaller option was used to construct general variant calling files for all the samples by invoking -ERC:GVCF. All of gVCF files were combined using GenotypeGVCFs option to form a single variant calling file. To obtain high-quality SNPs and Indels, we used the GATK hard filter to filter the merged VCF data with the best practices recommended parameters36. After quality filtering, 2,574,885 high-quality INDELs and 14,988,840 high-quality SNPs were identified. Following the removal of SNPs and INDELs using vcftools v0.1.1637 with the parameters ‘–mac 3–maf 0.05–min-meanDP 5–minQ 30–max-missing 0.95’, a total of 6,991,669 SNPs and 757,682 INDELs from 58 individuals were retained.
Data Records
The chukar genome assembly reported in this paper have been deposited in the Genbank under the project PRJNA780965 with the accession number JAXHPU00000000038. The variation files for this study are located under analysis ERZ22149693 at EVA (European Variation Archive), with the accession number PRJEB713396039. The PacBio sequencing data (SRR27640724) were specifically used to construct the genome assembly. For variant analysis, we utilized whole-genome resequencing data (SRR16961228-SRR16961264). Additionally, transcriptome data from (SRR26796665-SRR26796677 and SRR27640723) informed our genome annotation. These datasets, integral to our project, are available through NCBI BioProject PRJNA78096540.
Technical Validation
Jellyfish was used in conjunction with GenomeScope14 to calculate genome size and heterozygosity in the chukar genome using a k-mer frequency of 18. The genome was consistent with the genome size estimated (1.0 Gb) (Fig. 3a). The Hi-C heatmap revealed a well-organized interaction contact pattern along the diagonals within/around the chromosome (Fig. 3b), which indirectly confirmed the accuracy of the chromosome assembly. The pairwise genome alignments of the chukar genome and the chicken genome using NGenomeSyn41 demonstrate a high level of consistency, which also implies that the genome assembly is accurate and reliable (Fig. 3c). To further assess the quality of assembled chukar genome, we have undertaken an extensive quality assessment by mapping 58 genome resequencing samples to it. The resulting quality metrics, including a coverage of over 99%, a sequencing depth greater than 15X, and a mapping rate of more than 98%, are detailed in Table S3. These metrics not only confirm the technical soundness but also the high quality of the genome assembly. We also evaluated the completeness of two haplotype genomes via BUSCO v5.1.2 benchmarking using the aves_odb10 dataset20. BUSCO reported 96.8% and 96.5% complete universal single-copy orthologs and a low rate of duplication (0.3% and 0.5%) for the Hap1 scaffolds and Hap2 scaffolds, respectively (Fig. 3).
Code availability
The genome and transcriptome analyses were performed following the manuals and protocols of the cited bioinformatic sofware. No new codes were written for this study.
References
Robinson, A. C., Larsen, R. T., Flinders, J. T. & Mitchell, D. L. Chukar Seasonal Survival and Probable Causes of Mortality. The Journal of Wildlife Management 73, 89–97 (2009).
Barbanera, F. et al. Genetic structure of Mediterranean chukar (Alectoris chukar, Galliformes) populations: conservation and management implications. Naturwissenschaften 96, 1203–1212 (2009).
Iqbal, F. et al. A Bayesian approach for describing the growth of Chukar partridges. European Poultry Science 83, 284 (2019).
Yİlmaz, A. & Tepeli, C. Breeding performance of a captive chukar partridge (Alectoris chukar) flock. Journal of Animal and Veterinary Advances 8, 1584–1588 (2009).
Caglayan, T., Kirikci, K. & Aygun, A. Comparison of hatchability and some egg quality characteristics in spotted and unspotted partridge (Alectoris chukar) eggs. Journal of Applied Poultry Research 23, 244–251 (2014).
Sariyel, V., Aygun, A. & Keskin, I. Comparison of growth curve models in partridge. Poultry science 96, 1635–1640 (2017).
Dalloul, R. A. et al. Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): genome assembly and analysis. PLoS Biol 8 (2010).
Hillier, L. W. et al. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432, 695–716 (2004).
Warren, W. C. et al. A New Chicken Genome Assembly Provides Insight into Avian Genome Structure. G3 (Bethesda) 7, 109–117 (2017).
Huang, Y. et al. The duck genome and transcriptome provide insight into an avian influenza virus reservoir species. Nat Genet 45, 776–783 (2013).
Sedlazeck, F. J., Lee, H., Darby, C. A. & Schatz, M. C. Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nat Rev Genet 19, 329–346 (2018).
Chen, Y. et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 7, 1–6 (2018).
Chin, C. S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13, 1050–1054 (2016).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963 (2014).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat Plants 5, 833–845 (2019).
Alonge, M. et al. RaGOO: fast and accurate reference-guided scaffolding of draft genomes. Genome Biology 20, 224 (2019).
Hillier, L. W., Miller, W., Birney, E., Warren, W. & Hardison, R. C. Gallus gallus breed Red Jungle fowl, inbred line UCD001 isolate RJF #256, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:AADN00000000.00000005 (2018).
Ouchia-Benissad, S. & Ladjali-Mohammedi, K. Banding cytogenetics of the Barbary partridge Alectoris barbara and the Chukar partridge Alectoris chukar (Phasianidae): a large conservation with Domestic fowl Gallus domesticus revealed by high resolution chromosomes. Comp Cytogenet 12, 171–199 (2018).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: Assessing Genome Assembly and Annotation Completeness. Methods Mol Biol 1962, 227–245 (2019).
He, C. et al. Chromosome level assembly reveals a unique immune gene organization and signatures of evolution in the common pheasant. Mol Ecol Resour 21, 897–911 (2021).
Peona, V. et al. Identifying the causes and consequences of assembly gaps using a multiplatform genome assembly of a bird-of-paradise. Mol Ecol Resour 21, 263–286 (2021).
Vignal, A. et al. A guinea fowl genome assembly provides new evidence on evolution following domestication and selection in galliformes. Mol Ecol Resour 19, 997–1014 (2019).
Chattopadhyay, B. et al. Novel genome reveals susceptibility of popular gamebird, the red-legged partridge (Alectoris rufa, Phasianidae), to climate change. Genomics 113, 3430–3438 (2021).
Delcher, A. L., Phillippy, A., Carlton, J. & Salzberg, S. L. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res 30, 2478–2483 (2002).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob DNA 12, 2 (2021).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA 6, 11 (2015).
Zhang, G. et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346, 1311–1320 (2014).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Keilwagen, J., Hartung, F. & Grau, J. GeMoMa: Homology-Based Gene Prediction Utilizing Intron Position Conservation and RNA-seq Data. Methods Mol Biol 1962, 161–177 (2019).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9, R7 (2008).
Alamancos, G. P., Pagès, A., Trincado, J. L., Bellora, N. & Eyras, E. Leveraging transcript quantification for fast computation of alternative splicing profiles. Rna 21, 1521–1531 (2015).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498 (2011).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Zhou, H., Huang, X. H., Du, B. W., Song, S. & Meng, H. Alectoris chukar genome assembly. GenBank https://identifiers.org/ncbi/insdc:JAXHPU000000000 (2023).
European Variation Archive (EVA) https://identifiers.org/ena.embl:PRJEB71339 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP346448 (2021).
He, W. et al. NGenomeSyn: an easy-to-use and flexible tool for publication-ready visualization of syntenic relationships across multiple genomes. Bioinformatics 39 (2023).
Acknowledgements
The work was supported by the Animal Branch of the Southwest China Germplasm Bank of wildlife, Chinese Academy of Sciences (the Large Research Infrastructure Funding). We thank Dr. Yan Zhang (Carilion Clinic, Virginia, USA) for his help in the revision of this manuscript. We also thank Dr. Naifa Liu (Lanzhou University, China), Dr. Xiaolin Chen (Xiamen University, China), and Dr. Xinkang Bao (Lanzhou University, China) for their assistance in making the project possible.
Author information
Authors and Affiliations
Contributions
H.Z., X.H.H., B.W.D., S.S. and H.M. are the principal investigators and project managers in this research; X.H.H., Y.R.S., J.Y.Z., Z.X.W., Y.J.X., B.W.D., Q.W., M.Z. and S.S. provided the sample for the study. H.Z. and H.M. conducted data sequencing; C.Q., L.X.L., K.C.C., Y.M.Z. and S.Y.J. contributed to data presentation; H.Z., J.J.L., J.M.D., K.X., W.Q.Z., C. H., L.Y.Y. and J.S.Z. performed the sequencing data analysis; X.H.H., B.W.D. and S.S. evaluated study quality; H.Z. and H.M. wrote and edited the manuscript, with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhou, H., Huang, X., Liu, J. et al. De novo Phased Genome Assembly, Annotation and Population Genotyping of Alectoris Chukar. Sci Data 11, 162 (2024). https://doi.org/10.1038/s41597-024-02991-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-02991-0
