
Abstract
The type I interferon (IFN) response is the body’s typical immune defense against viruses. Previous studies linked high expression of genes encoding type I IFNs in the brain’s choroid plexus to cognitive decline under virus-free conditions in aging and neurodegeneration. Multiple reports have documented persisting cognitive symptoms following recovery from COVID-19. Cumulative evidence shows that the choroid plexus is one of the brain regions most vulnerable to infection with the coronavirus SARS-CoV-2, and manifests increased expression of genes encoding type I IFNs even in the absence of viral traces within the brain. In this Perspective, we propose that the type I IFN defensive immune response to SARS-CoV-2 infection in the choroid plexus poses a risk to cognitive function if not resolved in a timely manner.
Similar content being viewed by others


Main
The brain’s choroid plexus (Box 1) serves as the interface within the brain’s ventricles between the cerebrospinal fluid (CSF) bathing the brain and the circulation. This interface is responsible for allowing passage of nutrients and other molecules that the brain needs for its lifelong health, while preventing entry of microorganisms such as viruses and bacteria to the brain. Here, based on the understanding that this anatomical barrier can express an antiviral immunological signature under virus-free conditions1,2,3,4, we propose that although the choroid plexus is endowed with an active protective role that can destroy viruses before they infect the brain, its defensive activities could also have negative consequences for the brain if not fully resolved in a timely manner. Multiple reports document cases of broad-spectrum neurological symptoms in individuals with COVID-19 after recovery, including persistent cognitive impairment5,6. In principle, cognitive decline following viral infection might result from a direct damage to the brain caused by the virus itself, or from the body’s response to the virus, if not optimally controlled. Given the global scale of the COVID-19 pandemic, the elucidation of the mechanisms potentially driving cognitive deterioration and the identification of predisposing factors are a pressing medical need. In this Perspective, we propose a link between impaired cognitive ability after severe infection with SARS-CoV-2 and the type I interferon (IFN) antiviral response in the choroid plexus epithelium. While such a response is part of the body’s natural immune defense against viruses, when persistent, it was shown to be detrimental to brain function1,7.
SARS-CoV-2 neurotropism in the choroid plexus
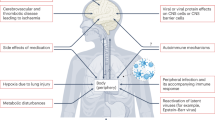
The choroid plexus has been proposed as one of the potential sites through which SARS-CoV-2 virus might enter the brain8,9. However, direct SARS-CoV-2 neuroinvasion is debatable, as traces of SARS-CoV-2 RNA or proteins are not always detected in brain autopsy samples of individuals with COVID-19 disease10. In addition, in cases where traces of SARS-CoV-2 RNA or proteins were found within the brain, they did not correlate with neurological symptoms10. Moreover, in mice, SARS-CoV-2 spike protein accumulates in several regions of the body, including the choroid plexus, but is not detected in the brain parenchyma or CSF11. It is thus very unlikely that direct viral damage to the brain is the leading cause of cognitive deficits following COVID-19. Human brain organoids exposed to SARS-CoV-2 spike pseudovirus or SARS-CoV-2 viral isolates had little to no presence of viral proteins in neuronal and glial cells, but high infection of choroid plexus epithelial cells12,13. These observations in human brain organoids were linked to the higher expression of angiotensin-converting enzyme 2 (ACE2), one of the receptors for SARS-CoV-2, in the choroid plexus epithelial cells relative to neuronal and glial cells12 (Fig. 1a). Consistent with these results, analysis of publicly available human and mouse brain transcriptome databases showed that while ACE2 gene expression is displayed by many neuronal and nonneuronal cell types, it is higher in some brain areas, including the choroid plexus9. Similarly, histological examination of postmortem human brains showed more intense ACE2 protein staining in the choroid plexus epithelium than in the brain parenchyma14. Conceivably, due to its relatively high expression of ACE2, the choroid plexus epithelium may be a site with high risk of attack by the SARS-CoV-2 virus (Fig. 1b) and, at the same time, as we propose here, the site that intercepts the virus and mounts an antiviral response that blocks viral entry into the brain parenchyma.

a, In the healthy state, the choroid plexus epithelium responds to type I IFN (IFN-I) signals and IFN-γ released by effector T cells in the circulation. The balance between type I IFN and IFN-γ responses in the choroid plexus (CP) epithelium regulates homeostatic choroid plexus functions such as barrier integrity, immune cell trafficking to the brain and release of neurotrophins, which are crucial for the maintenance of normal brain function27. b, During acute SARS-CoV-2 infection, type I IFN and IFN-γ responses in the choroid plexus epithelium increase as a result of elevated type I IFN and IFN-γ signals in the circulation, and possibly also due to a direct response to viral infection by choroid plexus epithelial cells, which express several molecules required for SARS-CoV-2 entry, notably ACE2. Consequently, the choroid plexus epithelium releases pro-inflammatory mediators such as IFN-β, complement (for example, C1S, C3 and C7)15 and CCL/CXCL family chemokines (for example, CCL2, CCL11 and CXCL10)19, which are in turn sensed by microglia and astrocytes, leading to their activation. c, Pro-inflammatory cytokines, including IFN-β, released by activated microglia and astrocytes, as well as debris from damaged cells, maintain the choroid plexus epithelium in an inflamed state. Following severe COVID-19, the host’s systemic immune effector functions, including type I IFN and IFN-γ production, are reduced. T cell populations with an exhausted phenotype, characterized by high expression of inhibitory immune checkpoint receptors (IICRs) like PD-1, increase in frequency. Increased inflammation within the brain and reduced IFN-γ from the periphery sustain elevated activation of type I IFN signaling in the choroid plexus epithelium, leading to progressive loss of choroid plexus function, microglial and astrocyte dysfunction, deterioration of synapses, and cognitive impairment.
Studies with human brain organoids infected with SARS-CoV-2 (refs. 12,13) and single-nucleus transcriptome analysis of postmortem choroid plexus samples from individuals with COVID-19 (refs. 15,16) collectively show prominent choroid plexus alterations, including impairment of blood–CSF barrier properties, increased cell death, cell functional deficits, aberrant leukocyte infiltration and inflammation. In agreement, a retrospective analysis of clinical findings in the CSF of patients with COVID-19 and neurological symptoms revealed that the majority of individuals had a high CSF/serum albumin ratio, an indicator of blood–CSF barrier dysfunction17. Given the broad importance of choroid plexus functions for brain homeostasis (Box 1), their impairment as an outcome of SARS-CoV-2 infection likely plays a role in COVID-19 neuropathogenesis.
Type I IFN and cognitive impairment
Type I IFN (for example, IFN-α and IFN-β) signaling is a distinctive immunological defense response to viral infection. Type I IFN responses at the brain’s barriers, including the choroid plexus epithelium, have been linked to cognitive impairment in a mouse model of infection with the vesicular stomatitis virus M2 (ref. 18). Of note, however, bulk RNA-sequencing analyses of the choroid plexus transcriptome showed enhanced type I IFN signaling also under noninfectious conditions, such as normal aging1 and neurodegenerative diseases of diverse etiology that are also associated with reduced cognitive ability in mice and humans, such as Alzheimer’s disease2,3 and Niemann–Pick’s disease type C4. In particular, type I IFN signaling at the choroid plexus is associated with cognitive impairment and reduced hippocampal neurogenesis in aged mice1. It is also associated with altered expression of several genes involved in the maintenance of blood–CSF barrier integrity and CSF composition in the J20 mouse model of Alzheimer’s disease2. Intracerebroventricular injection of an antibody blocking the type I IFN receptor (IFNAR) restored cognition in aged mice1, while overexpression of IFN-β in the choroid plexus of young mice induced an aging-like microglial phenotype and cognitive impairment7, suggesting a direct link between type I IFN signaling at the choroid plexus and cognitive dysfunction.
Single-nucleus transcriptome analysis of postmortem brain samples from individuals who died with COVID-19 showed strong expression of type I IFN signatures (for example, IFITM3 and STAT3) in the choroid plexus in the absence of both RNA and protein traces of SARS-CoV-2 in the brain15. Analysis of the cortices of the same postmortem brains identified subpopulations of microglia and astrocytes that expressed genes previously linked with Alzheimer’s disease (for example, C1Q and CD14) and inflammation or astrogliosis (for example, IFITM3 or GFAP), respectively15. In addition, broad dysregulation of genes associated with synaptic transmission was found in upper-layer excitatory neurons (for example, VAMP2, SNAP25 and ATP6V0C) as well as in proximal inhibitory neurons, potentially suggestive of cognitive deficits15. Furthermore, the in silico reconstruction of the choroid plexus-to-cortex signaling networks showed enhanced complement and CCL/CXCL chemokine pro-inflammatory pathways15. In support, the postmortem choroid plexus of individuals who were infected with SARS-CoV-2 expressed the C3, C7 and C1S complement genes15. In a mouse model of mild respiratory COVID, chemokines including CCL11, CCL2 and CXCL10 were persistently elevated in the CSF up to 7 weeks after infection19. Increased expression of complement components and CCL/CXCL chemokines in the choroid plexus has been associated with brain diseases. The postmortem examination of the choroid plexus of patients with Alzheimer’s disease revealed the presence of lipid deposits containing complement components (for example, C1q, C3 and C5) correlating with cognitive decline20. Similar deposits were also observed in the choroid plexus of ApoE−/− and ApoE4 knock-in (ApoE4-KI) mice fed a high-fat diet, concomitantly with high expression of genes encoding type I IFNs (also present in ApoeE4-KI mice fed normal chow)20. Of note, Ccl11 chemokine gene expression in the choroid plexus was found to increase in mice during aging and was connected with type I IFN signaling1. Moreover, systemic administration of CCL11 in mice impairs hippocampal neurogenesis and induces cognitive deficits19,21. Finally, expression of the chemokines encoded by Ccl2 and Cxcl10 in the choroid plexus was linked to myeloid cell homing to the brain under neurodegenerative conditions, and their reduced expression at the choroid plexus is associated with disease progression in the 5xFAD mouse model of Alzheimer’s disease22. Yet excess Ccl2 and Cxcl10 transcripts in the choroid plexus of mouse embryos are associated with disruption of the blood–CSF barrier following maternal immune activation during gestation, an experimental model for postnatal neurodevelopmental disorders23.
Taken together, it is possible that, upon SARS-CoV-2 infection, the choroid plexus relays inflammatory signals to the brain in a manner that, at least in part, may involve local type I IFN responses, and that these mediators have the potential to cause cognitive impairment. As such, it is conceivable that, in patients infected with SARS-CoV-2, type I IFN signaling in the choroid plexus epithelium constitutes a defensive response to arrest viral propagation to the brain (Fig. 1b). This physiological defensive response, however, if persistent, may negatively affect the brain parenchyma, especially microglia and astrocytes7,15,24,25 (Fig. 1b,c). Moreover, the type I IFN responses in microglia and astrocytes can further fuel the type I IFN responses in the choroid plexus epithelium through their production of pro-inflammatory cytokines such as IFN-β, tumor necrosis factor, interleukin (IL)-6 and IL-1β1,24,25 (Fig. 1c). Thus, the protective type I IFN antiviral response in the choroid plexus epithelium might trigger an inflammatory reaction within the brain parenchyma that perpetuates the choroid plexus type I IFN response and endangers brain function.
IFN-γ and cognitive loss
Type I IFN responses in the choroid plexus epithelium may not only be the consequence of direct signals from within the brain parenchyma, but also be amplified by reduction of type II IFN (or IFN-γ) signals from the periphery1,2. IFN-γ is necessary for normal brain function, including cognitive performance and social behavior1,26. In addition, IFN-γ is required for the expression by the choroid plexus epithelium of leukocyte trafficking molecules (for example, CCL2, CXCL10, ICAM-1 and VCAM-1) that recruit immune cells from the circulation, such as monocyte-derived macrophages, that could resolve brain damage27 (Fig. 1a). Reduced IFN-γ signaling at the choroid plexus has been associated with aging1 and chronic neurodegeneration2,22, and decreased expression of IFN-γ in the plasma of patients with Alzheimer’s disease correlates with cognitive decline28. Reduced production of IFN-γ in the periphery may result from decreased systemic immune effector functions27. Immunological features compatible with systemic immune exhaustion and suppression have been documented in patients with COVID-19, including impaired IFN-γ activity29, increased expression of inhibitory immune checkpoints30 (Fig. 1c), lymphopenia31 and increased proportions of regulatory T (Treg) cells with enhanced suppressive capabilities32. Of note, increased numbers of Treg cells may be a direct consequence of increased expression of type I IFN in the periphery33.
Because the periphery represents a more accessible compartment for intervention than the brain, the revitalization of systemic immunity after resolution of SARS-CoV-2 infection might be a potential strategy to prevent cognitive vulnerability. Such treatment should be administered after the acute phase of the infection to avoid any potential adverse synergy between immunotherapy-related and COVID-19-related immune effects. It was reported that enhanced IFN-γ signaling at the choroid plexus can be achieved by targeting inhibitory immune checkpoints, such as the 1PD-1–PD-L1 pathway, in the periphery as a strategy to fight neurodegeneration34. The possibility of deploying the same strategy to treat late sequelae of COVID-19 is currently being investigated in several clinical trials31.
Concluding remarks
The mechanisms underlying the cognitive deficits observed in some individuals recovering from COVID-19 are a matter of intense investigation. While the uncontrolled antiviral defense response at the choroid plexus may not be the sole factor inducing cognitive dysfunction after severe SARS-CoV-2 infection35, it is very likely an important component of this pathway. We base this contention on the well-established negative effects of chronic type I IFN signaling in the choroid plexus epithelium in aging and chronic neurodegeneration, in mice and humans, which impacts microglial and astrocytic activities that may impair cognitive function. While further studies are needed, we argue that the site, level and duration of the immune response to SARS-CoV-2, and perhaps to other viruses, critically affect the fate of the brain, and potentially other tissues, in the infected host.
References
Baruch, K. et al. Aging. Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93 (2014).
Mesquita, S. D. et al. The choroid plexus transcriptome reveals changes in type I and II interferon responses in a mouse model of Alzheimer’s disease. Brain. Behav. Immun. 49, 280–292 (2015).
Stopa, E. G. et al. Comparative transcriptomics of choroid plexus in Alzheimer’s disease, frontotemporal dementia and Huntington’s disease: implications for CSF homeostasis. Fluids Barriers CNS 15, 18 (2018).
Van Hoecke, L. et al. Involvement of the choroid plexus in the pathogenesis of Niemann–Pick disease type C. Front. Cell. Neurosci. 15, 757482 (2021).
Liu, Y.-H. et al. One-year trajectory of cognitive changes in older survivors of COVID-19 in Wuhan, China: a longitudinal cohort study. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2022.0461 (2022).
Hampshire, A. et al. Cognitive deficits in people who have recovered from COVID-19. EClinicalMedicine 39, 101044 (2021).
Deczkowska, A. et al. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat. Commun. 8, 717 (2017).
Piras, M. et al. Strong ACE-2 expression in the choroidal vessels: do high choroid plexuses serve as a gateway for SARS-CoV-2 infection on the human brain? Eur. Rev. Med. Pharmacol. Sci. 26, 3025–3029 (2022).
Chen, R. et al. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front. Neurol. 11, 573095 (2020).
Cosentino, G. et al. Neuropathological findings from COVID-19 patients with neurological symptoms argue against a direct brain invasion of SARS-CoV-2: a critical systematic review. Eur. J. Neurol. 28, 3856–3865 (2021).
Brady, M. et al. Spike protein multiorgan tropism suppressed by antibodies targeting SARS-CoV-2. Commun. Biol. 4, 1318 (2021).
Pellegrini, L. et al. SARS-CoV-2 infects the brain choroid plexus and disrupts the blood–CSF barrier in human brain organoids. Cell Stem Cell 27, 951–961 (2020).
Jacob, F. et al. Human pluripotent stem cell-derived neural cells and brain organoids reveal SARS-CoV-2 neurotropism predominates in choroid plexus epithelium. Cell Stem Cell 27, 937–950 (2020).
Deffner, F. et al. Histological evidence for the enteric nervous system and the choroid plexus as alternative routes of neuroinvasion by SARS-CoV-2. Front. Neuroanat. 14, 596439 (2020).
Yang, A. C. et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 595, 565–571 (2021).
Fullard, J. F. et al. Single-nucleus transcriptome analysis of human brain immune response in patients with severe COVID-19. Genome Med. 13, 118 (2021).
Jarius, S. et al. Cerebrospinal fluid findings in COVID-19: a multicenter study of 150 lumbar punctures in 127 patients. J. Neuroinflammation 19, 19 (2022).
Blank, T. et al. Brain endothelial- and epithelial-specific interferon receptor chain 1 drives virus-induced sickness behavior and cognitive impairment. Immunity 44, 901–912 (2016).
Fernández-Castañeda, A. et al. Mild respiratory COVID can cause multi-lineage neural cell and myelin dysregulation. Cell 185, 2452–2468 (2022).
Yin, C. et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat. Med. 25, 496–506 (2019).
Villeda, S. A. et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94 (2011).
Baruch, K. et al. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nat. Commun. 6, 7967 (2015).
Cui, J. et al. Inflammation of the embryonic choroid plexus barrier following maternal immune activation. Dev. Cell 55, 617–628 (2020).
Cox, D. J. et al. DNA sensors are expressed in astrocytes and microglia in vitro and are upregulated during gliosis in neurodegenerative disease. Glia 63, 812–825 (2015).
Nazmi, A. et al. Chronic neurodegeneration induces type I interferon synthesis via STING, shaping microglial phenotype and accelerating disease progression. Glia 67, 1254–1276 (2019).
Filiano, A. J. et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behaviour. Nature 535, 425–429 (2016).
Deczkowska, A., Baruch, K. & Schwartz, M. Type I/II interferon balance in the regulation of brain physiology and pathology. Trends Immunol. 37, 181–192 (2016).
Yang, H.-S. et al. Plasma IL-12/IFN-γ axis predicts cognitive trajectories in cognitively unimpaired older adults. Alzheimers Dement. 18, 645–653 (2022).
Ruetsch, C. et al. Functional exhaustion of type I and II interferons production in severe COVID-19 patients. Front. Med. 7, 603961 (2021).
Diao, B. et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front. Immunol. 11, 827 (2020).
Pezeshki, P. S. & Rezaei, N. Immune checkpoint inhibition in COVID-19: risks and benefits. Expert Opin. Biol. Ther. 21, 1173–1179 (2021).
Galván-Peña, S. et al. Profound Treg perturbations correlate with COVID-19 severity. Proc. Natl Acad. Sci. USA 118, e2111315118 (2021).
Kasper, L. H. & Reder, A. T. Immunomodulatory activity of interferon-beta. Ann. Clin. Transl. Neurol. 1, 622–631 (2014).
Baruch, K. et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat. Med. 22, 135–137 (2016).
Aschman, T., Mothes, R., Heppner, F. L. & Radbruch, H. What SARS-CoV-2 does to our brains. Immunity 55, 1159–1172 (2022).
Acknowledgements
We thank S. Schwarzbaum (Weizmann Institute) for proofreading the manuscript, and G. Brodsky (Weizmann Institute) for help with the artwork. M.S. is supported by the Advanced European Research Council grants 232835 and 741744, the European Seventh Framework Program HEALTH-2011 (279017), the Israel Science Foundation (ISF)-research grant no. 991/16, the ISF-Legacy Heritage Bio-medical Science Partnership research grant no. 1354/15, the Thompson Foundation and the Adelis Foundation.
Author information
Authors and Affiliations
Contributions
All the authors contributed to the writing of the manuscript and the design of the figure.
Corresponding author
Ethics declarations
Competing interests
M.S. is an inventor of the intellectual property that forms the basis for development of PD-L1 immunotherapy for AD.
Peer review
Peer review information
Nature Immunology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Ioana Visan, in collaboration with the Nature Immunology team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Suzzi, S., Tsitsou-Kampeli, A. & Schwartz, M. The type I interferon antiviral response in the choroid plexus and the cognitive risk in COVID-19. Nat Immunol 24, 220–224 (2023). https://doi.org/10.1038/s41590-022-01410-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41590-022-01410-z
This article is cited by
-
Cullin5 drives experimental asthma exacerbations by modulating alveolar macrophage antiviral immunity
Nature Communications (2024)
-
Type I Interferon Signalling and Ischemic Stroke: Mechanisms and Therapeutic Potentials
Translational Stroke Research (2024)
-
Brain Pathology in COVID-19: Clinical Manifestations and Potential Mechanisms
Neuroscience Bulletin (2024)
