
Abstract
Coffea arabica, an allotetraploid hybrid of Coffea eugenioides and Coffea canephora, is the source of approximately 60% of coffee products worldwide, and its cultivated accessions have undergone several population bottlenecks. We present chromosome-level assemblies of a di-haploid C. arabica accession and modern representatives of its diploid progenitors, C. eugenioides and C. canephora. The three species exhibit largely conserved genome structures between diploid parents and descendant subgenomes, with no obvious global subgenome dominance. We find evidence for a founding polyploidy event 350,000–610,000 years ago, followed by several pre-domestication bottlenecks, resulting in narrow genetic variation. A split between wild accessions and cultivar progenitors occurred ~30.5 thousand years ago, followed by a period of migration between the two populations. Analysis of modern varieties, including lines historically introgressed with C. canephora, highlights their breeding histories and loci that may contribute to pathogen resistance, laying the groundwork for future genomics-based breeding of C. arabica.
Similar content being viewed by others


Main
Polyploidy is a powerful evolutionary force that has shaped genome evolution across many eukaryotic lineages, possibly offering adaptive advantages in times of global change1,2. Such whole-genome duplications (WGDs) are particularly characteristic of plants3, and a great proportion of crop species are polyploid4,5,6,7,8,9,10,11. Our understanding of genome evolution following WGD is still incomplete, but outcomes can include genomic shock, in terms of activation of cryptic transposable elements (TEs), subgenome-partitioned gene regulation or fractionation, homoeologous exchange (HE), meiotic instability and even karyotype variation8,12,13,14,15,16. Alternatively, few or none of the above phenomena can materialize, and the two subgenomes can coexist harmonically, gradually adapting to new ploidy levels17. Regardless, the most common fate of polyploids appears to be fractionation and eventual reversion to the diploid state18.
With an estimated production of 10 million metric tons per year, coffee is one of the most traded commodities in the world. The most broadly appreciated coffee is produced from the allotetraploid species Coffea arabica, especially from cultivars belonging to the Bourbon or Typica lineages and their hybrids19. C. arabica (2n = 4x = 44 chromosomes) resulted from a natural hybridization event between the ancestors of present-day Coffea canephora (Robusta coffee, subgenome CC (subCC)) and Coffea eugenioides (subgenome EE (subEE)), each with 2n = 2x = 22. The founding WGD has previously been dated to between 10,000 and 1 million years ago20,21,22,23, with the Robusta-derived subgenome of C. arabica most closely related to C. canephora accessions from northern Uganda24. Arabica cultivation was initiated in fifteenth- to sixteenth-century Yemen (Extended Data Fig. 1). Around 1600, the so-called seven seeds were smuggled out of Yemen25, establishing Indian C. arabica cultivar lineages. A century later, the Dutch began cultivating Arabica in Southeast Asia—thus setting up the founders of the contemporary Typica group. One plant, shipped to Amsterdam in 1706, was used to establish Arabica cultivation in the Caribbean in 1723. Independently, the French cultivated Arabica on the island of Bourbon (presently Réunion)26, and the descendants of a single plant that survived by 1720 form the contemporary Bourbon group. Contemporary Arabica cultivars descend from these Typica or Bourbon lineages, except for a few wild ecotypes with origins in natural forests in Ethiopia. Due to its recent allotetraploid origin and strong bottlenecks during its history, cultivated C. arabica harbors a particularly low genetic diversity20 and is susceptible to many plant pests and diseases, such as coffee leaf rust (Hemileia vastatrix). As a result, the classic Bourbon–Typica lineages can be cultivated successfully in only a few regions around the world. Fortunately, a spontaneous C. canephora × C. arabica hybrid resistant to H. vastatrix was identified on the island of Timor27 in 1927. Many modern Arabicas contain C. canephora introgressions derived from this hybrid, ensuring rust resistance, but having also unwanted side effects, such as decreased beverage quality28.
Modern genomic tools and a detailed understanding of the origin and breeding history of contemporary varieties are vital to developing new Arabica cultivars, better adapted to climate change and agricultural practices29,30,31. Here, we present chromosome-level assemblies of C. arabica and representatives of its progenitor species, C. canephora (Robusta) and C. eugenioides (hereafter Eugenioides). Whole-genome resequencing data of 41 wild and cultivated accessions facilitated in-depth analysis of Arabica history and dissemination routes, as well as the identification of candidate genomic regions associated with pathogen resistance.
Results
The genomes of C. arabica, C. canephora and C. eugenioides
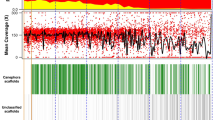
As reference individuals, we chose the di-haploid Arabica line ET-39 (ref. 32), a previously sequenced doubled haploid Robusta33 and the wild Eugenioides accession Bu-A, respectively. Long- and short-read-based hybrid assemblies were obtained (Methods and Supplementary Sections 2.1 and 2.2), spanning 672 megabases (Mb) (Robusta), 645 Mb (Eugenioides) and 1,088 Mb (Arabica), respectively. Upon Hi-C scaffolding, the Robusta and Arabica assemblies consisted of 11 and 22 pseudochromosomes, and spanned 82.7% and 62.5%, respectively, of the projected genome sizes (Table 1). To improve the Arabica assembly, we generated a second assembly using Pacific Biosciences (PacBio) HiFi technology followed by Hi-C scaffolding (Methods and Supplementary Sections 2.2 and 2.3). This assembly was 1,198 Mb long, of which 1,192 Mb (93.1% of the predicted genome size based on cytological evidence34) was anchored to pseudochromosomes (Table 1). Gene space completeness, assessed using Benchmarking Universal Single-Copy Orthologs (BUSCOs)35, was >96% for all assemblies. Importantly, 93.2% of the BUSCO genes were duplicated in the HiFi assembly (Table 1), indicating that most of the gene duplicates from the allopolyploidy event were retained.
The Robusta and Eugenioides genomes contained, respectively, 67.5% and 59.7% TEs (Supplementary Section 3.2), with Gypsy long terminal repeat (LTR) retrotransposons accounting for most of the difference between the two species. This difference was greatly reduced (63.1% and 63.8%) in the two Arabica subgenomes (subCC and subEE, stemming from Robusta and Eugenioides ancestors, respectively), possibly indicating TE transfer via HE. Robusta contained considerably more recent LTR TE insertion elements than Eugenioides. Again, the two Arabica subgenomes showed greater similarity to each other in recent LTR TE insertions than the two progenitor genomes. No major evidence was found for LTR TE mobilization following Arabica allopolyploidization, in contrast to what has been observed in tobacco36, but similar to Brassica synthetic allotetraploids37. Observed Arabica genome evolution instead more closely follows the ‘harmonious coexistence’ pattern38 seen in Arabidopsis hybrids17,39.
High-quality gene annotations, followed by manual curation of specific gene families (Supplementary Sections 3.1–3.4), resulted in 28,857, 32,192, 56,670 and 69,314 gene models for the Robusta, Eugenioides, PacBio Arabica and Arabica HiFi assemblies, respectively (Table 1). Altogether, ~97% of Robusta and 99.6% of Arabica HiFi gene models were placed on the pseudochromosomes, with 33,618 and 35,449, respectively, to subgenomes subCC and subEE (Table 1). Annotation completeness from BUSCO was ≥95% for Eugenioides and Robusta, and reached 97.3% for Arabica HiFi.
Genome fractionation and subgenome dominance
Comparison of Arabica subCC and subEE against their Robusta and Eugenioides counterparts revealed high conservation in terms of chromosome number, centromere position and numbers of genes per chromosome (Fig. 1 and Supplementary Section 4). Patterns of gene loss following the gamma paleohexaploidy event displayed high structural conservation between Robusta and Eugenioides during the 4–6 million years since their initial species split22,23 (Supplementary Section 4). Likewise, the structures of the two Arabica subgenomes were highly conserved between each other, with, since the Arabica-founding allotetraploidy event, only ~5% of BUSCO genes having reverted to the diploid state (Fig. 1a and Table 1). Syntenic comparisons revealed that genomic excision events, removing one or several genes at a time in similar proportions across the two subgenomes, have been the main driving force in genome fragmentation both before and after the polyploidy event (Fig. 1b and Supplementary Section 4). Fractionation occurred mostly in pericentromeric regions, whereas chromosome arms showed more moderate paralogous gene deletion (Fig. 1c and Supplementary Section 4). The Arabica allopolyploidy event seemingly did not affect the rate of genome fractionation, which remained roughly constant when comparing deletions in progenitor species versus Arabica subgenomes after the event. In support of the dosage-balance hypothesis40, subgenomic regions with high duplicate retention rates were significantly enriched for genes that originated from the Arabica WGD (Fisher exact test, P < 2.2 × 10−16). In contrast, low duplicate retention rate regions significantly overlapped with genes originating from small-scale (tandem) duplications (Supplementary Table 1). Genes with high retention rates were enriched in Gene Ontology (GO) categories such as ‘cellular component organization or biogenesis’, ‘primary metabolic process’, ‘developmental process’ and ‘regulation of cellular process’, while low retention rate genes were enriched in categories such as ‘RNA-dependent DNA biosynthetic process’ and ‘defense response’ (in both subgenomes), and ‘spermidine hydroxycinnamate conjugate biosynthetic process’ (involved in plant defense41) and ‘plant-type hypersensitive response’ (in subEE) (Supplementary Tables 2–5).

a, Corresponding syntenic blocks between CA subgenomes subCC (orange) and subEE (blue), and with the CC (orange) and CE (blue) genomes. b, The base pairs in intergenic DNA in synteny block gaps caused by fractionation in a subCC–subEE comparison, compared with numbers of base pairs in homoeologous unfractionated regions, as a function of numbers of consecutive genes deleted. c, Gene retention rates in synteny blocks plotted along subCC chromosome 2; subCC is plotted in orange and subEE in blue. The green box indicates the pericentromeric region. CA, C. arabica; CC, C. canephora; CE, C. eugenioides.
To study possible expression biases between subgenomes, we identified syntelogous gene pairs and removed the pairs showing HEs in the Arabica subgenomes (see under ‘Origin and domestication of Arabica coffee’ below)42 (Supplementary Section 5). Overall, no significant global subgenome expression dominance was observed (Supplementary Tables 6 and 7). However, gene families regularly displayed mosaic patterns of expression, including several encoding enzymes that contribute to cup quality, such as N-methyltransferase (NMT), terpene synthase (TPS) and fatty acid desaturase 2 (FAD2) families, all having some genes being more expressed in one of the two subgenomes (Extended Data Fig. 2), as per a recent study43. Similar gene family-wise patterns occur in other evolutionarily recent polyploids such as rapeseed10 and cotton44, which are also at their early stages of transitioning back to a diploid state.
Origin and domestication of Arabica coffee
To obtain a genomic perspective on the evolutionary history of Arabica, we sequenced 46 accessions, including three Robusta, two Eugenioides and 41 Arabica. The latter included an eighteenth-century type specimen, kindly provided by the Linnaean Society of London, 12 cultivars with different breeding histories, the Timor hybrid and five of its backcrosses to Arabica, and 17 wild and three wild/cultivated accessions collected from the Eastern and Western sides of the Great Rift Valley45,46 (Supplementary Table 8 and Fig. 2a).

a, Geographic origin of resequenced wild C. arabica accessions (red placeholders). Accession names are given in c. The red arrow indicates the probable route of migration to Yemen in historical times. b, Ancestral population assignments of C. arabica accessions for subCC (left) and subEE (right). Relationships among individuals are illustrated with phylogenetic trees obtained from independent SNPs. For magnified views of the trees, see Supplementary Fig. 37. c, Magnification of the bottom left part of a, showing the admixture values for each of the accessions in subCC (top) and subEE (bottom); the colors correspond to the analysis in b. d, Population sizes of wild and cultivated accessions, inferred using SMC++, suggest genetic bottlenecks at ~350 and 1 ka (limited to nonadmixed wild individuals). e, FastSimcoal2 output, suggesting a population split ~30.5 ka, followed by a period of migration between the populations until ~8.9 ka. This timing corresponds with increased population diversity in cultivars at a similar time, calculated using SMC++. Green rectangles along the timeline show ‘windows of opportunity’, times when Yemen was connected with the African continent wherein human migrations to the Arabian Peninsula may have occurred. The purple rectangle shows the last ice age. M, migration; OAE, out-of-Africa event. f, Directional gene flow analysis using Orientagraph suggests two hypotheses: gene flow from the shared ancestral population of all cultivars to the Ethiopian wild individuals (subCC), or gene flow from the Typica lineage to Ethiopia (subEE). Maps in a and c were generated with Google Earth and Google Maps, respectively.
HE between subgenomes has been observed in several recent polyploids8,10,42. Arabica generally displays bivalent pairing of homologous chromosomes and disomic inheritance47, but since the subgenomes share high similarity, occasional homoeologous pairing and exchange may also occur. We therefore explored the extent of HE among Arabica accessions and its possible contribution to genome evolution. Overall, all accessions shared a fixed allele bias toward subEE at one end of chromosome 7, which contained genes enriched for chloroplast-associated functions (Extended Data Fig. 3a, Supplementary Section 5 and Supplementary Table 9). Since the Arabica plastid genome is derived from Eugenioides48, HE in this region was likely selected for, due to compatibility issues between nuclear and chloroplast genes encoding chloroplast-localized proteins49. Surprisingly, all but one accession (BMJM) showed significant (Bonferroni-adjusted P values < 0.0005; chi-squared test, each d.f. = 1) 3:1 allelic biases toward subCC. The highly concordant HE patterns, present in both wild and cultivated Arabicas (Extended Data Fig. 4), suggested that (1) the allelic bias is an adaptive trait not associated with breeding and (2) it originated in a common ancestor of all sampled accessions, possibly immediately after the founding allopolyploidy event. Some exchanges, shared by only a few accessions, probably originated more recently (Extended Data Fig. 3b). More recent HE events were also found in some cultivars and also showed a bias toward subCC, except for BMJM, which showed bias toward subEE due to a single large crossover in chromosome 1 (Extended Data Fig. 3a). An interesting hypothesis for future investigation is that in a low-diversity polyploid species such as Arabica, HE could be a major contributor to phenotypic variation observed among closely related accessions50.
We next studied population genetic statistics for each of the subgenomes (Supplementary Table 10). The 17 wild samples demonstrated low genomic diversities, indicative of small effective population sizes, while negative Tajima’s D suggested an expanding population, possibly following one or more population bottlenecks. The cultivars and wild population samples had similar genetic diversities, as demonstrated by low fixation index (FST) values. In cultivars, nucleotide diversities were only slightly lower than in wild populations and Tajima’s D scores were less negative, suggesting that only minor bottlenecks and subsequent population expansions occurred during domestication.
SNP tree estimation and ADMIXTURE analyses (Fig. 2b) identified a three-population solution for subCC: Typica–Bourbon cultivars (Population 1), wild accessions (Population 2), and Timor hybrid-derived cultivars (Population 3). The old BMJM and the recently established Geisha cultivars showed admixed states on both subgenomes, similar to about half of the wild accessions. Indian varieties encompassed both Typica and Bourbon variation, in agreement with previous studies20. The Linnaean sample grouped with the cultivars, supporting its hypothesized origin from the Dutch East Indies25. A complementary principal component analysis (PCA) (Extended Data Fig. 5) was in agreement with ADMIXTURE analysis.
In wild accessions, both subgenomes concordantly showed two population bottlenecks (Fig. 2d) in the SMC++ (ref. 51) modeling. Assuming a 21-year generation time52, the oldest bottleneck initiated abruptly around 350 thousand years ago (ka) and ended around 15 ka, at the start of the African humid period53, when climatic conditions were more favorable for Arabica growth. The more recent bottleneck initiated more gradually around 5 ka and lasts to this day. Cultivated accessions, however, exhibited the oldest, but not the more recent, bottleneck. In part due to these differences, we also modeled Arabica population history using FastSimcoal2 (ref. 54), modeling the wild population and cultivars as two separate lineages. In the best-fitting model (Fig. 2e), the wild population was predicted to split from the cultivar founding population 1,450 generations ago (~30 ka), that is, before the last glacial maximum. The original founding event was analyzed using the nonadmixed wild individuals, revealing an ancestral population bottleneck at 350 ka (Extended Data Fig. 6a). Divergence estimates based on gene fractionation, the distribution of nonsynonymous mutations (Extended Data Fig. 6b) and calibrated SNP trees (Fig. 2b) suggested the allopolyploid founding event occurred at 610 ka, which is close to previous estimates22,23. The 350 ka bottleneck, on the other hand, corresponds to that found in the SMC++ analyses (Fig. 2d). We therefore consider 610–350 ka a likely time range for the polyploidization event (Fig. 2e). The wild and pre-cultivar lineages maintained some gene flow (in terms of migration) until ~8–9 ka, which may have contributed to the modeled increase in effective population size (Fig. 2d,e).
While these data were not able to identify the precise place of origin of the modern cultivated population (see also the following section), the extended period of migration between wild and cultivated accessions suggests that they were separated only by a relatively small geographic distance, such as along the two sides of the African Great Rift Valley (Fig. 2a–c). It is also possible that the cultivated lineage could have extended as far as Yemen and that the end of migration between the two populations could have been caused by the widening of the Bab al-Mandab strait (separating Yemen and Africa) due to rising sea levels55 at the end of the African humid period. A native Arabica population exists in Yemen56, which could support this hypothesis. The Linnaean sample, together with the Typica and Bourbon cultivars, originates from this second population, which was also used to establish cultivation in Yemen, as suggested by the SNP, ADMIXTURE and PCA analyses (Fig. 2b and Extended Data Fig. 5).
In conclusion, our analyses suggest that the Arabica allopolyploidy event occurred between 610 and 350 ka, when considering that inbreeding present in Coffea populations would accelerate coalescence estimation57,58. Earlier work proposing more recent timings, such as 20 ka (ref. 20), could be underestimates stemming from confounding effects of population bottlenecks in cultivated and wild lineages.
Origin of modern cultivars
The known breeding history of several of our Arabica cultivars provided us with a gold standard set for deducing the Arabica pedigree using Kinship-based INference for Gwas (KING)59 (Fig. 3). The method correctly identified the relationships between Bourbon and Typica group cultivars and the Bourbon–Typica crosses in subCC. In contrast, the subEE pedigree showed lower (second) order relationships, possibly due to HE in that subgenome (Extended Data Fig. 7). Timor hybrid-derived accessions did not show significant relationships to mainline cultivars in subCC (likely due to Robusta introgressions in this subgenome that broke the haplotype blocks; see below), while subEE showed second-degree relationships to both the Typica and Bourbon groups (Fig. 3 and Extended Data Fig. 7), confirming that subEE has not received substantial introgression.

The degree of relatedness was estimated using KING and describes the number of generations between the related accessions. Thumbnail images show FDR-corrected F3 tests of introgression for each of the target individuals. Each cell in the matrix illustrates an F3 test result for the target accession containing introgression from two different sources (x and y axes); blue color illustrates significant adjusted Z-score (Z adj; value indicated by color key), indicative of gene flow (or allele sharing via identity by descent78) from the two source accessions to the target, while red color illustrates no support for gene flow. See Extended Data Fig. 7 for corresponding analyses in subEE. In the wild accessions, the dark green background highlights the admixed individuals (Fig. 2b), while the nonadmixed individuals are highlighted with red background. Relationships follow standard nomenclature (for example, second degree refers to an individual’s grandparents, grandchildren and so on, whereas third degree refers to great-grandparents, great-grandchildren and so on).
Interestingly, the Typica, Bourbon and JK1 individuals were also first degree related, suggesting direct parent–offspring relationships. Besides confirming their shared Yemeni origins, this finding also underscores the Yemeni germplasm’s limited genetic diversity. Further, the old cultivar lines JK1 (Indian), Erecta (Indonesian Typica), BMJM (Caribbean Typica), TIP1 (Brazilian Typica) and BB1 (Brazilian Bourbon) showed second- or higher-degree relationships with a cluster of closely related wild admixed accessions, centered on E016/136 (Fig. 2b). The recently established Geisha cultivar showed similar relationships to the wild admixed individuals and the Bourbon and Typica groups, suggesting common origins. Interestingly, admixed wild accession E016/136 was closely related to both wild and cultivated populations.
In a comparison of geographic origins, wild individuals from the Eastern side of the Great Rift Valley had some levels of admixture and were closely interrelated, while on the Western side, the admixed, related individuals were mostly concentrated around the Gesha region (Figs. 2c and 3). The E016/136 admixed accession, closest to cultivars, demonstrated a first-degree relationship with several wild accessions, of which only Ar35-06 and Eth28.2 were pure representatives of the wild population (Fig. 2b). Therefore, these two accessions are genetically closest, in our sample, to the hypothetical true wild parent of cultivated Arabica, with E016/136 representing an intermediate form. Ar35-06 was collected near Gesha mountain, close to the origin of the modern Geisha cultivar. Altogether, these data point to the Gesha region as a hotspot of wild accessions amenable to domestication.
Admixed wild samples may have originated from a recent hybridization event that occurred before or after their collection from the wild. A third alternative is that the Yemeni population (and hence the cultivars) originated from an admixed population from the Eastern side of the Great Rift Valley or the Gesha region. Analysis of admixture patterns with Orientagraph60 (Fig. 2f) suggested hybridization with the common ancestor of the Bourbon and Typica lineages in subCC, and of Typica in subEE. In the case of recent hybridization, introduced haplotypes would exist as long contiguous blocks (as in the Timor hybridization, which occurred 100 years ago), while for older events, the blocks would be more fragmented due to crossing-over. Analysis using the distance fraction (df) statistic61 showed the latter to be the case (Extended Data Fig. 8), indicating that admixture events among wild accessions were not very recent, supporting our third hypothesis.
Domestication and cultivation usually involve strong population bottlenecks based on high wild diversity, resulting in reduced genetic diversity in cultivars62. However, Arabica nucleotide diversity was already very low in the wild, probably as a result of earlier bottlenecks (Fig. 2d,e), but only marginally reduced in the pre-cultivated lineage (Extended Data Fig. 9a). Bourbon had lower diversity than Typica, probably resulting from the known single-individual bottleneck in this group. Also, the inbreeding coefficients in the wild and cultivated accessions were similar (Extended Data Fig. 9b), differing from general expectations for a domesticated species62.
To look for pathways under purifying selection in cultivars, we identified genes with high FST (95% quantile) between cultivars and wild accessions. This resulted in a set of 1,908 genes that were enriched for the GO categories ‘cellular response to nitrogen starvation’, ‘regulation of innate immune response’ and ‘regulation of defense response’ (Supplementary Table 11), and contained homologs of ammonium transporters AMT1 and AMT2, important for nitrogen uptake in Coffea63; a homolog of the salicylic acid receptor NONEXPRESSER OF PR GENES 1 (NPR1), required in salicylic acid signaling and systemic acquired resistance64; as well as a homolog of the Arabidopsis LSU2 gene, previously identified as a hub convergently targeted by effectors of pathogens from different kingdoms65. A second screen, focused on genes with a large number of high-impact nonsynonymous mutations shared among cultivars (>40% individuals having the mutation), generated a list of 556 genes that were significantly enriched for only one GO category, ‘defense response’ (Supplementary Table 12). From the 22 genes in this category, 16 were NB-ARC domain-containing resistance (R) genes, and two were members of the leucine-rich repeat (LRR) defense gene family. High diversity in immune-related responses is one possible pathogen resistance mechanism in plant communities66, and therefore reduced diversity may have compromised modern Arabica cultivar immunity.
The high level of conservation between the Arabica subgenomes and their diploid progenitors may have facilitated spontaneous interspecific hybridization events. This was the case for the Timor hybrid, a spontaneous Robusta × Arabica hybrid resistant to H. vastatrix27. Our sample set included five descendants of the original Timor hybrid, obtained by backcrossing to Arabica. As expected, the hybridization affected subCC more profoundly, with much higher levels of nucleotide divergence apparent (FST = 0.185) than in subEE (FST = 0.0897), when comparing cultivars and hybrids. The divergence from wild populations was even greater, with FST = 0.254 for subCC and FST = 0.138 for subEE, illustrating that introgression occurred almost exclusively within subCC.
In the Timor hybrids, the regions found with df statistics61 largely overlapped the introgressed loci identified using FST scans (Fig. 4a) and were found in large blocks, reflecting recent hybridization, and covering 7–11% of the genome (Fig. 4a and Extended Data Fig. 8). Transposon insertion polymorphisms (TIPs) also overlapped with introgressed regions (Gypsy P = 0.0002; Copia P = 0.035; Fisher exact test), confirming their recent origin from Robusta (Fig. 4b). The introgressed regions overlapped with regions of higher subgenome fractionation (P = 0.001873; Supplementary Table 13), possibly due to heterologous recombination between subCC and Robusta, resulting in unequal crossing-over.

a, Introgression df statistic estimated for different Timor hybrid derivatives. Colored lines above the axis mark regions of significant introgression in the line under inspection, and are colored by chromosome. The shared introgressed region on chromosome 4 is colored in purple and boxed. TIPs are represented as lines below the x axis and exhibit overlap with introgressed regions. b, The shared introgressed genomic region on subCC chromosome 4 contains a cluster of R genes (RPP8), a cluster of homologs of a negative regulator of R genes (CPR1) and a cluster of homologs of Leaf rust resistance 10 kinases (LRK10L) (bottom). The heatmap shows, from the bottom up, (1) log fold change of gene expression after H. vastatrix inoculation, when comparing resistant Timor hybrid lineage against a susceptible cultivar; red color means elevated expression in the hybrid, and blue decreased expression. (2) Fixation index (FST) values for the introgressed lines versus cultivars and between cultivars and wild accessions. (3) Nucleotide diversity for the wild and cultivated accessions for each gene coding region, plus the flanking 2 kb upstream and downstream of the region. FC, fold change.
An introgressed region shared by all Timor hybrid lines was evident on chromosome 4 (Fig. 4a). We identified a set of 233 genes shared by all hybrids (Supplementary Table 14). The set contained members of three colocalized tandemly duplicated blocks of resistance-related genes on chromosome 4, subCC, and showed high FST values between cultivars and introgressed lines. A tandem array of five genes were homologs of Arabidopsis RPP8, a NOD-like receptor resistance locus conferring pleiotropic resistance to several pathogens67,68. RPP8 shows a great amount of variation in Arabidopsis alone, where intrachromosomal gene conversion combined with balancing selection contributes to its exceptional diversity69. The same subCC region also included a tandem array of ten homologs of CONSTITUTIVE EXPRESSER OF PR GENES 1 (CPR1), a negative regulator of defense response that targets resistance proteins70,71. Finally, we identified three duplicates encoding Leaf rust 10 disease-resistance locus receptor-like protein kinases (LRK10L). The LRK10L are a gene family that is widespread across plants. First identified as a protein kinase in a locus contributing leaf rust resistance in wheat72, they were found to be upregulated during various biotic and abiotic stresses73 and were confirmed as positive regulators of wheat hypersensitive resistance response to stripe rust fungus73 and powdery mildew74.
The high FST values between cultivated and introgressed, but not wild, individuals (Fig. 4b) indicate that the wild population cannot be the source for allelic asymmetries. Nucleotide diversities further illustrate this point; some genes demonstrate lower nucleotide diversity in wild individuals, suggesting these genes to have experienced selective sweeps. To further narrow down candidate genes involved in leaf rust resistance, we reanalyzed comparative gene expression data from susceptible and resistant accessions after H. vastatrix inoculation75. This analysis identified 723 differentially expressed genes, most of which were associated with defense responses (Fig. 4b and Supplementary Tables 14 and 15). The combination of high FST values, nucleotide diversities and differential expression data highlights several strong candidate genes (one RPP8, six CPR1 and one LRK10L) at this locus.
Discussion
Besides providing genomic resources for molecular breeding of one of the most important agricultural commodities, our Arabica, Robusta and Eugenioides genomes provide a unique window into the genome evolution of a recently formed allopolyploid stemming from two closely related species. Our Arabica data did not suggest a genomic shock induced by allopolyploidy, but, instead, only higher LTR transposon turnover rate. Genome fractionation rates remained basically unaltered before and after the allopolyploidy event. Likewise, no global subgenome dominance in gene expression was observed, but rather a mosaic-type pattern as in other recent polyploids10,44, affecting the expression of individual gene family members. However, similar to octoploid strawberry8, we detected genome dominance in terms of biased HEs favoring subCC. Since Robusta has one of the widest geographic ranges in the Coffea genus, whereas Eugenioides is more range-limited, this biased HE might be adaptive. This hypothesis was supported by the site frequency spectrum of HE loci, showing signs of directional selection (Extended Data Fig. 3). Intriguingly, transposable insertion polymorphisms significantly overlapped with tandem gene duplications and biosynthetic gene clusters, hinting at their possible roles in cluster evolution.
Domestication of perennial species such as Arabica coffee differs markedly from that of annual crops, consisting instead of three phases: selection of outstanding genotypes from wild forests, clonal propagation and cultivation, and then breeding and diversification76. In addition to being a perennial crop, Arabica is also a predominantly autogamous allopolyploid, which puts it in a class of its own. We show here that genetic diversity was already very low among wild accessions, due to multiple pre-domestication bottlenecks, and that the genotypes selected for cultivation by humans (both the ancient cultivated Ethiopian landraces and the recent Geisha cultivar) already were somewhat admixed between divergent lineages. The resequenced accessions displayed a geographic split along the Eastern versus Western sides of the Great Rift Valley, with cultivated coffee variants all placed with the Eastern population. Such admixture has played a large role in breeding many fruit-bearing crops, the nonpolyploid allogamous perennial lychee being one of the most extreme cases58.
The prevalent autogamy of Arabica, combined with the multiple genetic bottlenecks it underwent in the wild, may have selectively purged deleterious alleles, explaining the capacity of the species to survive single-plant bottlenecks that occurred during its cultivation. An additional element buffering deleterious alleles was probably Arabica’s allopolyploidy itself, which provided some level of heterosis77. However, the narrow genetic basis of both cultivated and wild modern Arabica constitutes a major drawback, as well as an obstacle for its breeding using wild genepool diversity. On the other hand, the extensive collinearity of its CC and EE subgenomes with those of its Robusta and Eugenioides progenitors is likely to facilitate introgression of interesting traits from these species, as already happened historically in the Timor spontaneous hybrid. The high-quality genome sequences of the three species provided in this work, together with the identification of the genomic region conferring resistance to coffee leaf rust, constitute a cornerstone for the breeding of novel Arabica varieties with superior adaptability and pathogen resistance.
Methods
Genome sequencing
For the three Coffea species, genomic DNA was extracted from leaf tissue. A Qiagen kit was used for DNA extraction for Illumina sequencing. Illumina short reads and PacBio 20-kilobase (kb) libraries were prepared following the manufacturer’s instructions. Sequencing was performed on a HiSeq2000 instrument for the short reads, and the PacBio RSII platform for long reads (specifications given in Supplementary Table 16). For the generation of HiFi reads, DNA was extracted from C. arabica leaf tissue following nuclei purification by centrifugation followed by lysis, phenol–chloroform extraction and isopropanol precipitation. DNA was fragmented to 20 kb using a Megaruptor 3. SMRTbell libraries were sequenced on a single SMRTcell on a Sequel IIe platform.
For the resequencing of 39 wild and cultivated C. arabica accessions, libraries were prepared using the KAPA HyperPrep Kits (Roche) following the manufacturer’s instructions, and paired-end (2 × 125) sequenced on an Illumina HiSeq2500 instrument to ~40× coverage. The Linnaean herbarium sample was sequenced to 46× coverage with Ion Torrent technology.
Assembly
Contig-level assembly for C. canephora was obtained with MHAP79 and scaffolded using BAC-end sequences and 454 paired-end sequences generated previously33. Both C. eugenioides and C. arabica were assembled with Falcon80, and C. arabica was subsequently phased using Falcon_unzip. All three genomes were error-corrected with Pilon81 using Illumina short reads (Supplementary Section 2.2). C. canephora and C. arabica were further scaffolded into pseudochromosomes using Dovetail Hi-C technology. For C. eugenioides no more material could be obtained for further improvement of the assembly contiguity, and the assembly was scaffolded into pseudomolecules using C. canephora as reference. Gaps in the scaffolds were filled with PBJelly82, after which six more rounds of polishing were done with Pilon using the Illumina shotgun sequenced genomic DNA as well as RNA sequencing (RNA-seq) reads.
The resulting chromosome assemblies for C. canephora were checked and corrected using an ultra-high-density linkage map83 generated during the project. To further improve the quality of the C. arabica assembly, Bionano genome maps were generated.
C. arabica HiFi assembly was carried out with hifiasm v.0.16.1 (ref. 84), followed by scaffolding using Hi-C data from Dovetail technology and ALLHiC85 pipeline. Final quality checks and manual adjustments of the assembly were carried out using 3d-DNA86 and juicebox87.
The completeness of the different assemblies was assessed using BUSCO v.5.2.2 (ref. 35) with the eudicots_odb10 database (2,326 genes; Table 1). Telomeric repeats were searched across the chromosomes using CoGeBLAST88.
To assess the phasing of both subgenomes from C. arabica, synonymous nucleotide substition (Ks) values were obtained from CoGe89 and compared between C. arabica and each of two diploid outgroups, C. canephora and C. eugenioides, using scripts in R.
Linkage map
A reference genetic map was constructed from a cross between a Congolese group genotype (BP409) and a Congolese × Guinean hybrid parent (Q121). The segregating population was composed of 93 F1 individuals90. The parents were sequenced to 60× and progeny to 20× coverage using the Illumina HiSeq2000 platform at Nestlé Research. Following quality control with FastQC and trimming with Trimmomatic v.0.36 (ref. 91), the reads were mapped against the C. canephora reference assembly using BWA-MEM v.0.7.15 (ref. 92). The linkage mapping was conducted with Lep-MAP3 (ref. 83). The markers were clustered into paternal and maternal linkage groups by using a logarithm of the odds score of 18 in a segregation distortion aware model. The final curation of the assembly, combining the two parental maps, solving conflicts as well as identification of haplotype alleles, was carried out manually.
TE annotation and analysis
EDTA93 was used to de novo identify TEs in the C. canephora, C. eugenioides as well as C. arabica subgenomes. Inpactor2 (ref. 94) was used to recover full-length LTR retrotransposons in the three genomes and to classify them at the lineage level. EDTA and Inpactor2 libraries were merged and clustered using cd-hit95. Clusters were manually inspected to remove nested and false predictions. After curation, libraries were used for annotation using Repeat Masker (default parameters). Annotations with length >200 base pairs (bp) were retained. The timing of LTR retrotransposon insertions was studied in the three genomes using individual sequences recovered by Inpactor2 and using an average base substitution rate of 1.3 × 10−8 (ref. 96), similar to Orozco-Arias et al.97.
Gene prediction
RNA-seq and IsoSeq reads were generated to support de novo gene prediction. A MAKER-P pipeline98 was used to combine several de novo gene callers with the IsoSeq and junction information from short-read RNA-seq. High-evidence gene models with Annotation Edit Distance score < 0.5 were selected for the annotation. For C. arabica HiFi assembly, the annotations were first transferred from CC, CE and the previous CA assembly using GeMoMa v.1.9 (ref. 99), and then combined. All genes of interest linked to coffee flavor were subjected to manual inspection and gene model curation. Following the annotation, BUSCO completeness scores were assessed for the CC, CE and CA predicted transcriptomes.
Gene expression
Three gene families, encoding terpene synthases (TPS), N-methyltransferases (NMT) and fatty acid desaturase 2 (FAD2), were further characterized and used to investigate the influence of the presence of the extra gene copies in the allopolyploid using previously published expression data100. The expression data presented here are the TPM (transcripts per million) normalized counts with log-scaling: log10(x + 1 × 10−4), where x is the TPM count from STARaligner101. For leaf rust differential expression analysis, previously published RNA-seq data75 were reanalyzed by mapping the reads on C. arabica HiFi assembly using STARaligner. Differential expression in Timor hybrid versus susceptible Caturra accession after inoculation with H. vastatrix was analyzed with DEseq2 (ref. 102) in R. False discovery rate (FDR) adjustment was carried out using the Benjamini–Hochberg method; adjusted P value < 0.05 was considered statistically significant.
Evolution of synteny and fractionation
Synteny information was obtained using the SynMap tool on the CoGe platform88,89. Only genes within synteny blocks were considered, not only gene pairs but also singleton genes in each genome that have lost their counterpart in the other genome due to fractionation or other gene loss.
We used the ‘peaks’ method103, as calculated by the R function geom_density, for the three events that generate duplicate genomes during genome evolution of C. arabica, that is, the gamma triplication at the origin of the core eudicots, the speciation underlying the CC/CE divergence and the allotetraploidization event.
HE
Syntenic genes between CE, CC, subCC and subEE were identified using the SynMap tool on the CoGe platform. Identification of allele biases was carried out by mapping the C. arabica short-read sequencing data against combined CE and CC assemblies using BWA-MEM92 and calculating sequencing coverages on syntenic genes using bedtools. Differential coverage across the chromosomes was visualized using custom R scripts. To reduce noise, a sliding window of ten genes was used to calculate the average coverage along chromosomes. The allele balance was calculated as A = 4 × ((CC/(CC + EE)) − 0.5), where CC and EE are the subCC and subEE syntelog coverages, respectively. Allele balances <−1.5 or >1.5 were considered homozygous for EE, or CC, respectively, while balances <0.5 and >−0.5 were considered equal.
SNP calling
Following quality control with FastQC104, Illumina short reads were trimmed using Trimmomatic v.0.36 (ref. 91) and mapped on the C. arabica reference assembly with BWA-MEM v.0.7.16a-r1181 (ref. 105). For the Linnaean sample, the reads were processed according to the protocols recommended for degraded DNA analysis in MapDamage v.2.0.8 (ref. 106). GATK (v.3.8.0) pipeline was used for SNP calling. Duplicates were marked and removed using Picard v.2.0.1 and genotype likelihoods were called into GVCF files using HaplotypeCaller (GATK). For the diploid progenitors, to allow interspecies comparisons, the mapping was done to each of the subgenomes separately, including chromosome zero, that is, contigs not assembled into pseudomolecules, in both mappings. Joint calling was carried out using GenotypeGVCFs (GATK)107 and snpEff v.4.3t was used to assess the impact of the SNPs108. To remove regions with cross-species mappings, we removed the SNPs that were called as heterozygous when mapping the di-haploid ET-39 sequencing data to the Arabica reference genome.
Genome-wide nucleotide diversity was calculated with vcftools v.0.1.17 (ref. 109), by calculating the mean of pi values from sliding windows of 100 kb with 10-kb step size. Similarly, genome-wide Tajima’s D was calculated from the mean of Tajima’s D values with window size of 100 kb. PCA was run using Plink v.1.90 (ref. 110). ADMIXTURE v.1.3.0 (ref. 111) was run for SNP data where the variants in repeat regions were filtered out and the outgroup species (diploid Coffea species) were excluded. The SNPs were filtered for linkage disequilibrium (LD) according to the recommendation in the ADMIXTURE manual with (--indep-pairwise 50 10 0.1) while allowing maximum 10% missing values (--geno 0.1). Admixture analysis was run using tenfold cross-validation. The solution giving lowest cross-validation score was selected as the best solution. Nonsynonymous nucleotide diversity, π0, and neutral, intergenic πs were calculated using the PiNSiR R package (https://github.com/jsalojar/PiNSiR) and ANGSD v.0.933 (ref. 112), similar to ref. 58.
Analysis of GBS data
Read data from 736 PstI GBS libraries of C. arabica20 were downloaded from the SRA repository (bioproject PRJNA554647). The samples were 100-bp single-end reads sequenced on an Illumina HiSeq2000 instrument. After trimming and quality filtering, the data were mapped onto the reference genome sequence of C. arabica using the BWA-MEM algorithm with default settings in BWA v.0.7.17 (ref. 105). SNPs were called using the Unified Genotyper in GATK v.3.7 (ref. 107).
F3 statistics
The Admixtools package113 was used to calculate the F3 statistics, and the obtained P values were subjected to FDR correction using the procedure developed by Salojärvi et al.114, where the Z-scores were converted into P values, subjected to FDR correction using Benjamini–Hochberg correction and then converted back to Z-scores.
SNP trees
The SNPs were filtered for repetitive regions, followed by filtering for LD > 0.4 and loci with >40% missing values, as well as minor allele prevalence <10%. The obtained fasta file of the selected sites was input for RAxML with -T 30 -m GTRGAMMA model, using 30 starting trees and 1,000 bootstrap samples115.
Pairwise sequentially Markovian coalescent modeling
For each individual, the reads were mapped against the full CA reference assembly. The mappings were then filtered for indels using bcftools and regions with <8× or >100× coverage. After filtering, the obtained pairwise sequentially Markovian coalescent (PSMC) fastq file was split into subCE and subCC specific parts and PSMC demography was estimated using standard parameter settings (-N25 -t15 -r5)116. The inferred history was then visualized using R and ggplot2 package.
Ancestral state estimation
The ancestral state was inferred from reads of two representatives of each of the diploid coffee species, C. canephora (BUD15, Q121) and C. eugenioides (BU-A, DA56), mapped against each of the subgenomes and the unassigned contigs. Subsequently, a majority vote was carried out to infer the ancestral allele using ANGSD v.0.933 (ref. 112) with options -doFasta 2 and -doCounts 1. The SNP calls in the VCF file were then flipped to the ancestral states using bcftools +fixref117.
SMC++
The input data for SMC++ comprised the VCF file where the ancestral state was used as reference (see above) and the SNPs in repeat regions were filtered out. For the cultivar population, the representatives of Bourbon and Typica lineages were included (TIP1, Bourbon, Mundo Novo, BMJM, Moka, Rubi, Topazio, Bourbon pointu, Catuai99, BB1, Erecta, JK1, Guatemalense, Amsterdam); Geisha was removed from the analysis because of its unknown pedigree. SMC++ parameter selection was carried out using threefold cross-validation (smc++ cv) implemented in SMC++ v.1.15.3 (ref. 51).
Kinship analysis
Before kinship analysis, the diploid species were removed from the SNP file and the kinship was estimated using KING software v.2.2.5. with --kinship option59. The results were visualized using Keynote, for each subgenome separately.
Introgression analyses
Orientagraph v.1.0 (ref. 60) was run for each of the subgenomes separately according to the developer recommendations by carrying out filtering for linkage as recommended for TreeMix118. PopGenome R package was used to calculate d_f statistics61. For the subCE introgression, BUD15 was used as outgroup, DA56 as the source of introgression and E383 as the nonadmixed wild representative. For subCC, DA56 was used as outgroup and BUD15 as the source of introgression. The statistic was calculated in 20-kb nonoverlapping windows using weighted jackknife to assess the significance of introgression. The results were visualized using R.
Population simulations
FastSimCoal v.2.6 was used for population simulations54. Site frequency spectrum was calculated using ANGSD112 with the VCF file containing wild individuals and repetitive regions filtered out. The ancestral states were estimated as described above. For each of the models, 100 parameter files were simulated. For each parameter file, 1,000,000 simulations were run; monomorphic sites were not used. Maximum composite likelihood estimation of parameters was carried out with 40 expectation-conditional maximization iterations.
Fixation index
Site-wise FST values between wild and cultivated individuals were calculated for each gene annotation and 2-kb flanking regions using vcftools109. Then, mean FST values were calculated for each gene model using the R package.
TE insertion polymorphisms
We studied LTR retrotransposon insertions via analysis of short-read whole-genome resequencing data using TIP_finder119, using the discordant mapping pair approach.
Biosynthetic gene clusters
Biosynthetic gene clusters were identified with the Plantismash web server (http://plantismash.secondarymetabolites.org/) following default analysis protocols120.
Statistical testing
Statistical significance of overlaps between various gene sets was assessed using Fisher exact test in R. Gene set enrichments were carried out by first assigning each gene to the GO category of the closest Arabidopsis homolog (using E-value threshold 1 × 10−5). Tests for enrichment were carried out using goatools121. Bonferroni-corrected P value of 0.05 was used as threshold for significance. Tests for the allele balance were carried out using chi-squared test; each test had d.f. = 1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Coffee genome assemblies are available at CoGe (https://genomevolution.org/): C. canephora: 50947; C. eugenioides: 67315; and C. arabica: 66663 (Pacbio HiFi) and 53628 (Pacbio). The genome data are also available at ORCAE (https://bioinformatics.psb.ugent.be/orcae/overview/Coara and https://bioinformatics.psb.ugent.be/gdb/coffea_arabica/). All sequencing data are available at NCBI under bioproject ID PRJNA698600, and our assemblies are accessioned there as JAZHSI000000000.1, JAZHGF000000000.1, JAZHGH000000000.1 and JAZHGG000000000.1. Genotyping data (VCF files) and syntenic alignments are available in Data Dryad: https://doi.org/10.5061/dryad.qnk98sfpt.
Code availability
R scripts for calculating the neutral and deleterious nucletide diversities (PiNSiR) are provided in Zenodo122 (https://zenodo.org/doi/10.5281/zenodo.5136526).
References
Van de Peer, Y., Mizrachi, E. & Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 18, 411–424 (2017).
Van de Peer, Y., Ashman, T.-L., Soltis, P. S. & Soltis, D. E. Polyploidy: an evolutionary and ecological force in stressful times. Plant Cell 33, 11–26 (2021).
Leebens-Mack, J. H. et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 574, 679–685 (2019).
Sun, H. et al. Chromosome-scale and haplotype-resolved genome assembly of a tetraploid potato cultivar. Nat. Genet. 54, 342–348 (2022).
Athiyannan, N. et al. Long-read genome sequencing of bread wheat facilitates disease resistance gene cloning. Nat. Genet. 54, 227–231 (2022).
Wu, S. et al. Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement. Nat. Commun. 9, 4580 (2018).
Wang, T. et al. A complete gap-free diploid genome in Saccharum complex and the genomic footprints of evolution in the highly polyploid Saccharum genus. Nat. Plants 9, 554–571 (2023).
Edger, P. P. et al. Origin and evolution of the octoploid strawberry genome. Nat. Genet. 51, 541–547 (2019).
Li, F. et al. Genome sequence of cultivated Upland cotton (Gossypium hirsutum TM-1) provides insights into genome evolution. Nat. Biotechnol. 33, 524–530 (2015).
Chalhoub, B. et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953 (2014).
Sattler, M. C., Carvalho, C. R. & Clarindo, W. R. The polyploidy and its key role in plant breeding. Planta 243, 281–296 (2016).
McClintock, B. The significance of responses of the genome to challenge. Science 226, 792–801 (1984).
Sha, Y. et al. Genome shock in a synthetic allotetraploid wheat invokes subgenome-partitioned gene regulation, meiotic instability, and karyotype variation. J. Exp. Bot. 74, 5547–5563 (2023).
Thomas, B. C., Pedersen, B. & Freeling, M. Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res. 16, 934–946 (2006).
Schnable, J. C., Springer, N. M. & Freeling, M. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc. Natl Acad. Sci. USA 108, 4069 (2011).
Gaeta, R. T., Pires, J. C., Iniguez-Luy, F., Leon, E. & Osborn, T. C. Genomic changes in resynthesized Brassica napus and their effect on gene expression and phenotype. Plant Cell 19, 3403–3417 (2007).
Burns, R. et al. Gradual evolution of allopolyploidy in Arabidopsis suecica. Nat. Ecol. Evol. 5, 1367–1381 (2021).
Conant, G. C., Birchler, J. A. & Pires, J. C. Dosage, duplication, and diploidization: clarifying the interplay of multiple models for duplicate gene evolution over time. Curr. Opin. Plant Biol. 19, 91–98 (2014).
Carvalho, A. et al. Melhoramento do cafeeiro: IV - Café Mundo Novo. Bragantia 12, 97–130 (1952).
Scalabrin, S. et al. A single polyploidization event at the origin of the tetraploid genome of Coffea arabica is responsible for the extremely low genetic variation in wild and cultivated germplasm. Sci. Rep. 10, 4642 (2020).
Cenci, A., Combes, M.-C. & Lashermes, P. Genome evolution in diploid and tetraploid Coffea species as revealed by comparative analysis of orthologous genome segments. Plant Mol. Biol. 78, 135–145 (2012).
Bawin, Y. et al. Phylogenomic analysis clarifies the evolutionary origin of Coffea arabica. J. Syst. Evol. 59, 953–963 (2020).
Yu, Q. et al. Micro-collinearity and genome evolution in the vicinity of an ethylene receptor gene of cultivated diploid and allotetraploid coffee species (Coffea). Plant J. 67, 305–317 (2011).
Merot-L’anthoene, V. et al. Development and evaluation of a genome-wide Coffee 8.5K SNP array and its application for high-density genetic mapping and for investigating the origin of Coffea arabica L. Plant Biotechnol. J. 17, 1418–1430 (2019).
Wellman, F. L. Coffee: Botany, Cultivation and Utilization (L. Hill, 1961).
Lécolier, A., Besse, P., Charrier, A., Tchakaloff, T.-N. & Noirot, M. Unraveling the origin of Coffea arabica ‘Bourbon pointu’ from La Réunion: a historical and scientific perspective. Euphytica 168, 1–10 (2009).
Clarindo, W. R., Carvalho, C. R., Caixeta, E. T. & Koehler, A. D. Following the track of ‘Híbrido de Timor’ origin by cytogenetic and flow cytometry approaches. Genet. Resour. Crop Evol. 60, 2253–2259 (2013).
Bertrand, B., Guyot, B., Anthony, F. & Lashermes, P. Impact of the Coffea canephora gene introgression on beverage quality of C. arabica. Theor. Appl. Genet. 107, 387–394 (2003).
Marie, L. et al. G × E interactions on yield and quality in Coffea arabica: new F1 hybrids outperform American cultivars. Euphytica 216, 78 (2020).
Bertrand, B., Villegas Hincapié, A. M., Marie, L. & Breitler, J.-C. Breeding for the main agricultural farming of Arabica coffee. Front. Sustain. Food Syst. 5, 709901 (2021).
Breitler, J.-C. et al. CRISPR/Cas9-mediated efficient targeted mutagenesis has the potential to accelerate the domestication of Coffea canephora. Plant Cell Tissue Organ Cult. 134, 383–394 (2018).
Berthaud, J. Etude cytogénétique d’un haploïde de Coffea arabica L. Cafe Cacao The 20, 91–96 (1976).
Denoeud, F. et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science 345, 1181–1184 (2014).
Pellicer, J. & Leitch, I. J. The Plant DNA C-values database (release 7.1): an updated online repository of plant genome size data for comparative studies. New Phytol. 226, 301–305 (2020).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Petit, M. et al. Mobilization of retrotransposons in synthetic allotetraploid tobacco. New Phytol. 186, 135–147 (2010).
Sarilar, V. et al. Allopolyploidy has a moderate impact on restructuring at three contrasting transposable element insertion sites in resynthesized Brassica napus allotetraploids. New Phytol. 198, 593–604 (2013).
Bird, K. A., VanBuren, R., Puzey, J. R. & Edger, P. P. The causes and consequences of subgenome dominance in hybrids and recent polyploids. New Phytol. 220, 87–93 (2018).
Göbel, U. et al. Robustness of transposable element regulation but no genomic shock observed in interspecific Arabidopsis hybrids. Genome Biol. Evol. 10, 1403–1415 (2018).
Birchler, J. A. & Veitia, R. A. The gene balance hypothesis: implications for gene regulation, quantitative traits and evolution. New Phytol. 186, 54–62 (2010).
Zeiss, D. R., Piater, L. A. & Dubery, I. A. Hydroxycinnamate amides: intriguing conjugates of plant protective metabolites. Trends Plant Sci. 26, 184–195 (2021).
Bird, K. A. et al. Replaying the evolutionary tape to investigate subgenome dominance in allopolyploid Brassica napus. New Phytol. 230, 354–371 (2021).
Combes, M.-C., Joët, T., Stavrinides, A. K. & Lashermes, P. New cup out of old coffee: contribution of parental gene expression legacy to phenotypic novelty in coffee beans of the allopolyploid Coffea arabica L. Ann. Bot. 131, 157–170 (2023).
Yoo, M. J., Szadkowski, E. & Wendel, J. F. Homoeolog expression bias and expression level dominance in allopolyploid cotton. Heredity 110, 171–180 (2013).
Meyer, F. G., Fernie, L. M., Narasimhaswami, R. L., Monaco, L. C. & Greathead, D. J. FAO Coffee Mission to Ethiopia, 1964–1965 (Food and Agriculture Organization of the United Nations, 1968).
Halle, F. Echantillonnage du matériel Coffea arabica récolté en Ethiopie. Bulletin IFCC 14, 13–18 (1978).
Krug, C. A. & Mendes, A. J. T. Cytological observations in Coffea – IV. J. Genet. 39, 189–203 (1940).
Cros, J. et al. Phylogenetic analysis of chloroplast DNA variation in Coffea L. Mol. Phylogenet. Evol. 9, 109–117 (1998).
Lashermes, P. et al. Molecular characterisation and origin of the Coffea arabica L. genome. Mol. Gen. Genet. 261, 259–266 (1999).
Wu, Y. et al. Genomic mosaicism due to homoeologous exchange generates extensive phenotypic diversity in nascent allopolyploids. Natl Sci. Rev. 8, nwaa277 (2021).
Terhorst, J., Kamm, J. A. & Song, Y. S. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 49, 303–309 (2017).
Moat, J., Gole, T. W. & Davis, A. P. Least concern to endangered: applying climate change projections profoundly influences the extinction risk assessment for wild Arabica coffee. Glob. Change Biol. 25, 390–403 (2019).
Kuper, R. & Kröpelin, S. Climate-controlled holocene occupation in the Sahara: motor of Africa’s evolution. Science 313, 803–807 (2006).
Excoffier, L. et al. fastsimcoal2: demographic inference under complex evolutionary scenarios. Bioinformatics 37, 4882–4885 (2021).
Lambeck, K. et al. Sea level and shoreline reconstructions for the Red Sea: isostatic and tectonic considerations and implications for hominin migration out of Africa. Quat. Sci. Rev. 30, 3542–3574 (2011).
Montagnon, C., Mahyoub, A., Solano, W. & Sheibani, F. Unveiling a unique genetic diversity of cultivated Coffea arabica L. in its main domestication center: Yemen. Genet. Resour. Crop Evol. 68, 2411–2422 (2021).
Nordborg, M. & Donnelly, P. The coalescent process with selfing. Genetics 146, 1185 (1997).
Hu, G. et al. Two divergent haplotypes from a highly heterozygous lychee genome suggest independent domestication events for early and late-maturing cultivars. Nat. Genet. 54, 73–83 (2022).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Molloy, E. K., Durvasula, A. & Sankararaman, S. Advancing admixture graph estimation via maximum likelihood network orientation. Bioinformatics 37, i142–i150 (2021).
Pfeifer, B. & Kapan, D. D. Estimates of introgression as a function of pairwise distances. BMC Bioinformatics 20, 207 (2019).
Gaut, B. S., Seymour, D. K., Liu, Q. & Zhou, Y. Demography and its effects on genomic variation in crop domestication. Nat. Plants 4, 512–520 (2018).
dos Santos, T. B., Baba, V. Y., Vieira, L. G. E., Pereira, L. F. P. & Domingues, D. S. The urea transporter DUR3 is differentially regulated by abiotic and biotic stresses in coffee plants. Physiol. Mol. Biol. Plants 27, 203–212 (2021).
Wang, W. et al. Structural basis of salicylic acid perception by Arabidopsis NPR proteins. Nature 586, 311–316 (2020).
Mukhtar, M. S. et al. Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 333, 596–601 (2011).
Jousimo, J. et al. Ecological and evolutionary effects of fragmentation on infectious disease dynamics. Science 344, 1289–1293 (2014).
Cooley, M. B., Pathirana, S., Wu, H. J., Kachroo, P. & Klessig, D. F. Members of the Arabidopsis HRT/RPP8 family of resistance genes confer resistance to both viral and oomycete pathogens. Plant Cell 12, 663–676 (2000).
Mohr, T. J. et al. The Arabidopsis downy mildew resistance gene RPP8 is induced by pathogens and salicylic acid and is regulated by W-box cis elements. Mol. Plant Microbe Interact. 23, 1303–1315 (2010).
MacQueen, A. et al. Population genetics of the highly polymorphic RPP8 gene family. Genes 10, 691 (2019).
Cheng, Y. T. et al. Stability of plant immune-receptor resistance proteins is controlled by SKP1-CULLIN1-F-box (SCF)-mediated protein degradation. Proc. Natl Acad. Sci. USA 108, 14694–14699 (2011).
Hedtmann, C. et al. The plant immunity regulating F-Box Protein CPR1 supports plastid function in absence of pathogens. Front. Plant Sci. 8, 1650 (2017).
Feuillet, C., Schachermayr, G. & Keller, B. Molecular cloning of a new receptor-like kinase gene encoded at the Lr10 disease resistance locus of wheat. Plant J. 11, 45–52 (1997).
Zhou, H. et al. Molecular analysis of three new receptor-like kinase genes from hexaploid wheat and evidence for their participation in the wheat hypersensitive response to stripe rust fungus infection. Plant J. 52, 420–434 (2007).
Xia, T. et al. Efficient expression and function of a receptor-like kinase in wheat powdery mildew defence require an intron-located MYB binding site. Plant Biotechnol. J. 19, 897–909 (2021).
Florez, J. C. et al. High throughput transcriptome analysis of coffee reveals prehaustorial resistance in response to Hemileia vastatrix infection. Plant Mol. Biol. 95, 607–623 (2017).
Gaut, B. S., Díez, C. M. & Morrell, P. L. Genomics and the contrasting dynamics of annual and perennial domestication. Trends Genet. 31, 709–719 (2015).
Chen, Z. J. Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci. 15, 57–71 (2010).
Lan, T. et al. Insights into bear evolution from a Pleistocene polar bear genome. Proc. Natl Acad. Sci. USA 119, e2200016119 (2022).
Berlin, K. et al. Assembling large genomes with single-molecule sequencing and locality-sensitive hashing. Nat. Biotechnol. 33, 623–630 (2015).
Chin, C.-S. et al. Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods 13, 1050–1054 (2016).
Walker, B. J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 9, e112963 (2014).
English, A. C. et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS ONE 7, e47768 (2012).
Rastas, P. Lep-MAP3: robust linkage mapping even for low-coverage whole genome sequencing data. Bioinformatics 33, 3726–3732 (2017).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat. Plants 5, 833–845 (2019).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Lyons, E. & Freeling, M. How to usefully compare homologous plant genes and chromosomes as DNA sequences. Plant J. 53, 661–673 (2008).
Lyons, E. et al. Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol. 148, 1772–1781 (2008).
Lefebvre-Pautigny, F. et al. High resolution synteny maps allowing direct comparisons between the coffee and tomato genomes. Tree Genet. Genomes 6, 565–577 (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://doi.org/10.48550/arXiv.1303.3997 (2013).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
Orozco-Arias, S. et al. Inpactor2: a software based on deep learning to identify and classify LTR-retrotransposons in plant genomes. Brief. Bioinform. 24, bbac511 (2023).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Ma, J. & Bennetzen Jeffrey, L. Rapid recent growth and divergence of rice nuclear genomes. Proc. Natl Acad. Sci. USA 101, 12404–12410 (2004).
Orozco-Arias, S. et al. Inpactor, integrated and parallel analyzer and classifier of LTR retrotransposons and its application for pineapple LTR retrotransposons diversity and dynamics. Biology 7, 32 (2018).
Campbell, M. S. et al. MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 164, 513–524 (2014).
Keilwagen, J., Hartung, F. & Grau, J. in Gene Prediction: Methods and Protocols (ed. Kollmar, M.) 161–177 (Springer, 2019).
Cheng, B., Furtado, A. & Henry, R. J. The coffee bean transcriptome explains the accumulation of the major bean components through ripening. Sci. Rep. 8, 11414 (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Sankoff, D. et al. Models for similarity distributions of syntenic homologs and applications to phylogenomics. IEEE/ACM Trans. Comput. Biol. Bioinform. 16, 727–737 (2019).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Jónsson, H., Ginolhac, A., Schubert, M., Johnson, P. L. F. & Orlando, L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29, 1682–1684 (2013).
Poplin, R. et al. Scaling accurate genetic variant discovery to tens of thousands of samples. Preprint at bioRxiv https://doi.org/10.1101/201178 (2018).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 6, 80–92 (2012).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, https://doi.org/10.1186/s13742-015-0047-8 (2015).
Alexander, D. H. & Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinf. 12, 246 (2011).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: analysis of next generation sequencing data. BMC Bioinf. 15, 356 (2014).
Patterson, N. et al. Ancient admixture in human history. Genetics 192, 1065–1093 (2012).
Salojärvi, J. et al. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nat. Genet. 49, 904–912 (2017).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Li, H. & Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 475, 493–496 (2011).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967 (2012).
Orozco-Arias, S. et al. TIP_finder: an HPC software to detect transposable element insertion polymorphisms in large genomic datasets. Biology 9, 281 (2020).
Kautsar, S. A., Suarez Duran, H. G., Blin, K., Osbourn, A. & Medema, M. H. plantiSMASH: automated identification, annotation and expression analysis of plant biosynthetic gene clusters. Nucleic Acids Res. 45, W55–W63 (2017).
Klopfenstein, D. V. et al. GOATOOLS: a Python library for Gene Ontology analyses. Sci. Rep. 8, 10872 (2018).
Salojärvi, J. jsalojar/PiNSiR: first release of PiNSiR. Zenodo https://doi.org/10.5281/zenodo.5136527 (2021).
Acknowledgements
We acknowledge the Natural History Museum in London for providing a sample of the C. arabica lectotype. Funding was provided from the Academy of Finland (decisions 318288 and 329441) and a Nanyang Technological University start-up grant (J. Salojärvi); Ecos-Nord grant no. C21MA01 and STIC-AmSud grant no. 21-STIC-13 (R.G. and S.O.-A.); the Academy of Finland, grant no. 343656 (P.R.); NAPI Bioinformática from Fundação Araucária and TELearning Project 2021–22 (grant no. 21-STIC-13) from STIC-AmSud (A.R.P.); Research Foundation – Flanders (FWO, grant no. G056517N) (Y.B.); the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant no. 833522) and Ghent University (Methusalem funding, grant no. BOF.MET.2021.0005.01) (Y.V.d.P.); the Horizon Europe program, PRO-GRACE project (grant no. 101094738) (G.G.); INCT-Café-CNPq/Fapemig (A.A.); São Paulo State Research Foundation (FAPESP), grant nos. 2016/10896-0 and 2017/01455-2 (D.D. and S.M.C.L.); NSERC and the Canada Research Chairs programs (D.S.); the United States National Science Foundation grant nos. 1442190 and 2030871 (V.A.A.); and Nestlé Research (P.D.). J. Salojärvi acknowledges the High Performance Computation Centre at NTU Singapore and University of Helsinki Linux administrators, as well as the CSC – IT Center for Science, Finland, for computational resources. R.G., P.H., E.C., C.T.D., V.P., A.d.K. and Unité Mixte de Recherche - Diversité, adaptation, développement des plantes (UMR DIADE) are grateful to The French National Research Institute for Sustainable Development (IRD).
Author information
Authors and Affiliations
Contributions
A.d.K., D.C. and P.D. conceived the study. A.A., A.N., C.K., E.C., G.H.S., H.R., L.B., L.F.P., L.P., M.S., M.T.B., O.G.F., P. Musoli, P. Marraccini, P.H. and U.S. provided genetic resources. A.C., C.F., D.M., G.L., J. Stolte, L.B., M.K., N.D., P.D. and S.M. carried out DNA sequencing. E.G. performed sequencing of the Linnaean accession. S.S., C.W., J. Salojärvi, S.P. and L.M. carried out genome assembly. P.R., M.R. and J. Salojärvi performed genetic mapping. A.R., S.S., L.M., J. Salojärvi, S. Rombauts, V.P., Z.Q.W., D.D., S.I.S., M.M., R.A., S.M.C.L., M.L., C.T.-D. and G.G. carried out genome annotation. A.R.P., J.E. and P.S. carried out annotation of noncoding RNA. S.O.-A., A.G. and R.G. performed transposable element annotation and analysis. V.A.A. and C.M.W. carried out telomere identification. Z.Y., C.Z., D.S.M., R.G., J.M., D.S., L.C.-P., T.L., T.J.K., V.A.A., S.O.-A., A.G. and J. Salojärvi analyzed genome evolution. Z.Q.W., V.P., D.D., G.G., S.J.F., V.A.A., S. Rajaraman and J. Salojärvi carried out gene family analysis. A.R., S.P., S. Rajaraman and J. Salojärvi performed RNA-seq data analysis. R.H. provided RNA-seq data. J. Salojärvi analyzed population data. Y.B. and R.G. analyzed GBS data. L.M., S. Rombauts and J. Salojärvi arranged online data access. J. Salojärvi wrote the first draft, which was completed with input from G.G., D.S., V.A.A., L.F.P., R.G., S. Rombauts, A.d.K., P.D., V.P., L.M., D.C., D.D., S.P. and A.A., as well as P. Marraccini, Y.B., T.R. and Y.V.d.P., and all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Genetics thanks France Denoeud and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Coffee dissemination routes.
Yellow: current Coffea arabica cultivation; black: current C. canephora cultivation; shaded black/yellow: current cultivation, both species. Solid lines over the Middle East: early spread of coffee consumption; dashed lines: main Bourbon routes; dotted lines: main Typica routes; Ethiopia (the center of origin of Arabica) and Timor Island (the origin of the Timor hybrid) are colored in red. The map was modified from an original available in the public domain from Wikimedia commons.
Extended Data Fig. 2 Composition and expression of exemplar Arabica gene families contributing to bean quality traits.
a, Schematic biosynthesis of caffeine (left), terpenoids (middle), and unsaturated fatty acids (right). b, Phylogenies and expression during fruit development of CA genes for N-methyltransferases (NMTs) mediating caffeine biosynthesis (left), terpene synthases (TPS) (middle), and fatty acid dehydrogenase 2 (FAD2) (right). RNA sequencing was carried out for three biological replicates from three different fruit maturation stages (green, yellow, and red) of the K7 cultivar. c, Genome-wide NMT (left), TPS (middle), and FAD2 (right) gene trees and expression patterns during fruit development. Genes located in the two subgenomes are indicated by font color; subCC (red) and subEE (blue). Arabidopsis genes are in brown. Grey wedges in the circular trees highlight the tree portions shown in b. XMT: xanthosine methyltransferase; MXMT: 7-methylxanthine methyltransferase; DXMT: 1,7-dimethylxanthine methyltransferase; MTL: N-methyltransferase-like; FS: (E,E)-a-farnesene synthase; GS: Geraniol synthase; IS: Isoprene synthase; MS: myrcene synthase; TS: (-)-a-terpineol synthase; FAD2: Fatty acid desaturase 2. Gene expression is shown by color scale, yellow (positive) through red to blue (negative), in units of log10 transcripts per million (log10 TPM); grey areas indicate the absence of expression data for Arabidopsis genes.
Extended Data Fig. 3 Homoeologous exchange.
a, Summary of homoeologous exchange between subgenomes. Blue bars indicate genes with 3:1 allele bias towards subCC, whereas yellow bars indicate genes with allele bias (1:3 or 0:4) towards subEE. b, Frequency spectrum of shared homoeologous exchanges at gene level.
Extended Data Fig. 4 Homoeologous exchange plots of chromosomes 2 (a) and 4 (b) overlaying all Coffea arabica accessions in this study.
The dark red region indicates 4:0 allele balance in favor of subCC, while the pink region illustrates 3:1, white 2:2, light blue 1:3 and dark blue 0:4 balances, respectively. The grey lines indicate the observed allele balances in syntelog gene pairs for the different Arabica accessions. For a view of all chromosomes and of the genes involved, see Supplementary Figure 35 and Supplementary Table 27.
Extended Data Fig. 5 PCA plots based on SNPs.
a, From SNP data called on the subCC (top) versus subEE (bottom) subgenomes. The rectangles highlight zoomed-in regions in panels b and c.
Extended Data Fig. 6 Arabica speciation time estimates based on population genetic modeling and rate of genome fractionation.
a, Summary of FastsimCoal2 models for historical effective population sizes (Ne). The effective population size (y-axis) is plotted against the number of generations before present (Gbp, x-axis). The bottlenecks were identified using 100 FastSimCoal runs with 10^6 simulations. Maximum composite likelihood estimation of parameters was carried out with 40 expectation-conditional maximization iterations. The plots summarize the best models for subgenomes CC and EE in the wild, non-admixed population. To convert generations to years, an estimate of 21 years/generation was used (Moat el al. 2019). b, Summary of the genome fractionation rate and divergence of syntenic gene models. The timing of the splits in the phylogeny (left) reflects the most recent estimates from (Bawin et al., 2020). The rate of gene loss (barplot) is presented as the percent of syntenic genes lost in the Eugenioides/subEE common ancestor (light blue) or only in subEE (blue). A similar analysis was carried out for Robusta-derived genomes, where the percent of genes lost in Robusta/subCC is shown in light red and genes lost only in subCC with dark red. The Ks peaks method (right) scales the divergence time between the subgenomes, estimated from numbers of synonymous mutations between syntenic genes to the timing of the speciation event.
Extended Data Fig. 7 Kinship analysis on subEE.
The degree of relatedness was estimated using Kinship-based INference for GWAS (KING); Thumbnail images show false discovery rate corrected F3 tests of introgression Z-statistics for each of the target individuals. Each cell in the matrix illustrates an F3 test result for the target accession containing introgression from two different sources (x- and y- axis); blue color illustrates significant adjusted Z-score (Z. adj; associated value indicated by color key), indicative of gene flow (or allele sharing via identity by descent; IBD) from the two source accessions to the target, while red color illustrates no support for gene flow. The green background in the wild accessions highlights the admixed individuals (Fig. 2b); the non-admixed individuals are highlighted with red. The corresponding analysis on subCC is shown in Fig. 3.
Extended Data Fig. 8 Timing of wild Arabica introgression.
Lengths of Robusta introgressed blocks in Timor hybrid accessions (a) and, as a control, of Typica introgressed blocks in wild Arabica accessions (b).
Extended Data Fig. 9 Population genetic statistics.
a, Non-synonymous (πn; left) and synonymous (πs; right) diversity in the different populations of C. arabica. b, F inbreeding coefficients for wild and cultivated lines, shown separately for subCC (left) and subEE (right).
Supplementary information
Supplementary Information
Supplementary Figs. 1–50 and text.
Supplementary Tables
Supplementary Tables 1–33.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Salojärvi, J., Rambani, A., Yu, Z. et al. The genome and population genomics of allopolyploid Coffea arabica reveal the diversification history of modern coffee cultivars. Nat Genet 56, 721–731 (2024). https://doi.org/10.1038/s41588-024-01695-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41588-024-01695-w
