
Abstract
The ability to switch between different lifestyles allows bacterial pathogens to thrive in diverse ecological niches1,2. However, a molecular understanding of their lifestyle changes within the human host is lacking. Here, by directly examining bacterial gene expression in human-derived samples, we discover a gene that orchestrates the transition between chronic and acute infection in the opportunistic pathogen Pseudomonas aeruginosa. The expression level of this gene, here named sicX, is the highest of the P. aeruginosa genes expressed in human chronic wound and cystic fibrosis infections, but it is expressed at extremely low levels during standard laboratory growth. We show that sicX encodes a small RNA that is strongly induced by low-oxygen conditions and post-transcriptionally regulates anaerobic ubiquinone biosynthesis. Deletion of sicX causes P. aeruginosa to switch from a chronic to an acute lifestyle in multiple mammalian models of infection. Notably, sicX is also a biomarker for this chronic-to-acute transition, as it is the most downregulated gene when a chronic infection is dispersed to cause acute septicaemia. This work solves a decades-old question regarding the molecular basis underlying the chronic-to-acute switch in P. aeruginosa and suggests oxygen as a primary environmental driver of acute lethality.
Similar content being viewed by others

Main
Many pathogenic bacteria can colonize their hosts and persist chronically. In some cases, bacteria disseminate from the initial infection sites to other body parts, resulting in acute systemic diseases. A central question in biology is to understand the molecular mechanisms and environmental cues controlling this lifestyle switch. The opportunistic human pathogen P. aeruginosa can cause both acute and chronic infections that are notoriously difficult to treat. P. aeruginosa exists as single cells (planktonic lifestyle) or matrix-enclosed aggregates (biofilm lifestyle), which are considered to favour acute and chronic infections, respectively. Therefore, laboratory studies have traditionally focused on probing the biofilm–planktonic transition of P. aeruginosa in vitro, with the goal to understand this pathogen’s lifestyle changes in humans. Decades of work have shown how global regulatory systems such as the second messenger cyclic di-GMP3,4,5 and the Gac–Rsm system2,6,7,8,9,10,11,12 control the biofilm–planktonic transition of P. aeruginosa in vitro, providing critical insights into the life history of this bacterium. However, so far, it has remained elusive how P. aeruginosa responds to environmental cues and accordingly switches infection lifestyles within the mammalian host. In this study, we addressed this gap in knowledge by leveraging P. aeruginosa transcriptomes acquired from human-derived samples to discover and mechanistically characterize a new small RNA, termed sRNA inducer of chronic infection X (SicX), that governs the P. aeruginosa chronic or acute decision during mammalian infection.
High expression of PA1414 in humans
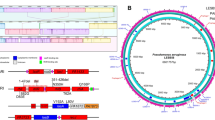
We previously obtained high-resolution P. aeruginosa transcriptomes from human infection samples, including sputum samples from patients with cystic fibrosis and debridement samples from patients with chronic wounds13,14. Using machine learning methods, we identified 30 P. aeruginosa genes whose expression levels collectively differentiate P. aeruginosa growth in humans from that in the laboratory13. More than half of these genes are uncharacterized, highlighting a substantial gap in knowledge of P. aeruginosa human infection biology. Included in these genes of unknown function is PA1414 (locus tag in P. aeruginosa strain PAO1), whose expression level in humans was 222-fold higher than that in the laboratory, and its expression level is the highest of the P. aeruginosa genes expressed during human infection (Fig. 1a). We next compared the relative abundance (transcripts per million (TPM)) of PA1414 transcripts with those of other protein-coding (5,893 genes) and non-coding (199 sRNAs)15 transcripts (Fig. 1b). On average, PA1414 transcripts represented 13.85% of the TPM in P. aeruginosa transcriptomes acquired from human chronic infection samples, and notably, they constituted almost 50% of the total TPM in some cases. However, the level of PA1414 expression was extremely low in P. aeruginosa grown under standard in vitro conditions. PA1414 is a small gene (234 base pairs long) of unknown function. Examination of 261 complete P. aeruginosa genomes identified 258 PA1414 orthologues (Fig. 1c and Extended Data Fig. 1), and no homologues were identified in other Pseudomonas species or other organisms. The DNA sequences of PA1414 orthologues including their upstream promoter regions are highly conserved (Fig. 1d), suggesting that PA1414 has a conserved function and the regulation of its expression is universal across P. aeruginosa isolates.

a, Differential gene expression of P. aeruginosa between human infections and common in vitro conditions. For each gene, the log2[fold change] is plotted against the Wald test P value. Bubbles with black borders highlight the 30 signature genes of P. aeruginosa human infection. Bubble size indicates mRNA read abundance during human infections. Blue bars on the y axis indicate genes (with negative log2[fold change] values) outside the scale range. Dashed grey lines indicate the cutoffs (−log10[adjusted P value] > 2, |log2[fold change]| > 1) for identifying differentially expressed genes. The upregulated (green) and downregulated (blue) genes in humans are colour-coded differently. Grey bubbles indicate genes that are not differentially expressed. b, Relative transcript abundance (TPM) of PA1414 compared to those of all other protein-coding sequences (CDSs) and non-coding sRNAs in 54 transcriptomes examined in a, sorted from the highest to the lowest PA1414 TPM. Average PA1414 TPM is indicated with dashed lines. c, PA1414 orthologues are found only in P. aeruginosa. Green and black bars indicate the presence and absence of orthologues, respectively. d, DNA sequence conservation of PA1414 and its neighbouring genes among 258 PA1414-containing P. aeruginosa isolates. Below, the percentage of orthologues identical to the PA1414 allele from PA14 at each nucleotide is shown.
Low oxygen induces PA1414 expression
P. aeruginosa can thrive in diverse ecological niches within the human body. To understand what environmental cues drive PA1414 expression, we examined 202 publicly available P. aeruginosa transcriptomes acquired across a range of environments (Fig. 2a and Supplementary Table 1). We found that PA1414 was expressed at high levels under low-oxygen and anaerobic conditions. Consistent with this finding, an early microarray study also showed that PA1414 is induced during anaerobic growth16. Moreover, a consensus binding motif for the global anaerobic transcriptional regulator, Anr16, was identified upstream of PA1414 (Extended Data Fig. 2a). Using a lacZ reporter that quantifies transcription of PA1414, we showed that indeed this gene was induced by 25-fold under anaerobic conditions, and mutagenesis of Anr or the binding motif abolished PA1414 expression (Fig. 2b). PA1414 was also expressed at an approximately 18-fold higher level in static cultivation (Fig. 2b), indicating that strict anaerobic conditions are not required for its induction. In addition, northern blot analysis provided direct evidence that PA1414 transcripts were highly abundant during anaerobic but not aerobic growth (Fig. 2c). To directly compare PA1414 expression levels in humans and in low-oxygen environments, we conducted RNA-sequencing (RNA-seq) experiments on P. aeruginosa during in vitro static growth (a low-oxygen condition), following the same library preparation method used in our study of P. aeruginosa transcriptomes in humans14. We found that although under low-oxygen conditions PA1414 transcript abundance (TPM) was sevenfold lower than that during human infections, PA1414 is among the three genes expressed at the highest levels (out of approximately 6,000 protein-coding and sRNA genes), supporting the notion that a low oxygen level is indeed a primary driver for its high expression level (Extended Data Fig. 2b). Given that P. aeruginosa often encounters low-oxygen environments during human chronic infection17,18, these data indicate that P. aeruginosa responds to this host environmental cue by increasing the expression level of PA1414.

a, PA1414 RNA abundance (TPM) and the corresponding ranking (out of all protein-coding genes and sRNAs) in 202 P. aeruginosa transcriptomes. Dashed lines indicate the cutoffs (PA1414 TPM > 103, PA1414 ranking < 102) for identifying transcriptomes with high PA1414 expression. b, β-galactosidase assay examining the induction of PA1414 under anaerobic and static conditions. PPA1414-lacZ, lacZ transcriptionally fused to PA1414 promoter; PPA1414*, PA1414 promoter containing mutations in the Anr-binding motif; MrT7, MAR2xT7 transposon. n ≥ 4 independent experiments. c, Northern blot analysis of PA1414 expression. Estimated size (nucleotides, nt) are indicated on the left. 5S rRNA served as a loading control. For gel source data, see Supplementary Fig. 1. d, Colony biofilm growth of ΔPA1414 and WT in different P. aeruginosa strain backgrounds. The presence and absence of oxygen are indicated. The CFU of ΔPA1414 was normalized against the CFU of the WT in each experiment (n = 4). A dashed line highlights the point where the CFU ratio = 1. e, Percentage of tetracycline-resistant (TetR) cells before and after daily passages under anaerobic conditions (n = 3). TetS, tetracycline sensitive; Δ, ΔPA1414. f, RNA-seq reads aligned to the PA1414 locus during standard in vitro growth and human infections. Blue shade indicates Rho-independent terminator in PA1414. g, Colony biofilm growth of ΔsicX harbouring different sicX mutations under anaerobic conditions. n = 4 independent experiments. h, Comparative proteomic study identifies the targets of SicX under both static and anaerobic conditions. i, β-galactosidase assay evaluating the translational control of SicX on ubiUVT under anaerobic conditions (n ≥ 4). j, β-galactosidase assay evaluating the transcription of ubiUVT in the absence of SicX or Anr under static conditions (n ≥ 8). k, Quantification of anaerobic UQ9 synthesis in different strains (n = 4). l, A model of dual regulation of ubiUVT expression by Anr and SicX. Error bars represent standard deviation from the mean. Significant differences (compared to the WT) are indicated with asterisks (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; two-tailed Mann–Whitney test).
We next considered whether PA1414 affects the anaerobic physiology of P. aeruginosa. For these experiments, we deleted PA1414 from the P. aeruginosa strain PA14 (the corresponding locus tag in this strain is PA14_46160) and compared the growth of ΔPA1414 to that of wild-type (WT) P. aeruginosa under aerobic and anaerobic conditions (Fig. 2d). Using a colony biofilm assay, we found that whereas deletion of PA1414 did not negatively affect the aerobic growth of P. aeruginosa, ΔPA1414 exhibited growth yields approximately half that of WT under anaerobic conditions with nitrate as the alternative electron acceptor (Fig. 2d). Similar observations were made in different P. aeruginosa strain backgrounds including a cystic fibrosis clinical isolate, B1 (Fig. 2d; ref. 19). Although we showed that PA1414 also played a minor role in anaerobic planktonic growth (Extended Data Fig. 2c), competition experiments in which WT and ΔPA1414 were co-cultured planktonically (at a 1:1 starting ratio) and passaged daily under anaerobic conditions revealed that WT outcompeted ΔPA1414 after day 3 (Fig. 2e).
PA1414 encodes an sRNA, SicX
We next investigated the molecular mechanism by which PA1414 controls anaerobic growth. Although annotated as a protein-coding gene, we discovered that during human infection PA1414 transcripts were restricted to the 5′ end (Fig. 2f), and a similar pattern was also observed under anaerobic conditions (Extended Data Fig. 2d). Clear enrichment of short transcripts encompassing the annotated start codon raised the question of whether PA1414 encodes a functional protein or a small regulatory RNA (sRNA). To address this question, we introduced different mutations in PA1414 and assessed whether they could restore the growth yield defect of the ΔPA1414 colony biofilm under anaerobic conditions (Fig. 2g and Extended Data Fig. 2e). We found that a start codon or frame-shift mutation (to prevent protein synthesis) did not affect the function of PA1414, whereas synonymous substitutions in the highly transcribed region (to disrupt potential interactions of sRNA with target mRNAs or RNA-binding proteins) abolished its function. Moreover, a truncated allele encompassing only the highly transcribed 5′ region was sufficient to restore the anaerobic growth defect in ΔPA1414, further highlighting this functional region of PA1414. Through northern blot analyses, we showed that the observed growth differences were not due to differential sRNA abundance (Extended Data Fig. 2f). Thus, we conclude that PA1414 functions as an sRNA (hereafter referred to as SicX) that is not essential for but promotes P. aeruginosa anaerobic growth in vitro.
SicX regulates anaerobic respiration
As sRNAs often exert their regulation post-transcriptionally20,21,22, we carried out quantitative shotgun proteomics to identify targets of SicX. Here, proteomes of WT P. aeruginosa and ΔsicX were obtained under two growth conditions in which sicX is expressed at a high level: low-oxygen (that is, static growth) and strict anaerobic conditions. We identified seven proteins (including three with known functions) exhibiting different abundances between WT and ΔsicX under both growth conditions (Fig. 2h and Supplementary Table 2). One of these proteins is UbiV, which is encoded by the ubiUVT operon23. This operon encodes proteins responsible for the anaerobic biosynthesis of ubiquinone, an essential electron carrier for respiratory metabolism in proteobacteria23. Although the other two proteins encoded by this operon, UbiU and UbiT, were not in the seven proteins identified with our stringent criteria, further investigation revealed that both of these proteins also showed significantly reduced levels in ΔsicX compared to WT P. aeruginosa during static growth (Supplementary Table 2). Using a lacZ translational reporter of the ubiUVT operon, we showed that sicX deletion indeed reduced the translational activity of ubiUVT by sixfold under anaerobic conditions, and genetic complementation with sicX in trans restored ubiUVT translation (Fig. 2i). Moreover, SicX did not affect the transcription of ubiUVT (Fig. 2j). To provide direct evidence of how SicX regulates anaerobic respiration, we quantified the ubiquinone (more specifically, UQ9) abundance in WT and ΔsicX under anaerobic conditions by liquid chromatography–mass spectrometry, and indeed, ΔsicX exhibited a twofold reduction in UQ9 compared to WT (Fig. 2k). Therefore, we concluded that SicX post-transcriptionally induces the expression of UbiUVT. Our finding also provides insights into the increased abundance of Dnr and HemF in ΔsicX (Fig. 2h), as Dnr can induce the denitrification pathway16 to alleviate the anaerobic stress in ΔsicX, and the expression of HemF is Dnr dependent24. Notably, we found that the transcription of ubiUVT is under the control of Anr (Fig. 2j). Together, these results support a model in which Anr activates the transcription of sicX and ubiUVT in response to oxygen deprivation. In turn, SicX post-transcriptionally induces ubiquinone biosynthesis to shape the anaerobic physiology of P. aeruginosa (Fig. 2l).
We next explored how SicX activates the translation of ubiUVT mRNA. In silico analysis through IntaRNA25 identified a putative base-pairing interaction between SicX and the 5′ untranslated region (UTR) of the ubiUVT operon (Extended Data Fig. 3a). Secondary structures predicted with Mfold26 indicate that a stem–loop within the 5′ UTR of ubiUVT occludes the ribosome-binding site of ubiU, whereas base pairing with SicX has the potential to liberate the ribosome-binding site for translation initiation. To experimentally test this, we carried out extensive mutational analyses of sicX and the 5′ UTR of ubiUVT, and the functional outcome of each point mutation (37 in total) was assessed through a lacZ reporter translationally fused to ubiUVT (Extended Data Fig. 3a,b). In concordance with in silico prediction, we identified mutations within the SicX seed region (for base pairing with the ubiUVT mRNA) and the target region in the 5′ UTR of ubiUVT that led to reduced translational activation of ubiUVT, whereas mutations in other regions generally had limited effects (Extended Data Fig. 3a,b). Moreover, a point mutation in the seed region of SicX that reduced ubiUVT translation can be partially restored by re-establishing base pairing in the 5′ UTR of ubiUVT (Extended Data Fig. 3b). However, it should be noted that the observed translational restoration may partly result from the mutation in the 5′ UTR of ubiUVT, which increased ubiUVT translation even in the absence of SicX (Extended Data Fig. 3c), probably by destabilizing the stem–loop. Finally, we showed that the RNA chaperone Hfq27 is not required for the induction of ubiUVT translation by SicX (Extended Data Fig. 3d). Although Hfq is known to facilitate the interactions between sRNAs and target mRNAs in many cases, Hfq-independent sRNA–mRNA interactions have been well documented28,29,30. On the basis of our genetic results, we propose an Hfq-independent base-pairing model for SicX stimulation of ubiUVT translation, which will be tested in the future. Notably, the predicted base-pairing regions in SicX and the 5′ UTR of ubiUVT are identical across all examined P. aeruginosa genomes (Extended Data Fig. 4a–c), and thus this mechanism of regulation may be universal in P. aeruginosa.
SicX governs the chronic-to-acute switch
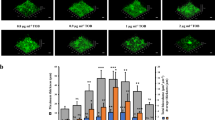
Although we now have a more granular understanding of SicX function in vitro, how SicX affects P. aeruginosa pathogenesis remains unknown. Here we addressed this question by introducing WT P. aeruginosa and ΔsicX into a mouse chronic wound model (Fig. 3a). This model was chosen because it has been quantitatively shown to accurately recapitulate P. aeruginosa gene expression in human chronic infections13, and SicX sRNA in this model is expressed similarly to that in humans (Fig. 2a and Extended Data Fig. 5a,b). The model involves infection of surgically created full-thickness dorsal wounds31 (Fig. 3a). It is considered a chronic, non-lethal model because P. aeruginosa generally persists at the infection site for weeks without causing systemic dissemination. At 10 days post-infection, we assessed the bacterial burden in wound tissues as well as dissemination to the spleen. In wound tissues, the bacterial levels were similar between WT P. aeruginosa and ΔsicX (Fig. 3b), indicating that lacking SicX does not affect colonization. However, whereas only 2 of 21 WT-infected mice showed dissemination to the spleen, 12 of 20 ΔsicX-infected mice showed systemic dissemination (two-tailed Fisher’s exact test, P = 0.0009; Fig. 3c). In addition, ΔsicX caused higher lethality (Fig. 3d), which is not commonly observed with WT P. aeruginosa in this model. Dissemination generally occurred when the wound burden was high (>108 colony-forming units (CFUs) g−1; Fig. 3c). However, among mice carrying a high wound burden (>108 CFUs g−1), ΔsicX caused more systemic infection (12 out of 15) compared to WT (2 out of 11), indicating that dissemination did not solely depend on the wound burden (two-tailed Fisher’s exact test, P = 0.0043; Fig. 3c). Moreover, these observations are not due to differential fitness of WT and ΔsicX in spleens. This was demonstrated using an acute skin infection model in which systemic infection occurs shortly after subcutaneous injection into the mouse inner thigh. In this model, WT and ΔsicX exhibited similar colonization and growth in spleens (Extended Data Fig. 6). Taken together, our data indicate that SicX is a key player in P. aeruginosa chronic infection, the high expression of which promotes chronic localized infection.

a, Schematic of the mouse chronic wound model (10-day infection). b, P. aeruginosa burden in wounds. Lines indicate means. c, P. aeruginosa burden in the spleen (y axis). Corresponding wound burden is indicated on the x axis. d, Survival curves of mice infected with WT (n = 24) and ΔsicX (n = 24). e, Restoration of ubiUVT translation in ΔsicX prevented dissemination, and deletion of ubiUVT in WT caused dissemination. f, P. aeruginosa spatial organization in wounds. C, core; E, edge. g, Schematic of the mouse pneumonia model (2-day infection). h, P. aeruginosa burden in lungs. Lines indicate means. i, P. aeruginosa burden in the spleen. Lines indicate means. Two-tailed Mann–Whitney test was used to compare the bacterial burden differences in b,c,h,i. Log-rank test and two-tailed Wilcoxon matched-pairs signed-rank test were used for d,f, respectively. Summary of n = 6 independent experiments for b–d; n = 3 independent experiments for e,f; n = 2 independent experiments for h,i. NS, not significant.
To determine whether the increased dissemination and lethality of ΔsicX infection is linked to reduced anaerobic ubiquinone production, we first decoupled the translation of ubiUVT from SicX by creating a P. aeruginosa strain (ΔsicX-ubiC) that constitutively translates UbiUVT, even in the absence of SicX (Extended Data Fig. 7a,b). This was accomplished by introducing point mutations to the 5′ UTR of ubiUVT that probably alter its secondary structure and enable active translation. ΔsicX-ubiC grew to similar yields to those of WT P. aeruginosa and significantly higher yields than those of ΔsicX under anaerobic conditions in vitro (Extended Data Fig. 7c), and this was due to the restoration of anaerobic ubiquinone synthesis (Fig. 2k). Next, using the mouse chronic wound model, we found that ΔsicX-ubiC did not exhibit systemic dissemination (Fig. 3e). We also deleted the ubiUVT operon from WT P. aeruginosa, and the resulting strain did not grow anaerobically (Extended Data Fig. 7d) owing to the defect in anaerobic ubiquinone synthesis23. Notably, although all strains exhibited similar bacterial burdens in the wound (Extended Data Fig. 5c), systemic dissemination was observed only in ΔsicX and ΔubiUVT infection (Fig. 3e), indicating that sufficient anaerobic ubiquinone synthesis contributes to chronic localized infection. Although the mechanism of how ubiquinone abundance influences infection lifestyles is unclear, we found that it affects the macroscale spatial distribution of P. aeruginosa cells in the wound: WT cells preferentially remained in the wound centre where the cells were originally inoculated, and ΔsicX-infected wounds contained equivalent cells in the centre and outer edges of the wounds (Fig. 3f). This is supported by our proteomic findings showing that ΔsicX exhibited increased levels of two proteins (encoded by PA0223 and PA0224) that are biomarkers of dispersed P. aeruginosa32 (Supplementary Table 2). Moreover, ΔubiUVT showed a similar spatial organization pattern to that of ΔsicX (Fig. 3f). Considering that the wound probably harbours oxygen gradients, we speculate that these different spatial distributions partly reflect varying oxygen availability across the wound. Collectively, these findings suggest that reduced anaerobic ubiquinone biosynthesis promotes the dissemination of ΔsicX.
P. aeruginosa is notorious for causing a range of lung infections, and therefore, we also assessed the role of SicX in colonization and dissemination using a mouse pneumonia model (Fig. 3g). Here, P. aeruginosa establishes infection in the lung after intranasal administration, which in turn can lead to systemic dissemination depending on the virulence of individual strains. We found that mice infected with sublethal doses of WT and ΔsicX variants of PA14, a highly virulent strain, had similar bacterial burdens in the lungs at 48 h post-infection (Fig. 3h). By contrast, bacterial burdens in the spleens of ΔsicX-infected mice were approximately 100-fold higher than that of WT-infected mice, and complementation with the truncated sicX allele (depicted in Fig. 2g) in trans reduced dissemination (Fig. 3i), indicating that lacking SicX promotes systemic infection. Moreover, we carried out experiments using another strain, PAO1, that is less acutely virulent than PA14. We found that WT and ΔsicX exhibited similar colonization in the lungs, and whereas only two of nine WT-infected mice showed dissemination to the spleen, eight of nine ΔsicX-infected mice showed systemic dissemination (two-tailed Fisher’s exact test, P = 0.015; Extended Data Fig. 8). These observations indicate that SicX promotes chronic localized infection in multiple mammalian infection models.
SicX is a biomarker of chronic–acute transition
As the presence or absence of SicX dictates distinct infection lifestyles, we next considered whether SicX sRNA abundance changes during the transition between chronic and acute infections. To test this, we used the mouse chronic wound model in which WT P. aeruginosa was first allowed to establish local infection and then treated with glycoside hydrolases (GHs) or cis-2-decenoic acid (cis-DA) to induce systemic dissemination, mimicking the chronic-to-acute transition (Fig. 4a). GHs and cis-DA were used as they offer distinct mechanisms that drive biofilm dispersal: GHs target biofilm exopolysaccharides by hydrolysing the glycosidic linkages to passively induce dispersal33,34,35, whereas cis-DA is a fatty acid signalling molecule that actively drives dispersal by regulating bacterial gene expression and physiology36,37,38. Importantly, it has been shown that GH-induced biofilm dispersal in this model can cause lethal septicaemia35. Here we compared P. aeruginosa transcriptomes before (that is, mature biofilms in wound tissues) and after (that is, dispersed cells) the treatment with GHs or cis-DA (Fig. 4b). Functional enrichment analyses support the notion that GHs and cis-DA induced different physiological and metabolic changes during in vivo dispersal (Extended Data Fig. 9). Strikingly, among the genes that were differentially expressed in response to both treatments, we found that sicX was the top downregulated gene (Fig. 4c), supporting the notion that SicX levels change markedly during the chronic-to-acute transition. We also found that genes involved in alginate synthesis (algA and algD) and export (algE) were downregulated during biofilm dispersal (Fig. 4d). Moreover, the denitrification pathway (nir, nar and molybdopterin cofactor genes in response to GHs; nir and nor genes in response to cis-DA) and high-oxygen-affinity oxidases (cbb3-type oxidase in response to both GHs and cis-DA) were downregulated, whereas low-oxygen-affinity oxidases (Cyo oxidase in response to GHs) were upregulated (Fig. 4d), indicating an increase of oxygen availability during dispersal. Collectively, our observations identify sicX as a biomarker for chronic-to-acute transition and suggest oxygen as a key environmental signal during this process.

a, Schematics of in vivo dispersal of mature biofilms in mouse chronic wounds. b, Venn diagram of genes differentially expressed before (n = 3 animals) and after GH (n = 2) or cis-DA (n = 2) treatment. c, sicX (orange) is the top downregulated gene among the 132 differentially expressed genes identified under both GH and cis-DA treatments. d, Heat map of differentially expressed (DE) genes involved in alginate synthesis and secretion, anaerobic respiration and aerobic respiration. Aerobic terminal oxidases with low or high oxygen affinities are indicated. Blank cells indicate genes not identified as differentially expressed. e, A working model of how SicX allows P. aeruginosa to establish chronic local infection in a host environment with low oxygen. No or low SicX promotes systemic infection.
In conclusion, by leveraging bacterial transcriptomes from human infections, we discovered that P. aeruginosa produces an oxygen-responsive sRNA, SicX, the abundance of which orchestrates the transition between chronic and acute infections (Fig. 4e). The ability to respond to stressful environments is crucial for the survival of many organisms, during which sRNAs play key roles in fine-tuning gene expression. For example, in Salmonella infection, the sRNA PinT is induced and regulates gene expression to facilitate the transition from the invasion stage to intracellular survival39. In another case, Helicobacter pylori silences the expression of the sRNA HPnc4160 during infection, which upregulates target genes to promote bacterial colonization in the host and potentially increase the risk of gastric carcinogenesis40. In this study, we provide a compelling case of an sRNA directing important lifestyle choices during P. aeruginosa infection. Moreover, SicX shows promise as a prognostic and diagnostic biomarker, as it is the most highly responsive gene during the chronic-to-acute transition.
Although decades of in vitro studies have defined key regulatory systems governing the biofilm–planktonic transition in P. aeruginosa, robust evidence substantiating their roles in the mammalian host is lacking. For example, the Gac–Rsm signalling cascade is widespread in many bacterial species, and it is known to modulate the expression of features associated with planktonic or biofilm lifestyle in P. aeruginosa. This cascade relies on the interactions between global post-transcriptional regulators (RsmA or RsmN (also known as RsmF)) and Rsm sRNAs to fine-tune gene expression6 (Extended Data Fig. 10a). However, we found that Rsm sRNAs are expressed only at low levels in patients chronically infected with P. aeruginosa (Extended Data Fig. 10b,c), indicating that they are minimally involved in maintaining long-term infection. Next, although studies have shown that inactivation of specific components of Gac–Rsm led to fitness or colonization defects in acute infection models7,41, compelling evidence supporting a lifestyle switch model during mammalian infection remains lacking. Moreover, we found that the regulation of SicX expression is independent from the GacS–GacA two-component system (Extended Data Fig. 10d). In addition, the primary P. aeruginosa strain used in this study (PA14) has a mutation in ladS, a sensor kinase that stimulates GacS–GacA activity and promotes biofilm features42, yet this strain still established chronic localized infection in the mouse chronic wound model (Fig. 3b). Like many other pathogenic bacteria, P. aeruginosa colonizes a wide range of ecological niches including host-free ecosystems, and as a result, certain regulatory features controlling the biofilm–planktonic transition may have been selected under conditions that are unrelated to human pathogenesis. Our study thus provides a framework for discovering important pathogenic traits by leveraging bacterial gene expression information from human infections.
Methods
Media and growth conditions
P. aeruginosa cells were routinely cultured in brain heart infusion (BHI) broth at 37 °C with shaking (250 r.p.m.) unless otherwise specified. Pseudomonas isolation agar (PIA) was used for selecting P. aeruginosa against other microbes. Escherichia coli cells were cultured in lysogeny broth. To prepare plates, 1.5% (w/v) agar was used. Antibiotics were added to the medium as necessary: 60 µg ml−1 gentamicin, 75 µg ml−1 Tet or 300 µg ml−1 carbenicillin for P. aeruginosa; 10 µg ml−1 gentamicin, 10 µg ml−1 Tet or 100 µg ml−1 carbenicillin for E. coli. For P. aeruginosa static or anaerobic cultivation, 50 mM KNO3 was added to BHI broth. The anaerobic condition was maintained in a vinyl anaerobic chamber (Coy Lab Products) with the following atmosphere: 85% N2, 10% CO2 and 5% H2.
Plasmids and bacterial strains
Strains, plasmids and primers used in this study are listed in Supplementary Table 3. To create transcriptional lacZ fusions (pPC100–102), desired promoter regions were PCR amplified (Q5, New England Biolabs) using PA14 genomic DNA as the template, and the resulting amplicons containing overlapping regions of about 25 base pairs (bp) were ligated into the mini-CTX-lacZ vector (linearized with EcoRI and KpnI) in Gibson Assembly Master Mix (New England Biolabs). To create sicX complementation plasmids (pPC103–106), the sicX promoter and sicX alleles (containing mutations as described in Supplementary Table 3) were ligated into mini-CTX1 vector (linearized with EcoRI and KpnI) through Gibson assembly. All sicX point mutations (pPC107–124) were constructed with the Q5 Site-Directed Mutagenesis Kit (New England Biolabs), using mini-CTX1-sicXTruncate (pPC106) as the template. To create translational lacZ fusions (pPC125–126), the promoter region of ubiU was PCR amplified and then ligated into pSW205 between the EcoRI and BamHI sites. All ubiUVT 5′ UTR point mutations (pPC127–145) were constructed with the Q5 Site-Directed Mutagenesis Kit (New England Biolabs), using pSW205-Pubi-lacZ (pPC125) as the template. To construct plasmids used for markerless gene deletion (pPC146–147, 149–150) and mutagenesis (pPC148), upstream and downstream regions (about 1,000 bp) flanking the target gene or region were PCR amplified (containing mutations in the overlapping region if needed) and ligated into pEXG2 (linearized with SacI and KpnI) through Gibson assembly. All plasmids were verified by PCR, restriction enzyme digestion and, if necessary, DNA sequencing.
Verified plasmids were then transformed into E. coli SM10 λpir, followed by conjugation into P. aeruginosa PA14, PAO1, B1 and their derivatives. The pSW205-derived plasmids were directly electroporated into P. aeruginosa. Appropriate antibiotics were used for selection as indicated in Supplementary Table 3. To create markerless in-frame deletion or mutagenesis, conjugants with the deletion cassette integrated at the target loci were counter-selected on low-salt lysogeny broth agar plates supplemented with 5% sucrose. The successful deletion was screened by PCR using flanking primers and verified by DNA sequencing if needed.
Homologue identification and sequence analysis
Blastn searches were carried out on pseudomonas.com and the National Center for Biotechnology Information website to identify orthologues of sicX and ubiUVT across the Pseudomonas genus and other organisms. The sicX sequence (for Fig. 1c and Extended Data Fig. 1), sicX with its neighbouring genes (Fig. 1d) and ubiUVT with its upstream intergenic region (for Extended Data Fig. 4) from the P. aeruginosa PA14 strain were used as queries. Orthologues identified from complete bacterial genomes with E values of <1 × 10−4, percentage identity >90% and query coverage values of >90% were retained for further analysis. Sequences of sicX and ubiUVT orthologues were aligned using MUSCLE43. To analyse the sequence conservation of sicX and ubiUVT orthologues, we first aligned sicX and ubiUVT orthologues using MUSCLE. Next, alignment ends were trimmed and gaps were removed, and percentages of orthologue sequences that are identical to sicX and ubiUVT queries were calculated at each nucleotide position in R. To create Extended Data Fig. 4b,c, the alignments were visualized in Jalview44 using the default nucleotide colour scheme.
β-galactosidase assay
P. aeruginosa cells were grown overnight in BHI broth supplemented with 50 mM KNO3. The overnight culture was diluted to an optical density at 600 nm (OD600nm) of about 0.05 in fresh BHI broth (with 50 mM KNO3) and then grown to the log phase (OD600nm around 0.5). Log-phase cells were diluted to OD600nm 0.2 and transferred to the anaerobic chamber for 3-h shaking (at 100 r.p.m.) cultivation at 37 °C. A 100 μl volume of the anaerobic culture was used to conduct the β-galactosidase assay (as described in ref. 45). Aerobic log-phase cultures were used as comparisons. For static cultivation, log-phase cultures were diluted to OD600nm 0.05 and incubated in a 96-well plate (500 μl in each 1-ml-deep well) statically at 37 °C for 3 h. The static cultures were then used for the β-galactosidase assay.
For making Extended Data Fig. 3a,b, strains harbouring ubiUVT-lacZ translational fusions were spotted (3 μl of OD600nm 0.1 cultures) onto 0.2-μm Nuclepore track-etched polycarbonate membranes (Whatman) that were placed (rough side facing upwards) on BHI agar plates containing 200 µg ml−1 X-gal, 50 mM KNO3 and 100 µg ml−1 carbenicillin. The plates were then incubated in the anaerobic chamber (protected from light) at 37 °C for 20 h before imaging. β-galactosidase activities were estimated as described previously46. Briefly, images of colony biofilms were converted to 8 bit in ImageJ. Thresholding was carried out to define colony objects for measurements. Mean pixel intensity was measured for each colony and subtracted with the mean pixel intensity of a colony of WT PA14.
Northern blot
P. aeruginosa cells were grown aerobically and anaerobically for total RNA extraction. To prepare aerobic cultures, log-phase cells were diluted to OD600nm 0.01 in BHI (with 50 mM KNO3) and incubated at 37 °C with shaking for 3 h. To prepare anaerobic cultures, log-phase cells were diluted to OD600nm 0.1 in BHI (with 50 mM KNO3) and incubated in the anaerobic chamber for 4 h with shaking at 37 °C. For total RNA extraction, P. aeruginosa cultures were first stored in RNAlater at 4 °C overnight. Next, cells were pelleted and resuspended in RNase-free TE buffer containing 1 mg ml−1 lysozyme. Samples were incubated at 37 °C for 30 min to enzymatically lyse cells. A 1 ml volume of RNA-Bee was then added, and cells were further mechanically lysed by bead beating three times for 30 s. A 200 μl volume of chloroform was added to the samples, and the aqueous and organic phases were separated by centrifugation at 13,000g for 30 min at 4 °C. The aqueous phase that contained RNA was mixed with 0.5 ml isopropanol and 20 μg linear acrylamide. After overnight incubation at −80 °C, RNA was pelleted and washed with 75% ethanol. Air-dried pellets were resuspended in 100 μl of RNase-free water. The RNA concentration for each sample was determined with a NanoDrop spectrophotometer (Thermo Fisher Scientific). Approximately 1 μg of total RNA was separated on a 15% polyacrylamide gel electrophoresis–urea gel and transferred to Hybond-N+ membranes (GE Healthcare). The membranes were then crosslinked with an ultraviolet crosslinker (UVP) and probed in hybridization solution (Takara) with the indicated DNA oligonucleotides (listed in Supplementary Table 3) that were radiolabelled with [γ-32P]ATP (Perkin Elmer) by T4 polynucleotide kinase (New England Biolabs). Probed membranes were exposed to a phosphor screen and visualized using a Typhoon Biomolecular Imager (GE Healthcare). When necessary, membranes were stripped by incubating them with boiled 0.1% SDS with agitation for 15 min three times. The sizes of RNA were estimated with the RiboRuler Low Range RNA Ladder (Thermo Fisher Scientific) as well as the migration of xylene cyanol (about 28 nt) and bromophenol blue (about 8 nt).
Colony biofilm assay
Bacterial cultures in BHI (with 50 mM KNO3) were grown to the log phase (OD600nm around 0.5) and then diluted to OD600nm 0.02 with fresh medium. Next, 10 μl diluted cultures were spotted onto 0.2-μm Nuclepore track-etched polycarbonate membranes (Whatman) that were placed (smooth side facing upwards) on BHI agar plates (with 50 mM KNO3). The plates were then incubated in the anaerobic chamber at 37 °C for 16 h. Membranes containing P. aeruginosa biofilms were transferred to the BeadBug prefilled tubes that contained 1 ml phosphate-buffered saline (PBS). Biofilms were mechanically disrupted by bead beating for 45 s. CFUs were determined by spreading the diluted cell suspensions on PIA plates.
Quantitative proteomics and data analysis
P. aeruginosa PA14 and ΔsicX cells were collected under both static and anaerobic growth conditions as described above (in the section entitled β-galactosidase assay).
For static growth condition, three biological replicates for each strain were collected. Cell pellets were resuspended in lysis buffer (100 mM Tris-HCl, 50 mM NaCl, 10% (v/v) glycerol, 1 mM Tris(2-carboxyethyl)phosphine) (TCEP), pH 7.5) with cOmplete Mini protease inhibitor cocktail (Roche). Following three rounds of sonication (3 × 10 s) on ice, supernatants were clarified by sedimentation (21,130g, 15 min, 4 °C). Aliquots (100 μg) of each sample were reduced with TCEP, alkylated with iodoacetamide and labelled with tandem mass tags (TMTs). TMT labelling was carried out according to the protocol specified by the manufacturer (Thermo Fisher).
Liquid chromatography–tandem mass spectrometry (LC–MS/MS) experiments were carried out using a Dionex Ultimate 3000 rapid separation LC (RSLC) nano-ultraperformance LC system (Thermo Fisher Scientific) and a Lumos Orbitrap mass spectrometer (Thermo Fisher Scientific). Separation of peptides was carried out by C18 reversed-phase chromatography at a flow rate of 300 nl min−1 using a Thermo Scientific reversed-phase nano EASY-spray column (Thermo Scientific PepMap C18; 2-mm particle size, 100-Å pore size, 75-mm i.d. by 50-cm length). Solvent A was water/0.1% formic acid, and solvent B was 80% (v/v) acetonitrile/20% water/0.1% formic acid. The linear gradient used was 2% to 40% solvent B over 93 min (the total LC run time was 120 min, including a high-organic-wash step and column re-equilibration).
The eluted peptides were sprayed into the mass spectrometer by means of an EASY-Spray source (Thermo Fisher Scientific). All m/z values representing eluting peptide ions were measured in an Orbitrap mass analyser (set at a resolution of 120,000) and were scanned at between m/z 380 and 1,500 Da. Data-dependent MS/MS scans (top speed) were used to automatically isolate and fragment precursor ions by collision-induced dissociation (normalized collision energy value, 35%) analysed in the linear ion trap. Singly charged ions and ions with unassigned charge states were excluded from selection for MS/MS, and a dynamic exclusion window of 70 s was used. The top 10 most abundant fragment ions from each MS/MS event were then selected for a further stage of fragmentation by synchronous precursor selection and MS/MS/MS47 in the high-energy-collision cell using high-energy collisional dissociation (normalized collision energy value of 65%). The m/z values and relative abundances of each reporter ion and of all fragments (mass range, 100 to 500 Da) in each MS3 step were measured in the Orbitrap analyser, which was set at a resolution of 60,000. This was carried out in cycles of 10 MS3 events, after which the Lumos instrument reverted to scanning the m/z ratios of the intact peptide ions and the cycle continued.
Proteome Discoverer v2.1 (Thermo Fisher Scientific) and Mascot (Matrix Science) v2.6 were used to process raw data files. Data were aligned to the sequences of P. aeruginosa UCBPP-PA14 (with common repository of adventitious proteins (cRAP) v1.0). The R package MSnbase v2.13 (ref. 48) was used for processing proteomics data. Protein differential abundances were evaluated using the Limma package v3.44.3 (ref. 49). Differences in protein abundances were statistically determined using Student’s t-test with variances moderated by the use of Limma’s empirical Bayes method. P values were adjusted for multiple testing by the Benjamini–Hochberg method50. Proteins were considered to have increased or decreased in abundance only when their log2[fold change] value was greater than 1 or less than −1, respectively, and when their P value was <0.01.
For anaerobic growth condition, two biological replicates for PA14 and three biological replicates for ΔsicX were collected as described above (in the section entitled β-galactosidase assay). Briefly, the proteins in each sample were reduced, alkylated and digested with trypsin according to the filter-aided sample preparation protocol51. The peptides were labelled with TMTs, separated by high-pH reversed-phase chromatography as described previously52. They were pooled into 12 fractions as described previously53. Each fraction was analysed by nano-LC–MS/MS, and peptides were identified as previously described54 with the following modifications. Reversed-phase chromatography was carried out using an in-house packed column (40 cm long × 75 μm inner diameter × 360 μm outer diameter, Dr. Maisch GmbH ReproSil-Pur 120 C18-AQ 1.9-µm beads) and a 120-min gradient. The Raw files were searched using the Mascot algorithm (v2.5.1) against a protein database constructed by combining the sequences of P. aeruginosa UCBPP-PA14 and a contaminant database (cRAP, downloaded 21 November 2016 from http://www.thegpm.org) through Proteome Discoverer v2.1. Only peptide spectral matches with expectation value of less than 0.01 (‘high confidence’) were used for our analyses as described above.
Lipid extractions and ubiquinone (UQ9) quantification
To prepare cells for lipid extractions, we first diluted log-phase cultures (OD600nm 0.5; grown aerobically) into the anaerobic BHI broth (supplemented with 50 mM KNO3) to a calculated OD600nm 0.05, and after 6-h anaerobic incubation with shaking, cells were collected, washed once with PBS and flash-frozen in liquid nitrogen. Cell numbers were estimated with OD600nm.
Frozen samples were thawed on ice. A 100 µl volume of ice-cold isopropanol with 10 nM coenzyme Q10-d9 (deuterated UQ10 as internal standard; Sigma-Aldrich) was added to 109 cells. With the addition of glass beads, samples were vortexed briefly and homogenized by TissueLyzer for 5 min twice, followed by centrifugation at 21,100g for 5 min. After centrifugation, supernatant was diluted tenfold and transferred to 4 °C. Ultraperformance LC–MS was carried out using an UltiMate 3000 fitted with an Accucore C30 column (2.1 × 150 mm, 2.6 µm particle size; Thermo Fisher) and coupled to an Orbitrap ID-X mass spectrometry system.
The chromatographic method for sample analysis involved elution with acetonitrile/water (60:40, v/v) with 10 mM ammonium formate and 0.1% formic acid (mobile phase A) and isopropanol/acetonitrile (90:10, v/v) with 10 mM ammonium formate and 0.1% formic acid (mobile phase B) at 0.4 ml min−1 flow rate using the following gradient programme: 0 min 20% B; 1 min 60% B; 5 min 70% B; 5.5 min 85% B; 8 min 90% B; 8.2 min 100% B hold to 10.5 min, then 10.7 min 20% B hold to 12 min. The column temperature was set to 50 °C, and the injection volume was 5 µl.
The targeted molecules UQ9 and deuterated UQ10 were fragmented by high-energy collisional dissociation at 20 collision energy in positive mode. MS/MS transitions for UQ9 and deuterated UQ10 are 812.66/197.08 and 889.77/206.18. Standard curves for UQ9 and deuterated UQ10 were generated. The amounts of UQ9 and deuterated UQ10 in the samples were calculated on the basis of the standard curve.
Planktonic growth and competition assays
For generating growth curves, bacterial cells in BHI (with 50 mM KNO3) were first grown aerobically to the log phase (OD600nm around 0.5) and then diluted to OD600nm 0.01 with either aerobic or anaerobic BHI (with 50 mM KNO3) broth. Aerobic and anaerobic growth of P. aeruginosa at 37 °C (with shaking) were monitored by measuring the OD600nm.
For long-term competition experiments, PA14, ΔsicX, TetR (resistance provided by mini-CTX1) PA14 and TetR ΔsicX were grown to the log phase (OD600nm around 0.5) in BHI (with 50 mM KNO3) before diluting to OD600nm 0.4 with fresh medium. PA14 and TetR ΔsicX were mixed at 1:1 ratio, and 100 μl of cell mixtures was transferred to 4 ml anaerobic BHI (with 50 mM KNO3), followed with shaking incubation at 37 °C under anaerobic conditions. After 24 h, 20 μl of the culture was transferred to another tube containing 4 ml anaerobic BHI (with 50 mM KNO3). The culture was then consecutively passaged daily in a similar fashion. The competition between TetR PA14 and ΔsicX was conducted similarly to that described above. CFU values of TetR and TetS cells were estimated by plating cultures (on each day) on PIA as well as PIA containing 75 µg ml−1 Tet.
In silico analyses of RNA folding and RNA–RNA interaction
RNA secondary structures were predicted using mfold26 with default parameters. IntaRNA 2.0 was used to predict base-pairing interactions between SicX sRNA and ubiUVT mRNA25 with default settings, and the minimum number of base pairs in the seed region was set as 7.
Mouse chronic wound model
Mouse chronic wound infections were carried out with female Swiss Webster mice (8–10 weeks old) essentially as described previously55 with a few modifications. Briefly, dorsal full-thickness skin excision was carried out in which a small circular wound (with a diameter of 1.5 cm) was surgically administered. After excision, the wound was immediately covered with a semipermeable polyurethane dressing. A total of 104 CFUs of P. aeruginosa cells were injected onto the wound bed underneath the dressing to establish infection. As bandages are required to reduce the contractile healing and minimize wound contamination with other bacteria, they were closely monitored and replaced when necessary throughout the infection course.
For assessing the bacterial burdens of WT and ΔsicX infection, wound tissues and spleens were collected at 10 days post-infection or at an early time point if the animal was found moribund (moribund mice that were not identified immediately were excluded from the CFU analysis). Tissues were homogenized in PBS for 45 s in BeadBug tubes with 2.8-mm steel beads (Sigma-Aldrich) using a Mini-Beadbeater-16 (BioSpec Products). To enumerate CFUs, homogenized samples were serially diluted and plated on PIA plates. For studying the dissemination outcomes of WT, ΔsicX, ΔubiUVT and ΔsicX-ubiC infection (Fig. 3e), at 10 days post-infection, distal organs including spleens, livers and gallbladders were collected for assessing the presence or absence of P. aeruginosa cells. For studying the macroscale organization of P. aeruginosa infection, the wound was excised at 4 days post-infection, and a biopsy punch of 10 mm diameter (Acu-Punch, Thermo Fisher Scientific) was used to separate the edge (about 0.98 cm2) and core (about 0.77 cm2) regions. The CFUs were then normalized by the surface area for comparison purposes.
In vivo biofilm dispersal
In vivo biofilm dispersal was carried out with female Swiss Webster mice (8–10 weeks old) essentially as described previously31 with a few modifications. Briefly, the mouse surgical excision wound was administered as demonstrated above. A total of 104 CFUs of P. aeruginosa cells (PAO1) were injected onto the wound bed underneath the dressing to establish infection. At 48 h post-infection (after the formation of mature biofilms in wounds), the established wound infections were treated through topical application of GHs or cis-DA to induce systemic dissemination. Specifically, GHs are composed of bacterial α-amylase (from Bacillus subtilis; MP Biomedicals) and fungal cellulase (from Aspergillus niger; MP Biomedicals). A 10% (w/v) α-amylase and cellulase (in a 1:1 combination) solution was prepared by dissolving powdered enzymes in PBS. cis-DA (Cayman Chemical Company) was diluted in PBS to a final concentration of 500 nM. GH and cis-DA solutions were prepared immediately before use. Wound beds were irrigated with a GH or cis-DA solution in three separate topical infusions with 30 min of dwell time for each. Topical infusions containing dispersed cells (n = 2 animals for each treatment) were aspirated and immediately placed in RNAlater (Thermo Fisher Scientific). In separate animals, P. aeruginosa-infected wound tissues (n = 3 animals) were carefully excised from the surrounding uninfected tissue and the underneath muscle layer at 48 h post-infection (without GH or cis-DA treatment) as a comparison. Infected wound tissues were immediately placed in RNAlater (Thermo Fisher Scientific).
Mouse pneumonia model
Mouse pneumonia infection was carried out with female BALB/c mice (6–8 weeks old) essentially as described previously56 with a few modifications. Briefly, mice were anaesthetized by intraperitoneal injection of 0.2 ml of a cocktail of ketamine (25 mg ml−1) and xylazine (12 mg ml−1). For infection with PA14-derived strains, mice were intranasally instilled with approximately 107 CFUs (a sublethal dose) P. aeruginosa cells (in 25 μl PBS). Mice were euthanized at 48 h post-infection. Whole lungs and spleens were collected aseptically, weighed and homogenized in 1 ml of PBS. Tissue homogenates were serially diluted and plated on PIA for CFU enumeration. For infection with PAO1-derived strains, 5 × 107 CFUs (a lethal dose) of P. aeruginosa cells were used as inocula, and tissues were collected at 24 h post-infection.
Mouse subcutaneous infection model
Mouse subcutaneous skin infection (also known as the mouse abscess model) was carried out with female Swiss Webster mice (8 weeks old) essentially as described previously55 with a few modifications. Briefly, the mouse inner thigh was shaved and any remaining hair was removed with a depilatory agent (Nair). A 100 μl volume of approximately 1 × 107 CFUs of P. aeruginosa cells (PA14 or ΔsicX) were subcutaneously injected to the inner thigh. Mice were euthanized at 16 h post-infection. Spleens were collected aseptically and homogenized in 1 ml PBS. Tissue homogenates were serially diluted and plated on PIA for CFU enumeration.
RNA-seq library preparation
RNA extractions were carried out essentially as described previously14. For in vitro static cultivation of PA14, cells were first grown as described above (in the section entitled β-galactosidase assay) and immediately mixed with five volumes of RNAlater. After storage at 4 °C overnight, cells were pelleted, resuspended in nuclease-free TE buffer (Acros Organics) containing 1 mg ml−1 lysozyme, and transferred to bead-beating tubes containing a mixture of large and small beads (2-mm zirconia and 0.1-mm zirconia/silica, respectively). For in vivo biofilm dispersal experiments (as described above), infected tissues were removed from RNAlater, mixed with TE buffer, and transferred to bead-beating tubes. In vitro as well as tissue samples in TE were briefly disrupted by homogenization using a Mini-Beadbeater-16. Samples were then incubated at 37 °C for 30 min to enzymatically lyse cells. Next, 1 ml RNA-Bee was added to 300-μl sample homogenate, and samples were further mechanically lysed by bead beating three times for 30 s. A 200 μl volume of chloroform per 1 ml RNA-Bee was added, and the tubes were shaken vigorously for 30 s and then incubated on ice for 5 min. The samples were then centrifuged at 13,000g for 30 min at 4 °C to separate the aqueous and organic phases. The aqueous phase was transferred to a new microcentrifuge tube to which 0.5 ml isopropanol (per 1 ml RNA-Bee) and 20 μg linear acrylamide were added. After overnight incubation at −80 °C, samples were thawed on ice and centrifuged at 13,000g for 30 min at 4 °C. Pellets were washed twice with 1 ml of 75% ethanol, air dried for 5 min and resuspended in 100 μl of RNase-free water. The RNA concentration for each sample was determined with a NanoDrop spectrophotometer (Thermo Fisher Scientific). rRNA was depleted using a MICROBExpress Kit (Thermo Fisher Scientific) for the in vitro samples and a QIAseq FastSelect Kit (QIAGEN) for mouse-derived samples, followed by ethanol precipitation. The depleted RNA was fragmented for 2 min with the NEBNext Magnesium RNA fragmentation module kit, and cDNA libraries were prepared using the NEBNext multiplex small RNA library prep kit (New England Biolabs) according to the manufacturer’s instructions. Libraries were sequenced at the Molecular Evolution Core at the Georgia Institute of Technology on an Illumina NextSeq500 using 75-bp single-end runs.
Analysis of RNA-seq datasets
In addition to the above-mentioned RNA-seq libraries prepared in this study, 202 publicly available P. aeruginosa RNA-seq datasets were analysed, including 28 P. aeruginosa transcriptomes derived from human samples, 28 from infection models and 146 from a range of well-defined in vitro conditions (listed in Supplementary Table 1). The quality of the raw sequencing reads was first confirmed with FastQC v0.11.7 (ref. 57). Next, RNA-seq reads were trimmed on the 3′ ends to remove the Illumina adaptor (AGA TCG GAA GAG CAC ACG TCT GAA CTC CAG TCA C) using Cutadapt 3.0 (ref. 58) with a minimum read length threshold of 22 bases. Trimmed reads were then mapped to the P. aeruginosa PA14 reference genome (available for download from pseudomonas.com) using Bowtie2 v2.4.2 with default parameters for end-to-end alignment59. As PA1414 (sicX) was originally annotated as a protein-coding gene, we first assigned reads to protein-coding genes with featureCounts Subread v2.0.1 (ref. 60; results shown in Fig. 1a). For the remaining analyses (Figs. 1b, 2a and 4b–d and Extended Data Figs. 2b and 10), reads assigned to both protein-coding genes as well as 199 sRNAs15 were tallied.
Differential expression was determined with DESeq2 (Figs. 1a and 4b–d; ref. 61). To compare sicX expression levels in different samples (Fig. 1a and Extended Data Fig. 5), raw reads were normalized using the varianceStabilizingTransformation() function in the DESeq2 package. To estimate the relative transcript abundance of various features (protein-coding genes, sRNAs, sicX and/or rsmYZ), we calculated TPM. First, raw counts were normalized by feature (gene) length to generate reads per kilobase (RPK). Next, all RPK values were counted up and divided by 1,000,000 to determine the scaling factor. Finally, RPK of each gene was divided by the scaling factor. The resulting TPM values were used for making Figs. 1b and 2a and Extended Data Figs. 2b and 10. To examine the alignment pattern of sicX reads, samtools v1.13 was used to measure the read depth encompassing the sicX locus at each nucleotide position62. The read depth was then normalized using the estimateSizeFactors() function in DESeq2 before plotting.
Phylogenetic analysis
A maximum-likelihood phylogenetic tree was constructed in PhyML 3.0 (ref. 63) using 365 16S rRNA sequences representing diverse Pseudomonas isolates. For better visualization of the tree, we included only approximately 50 16S sequences from the P. aeruginosa strains. The 16S sequences were aligned with MUSCLE and used as an input in PhyML 3.0. The best-fitting evolutionary models were predicted with the corrected Akaike information criterion. The tree was visualized and annotated in iTOL64.
Functional enrichment analysis
Gene Ontology enrichment analysis was conducted using Galaxy v2.0.1. Differentially expressed genes in response to GH or cis-DA treatment were analysed using the Gene Ontology file (obtained from http://geneontology.org/docs/download-ontology) and the P. aeruginosa PAO1 Gene Ontology term annotations (obtained from https://pseudomonas.com/goterms/list). A Benjamini–Hochberg test with P-value cutoff < 0.05 was carried out.
Statistics and reproducibility
All experiments were carried out independently at least three times (unless otherwise stated in the figure legends) with similar observations. The exact n values for animal studies as well as in vitro experiments are provided in the figures and legends. Statistical analyses were conducted using Prism GraphPad 9 and are specified in the figure legends. For northern blotting, each experiment was conducted at least twice independently (by using freshly extracted RNA samples) with similar observations, and representative images are shown.
Ethics statement
All mouse procedures were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animals were treated humanely and housed in temperature- and humidity-controlled facilities (18–22 °C; 40–50%) with 12 h light–dark cycles. The protocol for the mouse chronic wound infection was approved by the Institutional Animal Care and Use Committee (IACUC) of Texas Tech University Health Sciences Center (protocol number 07044) and by the IACUC of Georgia Institute of Technology (protocol number A100127). The protocol for the mouse subcutaneous infection model was approved by the IACUC of Georgia Institute of Technology (protocol number A100127). For the mouse pneumonia infection model, all experimental procedures were conducted according to the guidelines of the Emory University IACUC, under approved protocol number DAR-201700441.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The proteomics datasets generated in this study have been deposited in the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org/cgi/GetDataset), with the accession numbers are PXD032985 and PXD032986. Raw reads of our RNA-seq studies have been deposited in the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) under BioProject numbers PRJNA934328 and PRJNA904394. The 202 publicly available P. aeruginosa transcriptomes analysed in this study are summarized in Supplementary Table 1, and can be obtained from the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) with the corresponding accession number. The P. aeruginosa PA14 reference genome (BioSample accession SAMN02603591) is available for download from https://pseudomonas.com. Source data are provided with this paper.
References
Rossi, E. et al. Pseudomonas aeruginosa adaptation and evolution in patients with cystic fibrosis. Nat. Rev. Microbiol. 19, 331–342 (2021).
Valentini, M., Gonzalez, D., Mavridou, D. A. & Filloux, A. Lifestyle transitions and adaptive pathogenesis of Pseudomonas aeruginosa. Curr. Opin. Microbiol. 41, 15–20 (2018).
Valentini, M. & Filloux, A. Biofilms and cyclic di-GMP (c-di-GMP) signaling: lessons from Pseudomonas aeruginosa and other bacteria. J. Biol. Chem. 291, 12547–12555 (2016).
Ha, D.-G. & O’Toole, G. A. c-di-GMP and its effects on biofilm formation and dispersion: a Pseudomonas aeruginosa review. Microbiol. Spectr. 3, 3.2.27 (2015).
Pestrak, M. J. & Wozniak, D. J. in Microbial Cyclic Di-Nucleotide Signaling (eds Chou, S.-H., Guiliani, N., Lee, V. T. & Römling, U.) 471–486 (Springer Cham, 2020).
Brencic, A. et al. The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol. Microbiol. 73, 434–445 (2009).
Goodman, A. L. et al. A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa. Dev. Cell 7, 745–754 (2004).
Gebhardt, M. J., Kambara, T. K., Ramsey, K. M. & Dove, S. L. Widespread targeting of nascent transcripts by RsmA in Pseudomonas aeruginosa. Proc. Natl Acad. Sci. USA 117, 10520–10529 (2020).
Janssen, K. H., Diaz, M. R., Gode, C. J., Wolfgang, M. C. & Yahr, T. L. RsmV, a small noncoding regulatory RNA in Pseudomonas aeruginosa that sequesters RsmA and RsmF from target mRNAs. J. Bacteriol. 200, e00277-18 (2018).
Miller, C. L. et al. RsmW, Pseudomonas aeruginosa small non-coding RsmA-binding RNA upregulated in biofilm versus planktonic growth conditions. BMC Microbiol. 16, 155 (2016).
Broder, U. N., Jaeger, T. & Jenal, U. LadS is a calcium-responsive kinase that induces acute-to-chronic virulence switch in Pseudomonas aeruginosa. Nat. Microbiol. 2, 16184 (2016).
Wang, B. X. et al. Mucin glycans signal through the sensor kinase RetS to inhibit virulence-associated traits in Pseudomonas aeruginosa. Curr. Biol. 31, 90–102 (2021).
Cornforth, D. M. et al. Pseudomonas aeruginosa transcriptome during human infection. Proc. Natl Acad. Sci. USA 115, E5125–E5134 (2018).
Cornforth, D. M., Diggle, F. L., Melvin, J. A., Bomberger, J. M. & Whiteley, M. Quantitative framework for model evaluation in microbiology research using Pseudomonas aeruginosa and cystic fibrosis infection as a test case. MBio 11, e03042-19 (2020).
Trouillon, J., Han, K., Attrée, I. & Lory, S. The core and accessory Hfq interactomes across Pseudomonas aeruginosa lineages. Nat. Commun. 13, 1258 (2022).
Trunk, K. et al. Anaerobic adaptation in Pseudomonas aeruginosa: definition of the Anr and Dnr regulons. Environ. Microbiol. 12, 1719–1733 (2010).
Rossi, E., Falcone, M., Molin, S. & Johansen, H. K. High-resolution in situ transcriptomics of Pseudomonas aeruginosa unveils genotype independent patho-phenotypes in cystic fibrosis lungs. Nat. Commun. 9, 3459 (2018).
Schobert, M. & Tielen, P. Contribution of oxygen-limiting conditions to persistent infection of Pseudomonas aeruginosa. Fut. Microbiol. 5, 603–621 (2010).
Huse, H. K. et al. Parallel evolution in Pseudomonas aeruginosa over 39,000 generations in vivo. mBio 1, e00199-10 (2010).
Nelson, C. E. et al. Proteomic analysis of the Pseudomonas aeruginosa iron starvation response reveals PrrF small regulatory RNA-dependent iron regulation of twitching motility, amino acid metabolism, and zinc homeostasis proteins. J. Bacteriol. 201, e00754-18 (2019).
Han, K. & Lory, S. Toward a comprehensive analysis of posttranscriptional regulatory networks: a new tool for the identification of small RNA regulators of specific mRNAs. mBio 12, e03608-20 (2021).
Pusic, P., Sonnleitner, E. & Bläsi, U. Specific and global RNA regulators in Pseudomonas aeruginosa. Int. J. Mol. Sci. 22, 8632 (2021).
Vo, C.-D.-T. et al. The O2-independent pathway of ubiquinone biosynthesis is essential for denitrification in Pseudomonas aeruginosa. J. Biol. Chem. 295, 9021–9032 (2020).
Rompf, A. et al. Regulation of Pseudomonas aeruginosa hemF and hemN by the dual action of the redox response regulators Anr and Dnr. Mol. Microbiol. 29, 985–997 (1998).
Mann, M., Wright, P. R. & Backofen, R. IntaRNA 2.0: enhanced and customizable prediction of RNA–RNA interactions. Nucleic Acids Res. 45, W435–W439 (2017).
Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415 (2003).
Updegrove, T. B., Zhang, A. & Storz, G. Hfq: the flexible RNA matchmaker. Curr. Opin. Microbiol. 30, 133–138 (2016).
Sagawa, S., Shin, J.-E., Hussein, R. & Lim, H. N. Paradoxical suppression of small RNA activity at high Hfq concentrations due to random-order binding. Nucleic Acids Res. 43, 8502–8515 (2015).
Soper, T., Mandin, P., Majdalani, N., Gottesman, S. & Woodson, S. A. Positive regulation by small RNAs and the role of Hfq. Proc. Natl Acad. Sci. USA 107, 9602–9607 (2010).
Castillo-Keller, M., Vuong, P. & Misra, R. Novel mechanism of Escherichia coli porin regulation. J. Bacteriol. 188, 576–586 (2006).
Redman, W. K., Welch, G. S. & Rumbaugh, K. P. Assessing biofilm dispersal in murine wounds. J. Vis. Exp. 174, e62136 (2021).
Sauer, K. et al. Characterization of nutrient-induced dispersion in Pseudomonas aeruginosa PAO1 biofilm. J. Bacteriol. 186, 7312–7326 (2004).
Naumoff, D. Hierarchical classification of glycoside hydrolases. Biochemistry 76, 622–635 (2011).
Fleming, D., Chahin, L. & Rumbaugh, K. Glycoside hydrolases degrade polymicrobial bacterial biofilms in wounds. Antimicrob. Agents Chemother. 61, e01998-16 (2017).
Fleming, D. & Rumbaugh, K. The consequences of biofilm dispersal on the host. Sci. Rep. 8, 10738 (2018).
Davies, D. G. & Marques, C. N. A fatty acid messenger is responsible for inducing dispersion in microbial biofilms. J. Bacteriol. 191, 1393–1403 (2009).
Marques, C. N., Davies, D. G. & Sauer, K. Control of biofilms with the fatty acid signaling molecule cis-2-decenoic acid. Pharmaceuticals 8, 816–835 (2015).
Rahmani-Badi, A., Sepehr, S., Fallahi, H. & Heidari-Keshel, S. Dissection of the cis-2-decenoic acid signaling network in Pseudomonas aeruginosa using microarray technique. Front. Microbiol. 6, 383 (2015).
Westermann, A. J. et al. Dual RNA-seq unveils noncoding RNA functions in host–pathogen interactions. Nature 529, 496–501 (2016).
Kinoshita-Daitoku, R. et al. A bacterial small RNA regulates the adaptation of Helicobacter pylori to the host environment. Nat. Commun. 12, 2085 (2021).
Mulcahy, H. et al. Pseudomonas aeruginosa RsmA plays an important role during murine infection by influencing colonization, virulence, persistence, and pulmonary inflammation. Infect. Immun. 76, 632–638 (2008).
Mikkelsen, H., McMullan, R. & Filloux, A. The Pseudomonas aeruginosa reference strain PA14 displays increased virulence due to a mutation in ladS. PLoS ONE 6, e29113 (2011).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M. & Barton, G. J. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Pearson, J. P. et al. Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes. Proc. Natl Acad. Sci. USA 91, 197–201 (1994).
Mould, D. L., Botelho, N. J. & Hogan, D. A. Intraspecies signaling between common variants of Pseudomonas aeruginosa increases production of quorum-sensing-controlled virulence factors. MBio 11, e01865-20 (2020).
McAlister, G. C. et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158 (2014).
Gatto, L. & Lilley, K. S. MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 28, 288–289 (2012).
Smyth, G. K. in Bioinformatics and Computational Biology Solutions using R and Bioconductor (eds Gentleman, R., Carey, V. J., Huber, W., Irizarry, R. A. & Dudoit, S.) 397–420 (Springer, 2005).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 (1995).
Wisniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009).
Mertins, P. et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 13, 1632–1661 (2018).
Yang, F., Shen, Y., Camp, D. G. 2nd & Smith, R. D. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev. Proteomics 9, 129–134 (2012).
Rinker, T. E. et al. Microparticle-mediated sequestration of cell-secreted proteins to modulate chondrocytic differentiation. Acta Biomater. https://doi.org/10.1016/j.actbio.2017.12.038 (2017).
Michie, K. L. et al. Role of Pseudomonas aeruginosa glutathione biosynthesis in lung and soft tissue infection. Infect. Immun. 88, e00116-20 (2020).
Moustafa, D. A. et al. Peptide-conjugated phosphorodiamidate morpholino oligomers retain activity against multidrug-resistant Pseudomonas aeruginosa in vitro and in vivo. MBio 12, e02411-20 (2021).
Andrews, S. FastQC v0.11.7 (Babraham Bioinformatics, Babraham Institute, 2010).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Acknowledgements
We thank D. Cornforth and other members of the laboratory of M.W. for helpful discussion of the manuscript and assistance with the animal experiments. We thank G. Welch and H. Hui for help with the animal experiments. We thank S. Brown, S. Diggle and C. Baskett for critical reading of the manuscript. The proteomic and metabolomic studies were supported by the Georgia Institute of Technology Systems Mass Spectrometry Core Facility as well as the Cambridge Centre for Proteomics. This study was supported by National Institutes of Health grants R21AI154220 (to M.W.), R21AI137462 (to K.P.R.) and R21AI147178 (to C.S.S.), Cystic Fibrosis Foundation grants WHITEL20A0 and WHITEL22G0 (to M.W.) and Cystic Fibrosis Trust Foundation SRC017 (to M.W.). P.C. and S.K.D. are supported by Cystic Fibrosis Postdoctoral Fellowships CAO20F0 and DOLAN20F0, respectively.
Author information
Authors and Affiliations
Contributions
P.C. and M.W. conceived and designed the study, analysed data and wrote the manuscript; D.F., D.A.M., P.C., W.K.R. and F.L.D. contributed to various aspects of animal experiments; S.K.D. prepared samples and carried out data analyses for proteomics; K.H.S. carried out northern blotting and analyses; P.C., M.W. and A.R. constructed bacterial strains and carried out reporter assays; P.C. carried out all other experiments and data analysis; C.S.S., J.B.G. and K.P.R. provided technical guidance and analysed data.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 PA1414 orthologs are restricted in P. aeruginosa.
The maximum-likelihood tree is built with 16s rDNA sequences from representative strains across the Pseudomonas genus. Clusters of abundant species are indicated with colored boxes. Strains that were reported to have been isolated from human or hospital environments are highlighted.
Extended Data Fig. 2 The expression of PA1414 under low oxygen conditions.
a. Schematic of mutagenesis of the Anr binding motif. b. Figure 2a with the addition of five in vitro static growth transcriptomes (blue circles; the RNA-seq libraries were prepared following the same method used in our study of P. aeruginosa transcriptomes in humans). c. Planktonic growth (OD600) of WT and ΔPA1414 in the presence or absence of oxygen. Mean values from 3 independent experiments are shown. d. RNA-seq reads aligned to the PA1414 locus (sicX) during in vitro aerobic and anaerobic growth. e. Colony biofilm growth of ΔsicX harboring different sicX mutations (as illustrated in Fig. 2g) under aerobic conditions. Error bars represent standard deviation from the mean. n = 4 independent experiments. Two-tailed Mann-Whitney test comparing WT and each other strain was performed (n.s., not significant). f. Northern blot analyses on different sicX alleles as depicted in Fig. 2g. The sRNA abundance was estimated by first normalizing against the 5S rRNA (loading control) and next comparing to the WT level. Anaerobic growth fitness was indicated with + (no defect) and − (defect). For gel source data, see Supplementary Fig. 1.
Extended Data Fig. 3 A proposed base-pairing interaction between SicX and the 5’ UTR of ubiUVT.
a. Predicted base-pairing between SicX and ubiUVT mRNA. Nucleotides highlighted in bold were subject to point mutations (A → U, U → A, G → C, C → G), which were assessed via the ubiUVT-lacZ translational reporter. Colony biofilms grown under anaerobic conditions were assessed for β-galactosidase activities as described46. Heatmap displays the mean values. Nucleotides subject to compensatory base changes are highlighted with magenta borders. Predicted base-pairing regions are highlighted with dashed borders. b. β-galactosidase activities (individual values) of the experiment shown in (A) quantified as described46. Potential compensatory mutations are highlighted in blue. Green dashed lines indicate the mean β-galactosidase activities in WT and ΔsicX. A. U., arbitrary unit. n = 5 independent experiments. c. β-galactosidase assays evaluating whether 5’ UTR mutations can alter ubiUVT translation independently of SicX during static growth. WT and ΔsicX harboring the ubiUVT-lacZ reporter (grown as anaerobic colony biofilms) were also assessed using this assay. n ≥ 3 independent experiments. d. β-galactosidase assays indicating that the presence or absence of Hfq does not affect the induction of ubiUVT translation by SicX. n ≥ 5 independent experiments. Error bars represent standard deviation from the mean. Two-tailed Mann-Whitney test was performed for b–c (n.s., not significant).
Extended Data Fig. 4 DNA sequence conservation of the ubiUVT operon.
a. The percent identity of 258 ubiUVT orthologs (compared to the ubiUVT allele from PA14) at the single-nucleotide scale. The sicX binding site at the 5’ UTR is highlighted. b. DNA sequence alignment of the upstream intergenic region of 258 ubiUVT orthologs. c. DNA sequence alignment of 258 sicX orthologs (only the sRNA region is shown). The seed region in SicX and the target region in the 5’ UTR of ubiUVT are indicated.
Extended Data Fig. 5 SicX expression in mouse chronic wounds.
a. RNA-seq reads aligned to the PA1414 locus (sicX) in mouse chronic wound infection. b. sicX RNA level in the mouse chronic wound is comparable to that in humans. Lines indicate means. Two-tailed Mann-Whitney test was used to compare the bacterial burdens (n.s., not significant). c. Bacterial burdens in the wound (dissemination information shown in Fig. 3e). Lines indicate means. n = 9 animals (for each group) over three independent experiments. Two-tailed Mann-Whitney test was used to compare the bacterial burdens of WT and each other mutant (n.s., not significant).
Extended Data Fig. 6 WT and ΔsicX showed similar spleen colonization in a systemic infection model.
P. aeruginosa cells (~107 CFU) were subcutaneously injected into the mouse inner thigh to induce systemic infection. Bacterial burdens in spleens were assessed at 16 h post-infection. WT (n = 7) and ΔsicX (n = 8). Lines indicate means. Two-tailed Mann-Whitney test was used.
Extended Data Fig. 7 Decoupling the translation of ubiUVT from SicX.
a. Schematic of mutagenesis in the 5’ UTR of ubiUVT: SicX binding site, ribosome binding site, and start codon are colored differently; the stem-loop structure is highlighted; mutagenesis (ubiC) is indicated with an arrow. b. Restoring ubiUVT translation in ΔsicX. β-galactosidase assay was performed to assess the translational activity of ubiUVT with and without 5’ UTR mutation as described in A (n = 6). c–d. Growth yield of colony biofilms in the presence or absence of oxygen (n = 4). Error bars represent standard deviation from the mean. Significant differences (compared to the WT) are indicated with asterisks (*, P < 0.05; **, P < 0.01; Two-tailed Mann-Whitney test).
Extended Data Fig. 8 SicX (in PAO1 strain background) governs chronic-to-acute transition.
a. Schematic of the mouse pneumonia infection experiment. b. P. aeruginosa burden in the lung. Lines indicate means. c. P. aeruginosa burden in the spleen. Lines indicate means. Two-tailed Mann-Whitney test was used to compare bacterial burdens. Summary of two biological replicates for b and c.
Extended Data Fig. 9 Functional enrichment of differentially-expressed genes in P. aeruginosa after GH or cis-DA treatment.
Gene Ontology (GO) terms are labeled on the left. One-tailed Fisher’s exact test was performed. Down, downregulated; up, upregulated.
Extended Data Fig. 10 Relatively low expression of Rsm sRNAs during infections.
a. Schematics of the Gac-Rsm cascade in P. aeruginosa. Components that promote biofilm (green) and planktonic (blue) lifestyles are color-coded. b. Relative transcript abundance (TPM, transcripts per million) of sicX compared to all protein-coding sequences (CDS), rsmYZ sRNAs, and the remaining non-coding sRNAs in 202 transcriptomes examined in Fig. 2a. Transcriptomes are sorted from the highest to the lowest sicX TPM. c. A heatmap showing the mean TPM of CDS, sRNAs, rsmYZ, and sicX in P. aeruginosa transcriptomes. d. β-galactosidase assays demonstrating the expression of sicX is independent from GacS-GacA. PsicX-lacZ, lacZ transcriptionally fused to sicX promoter. MrT7, MAR2xT7 transposon. n = 4 in all experiments. Error bars represent standard deviation from the mean. Significant difference (compared to WT) was analyzed with a Two-tailed Mann-Whitney test (n.s., not significant).
Supplementary information
Supplementary Information
This file contains Supplementary Fig. 1, Tables 1–3 and References.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, P., Fleming, D., Moustafa, D.A. et al. A Pseudomonas aeruginosa small RNA regulates chronic and acute infection. Nature 618, 358–364 (2023). https://doi.org/10.1038/s41586-023-06111-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-023-06111-7
This article is cited by
-
Construction of programmed time-released multifunctional hydrogel with antibacterial and anti-inflammatory properties for impaired wound healing
Journal of Nanobiotechnology (2024)
-
In vivo RNA interactome profiling reveals 3’UTR-processed small RNA targeting a central regulatory hub
Nature Communications (2023)
-
Pathogen-driven degradation of endogenous and therapeutic antibodies during streptococcal infections
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
