
Abstract
Bridged medium-sized bicyclo[m.n.2] ring systems are common in natural products and potent pharmaceuticals, and pose a great synthetic challenge. Chemistry for making bicyclo[m.n.2] ring systems remains underdeveloped. Currently, there are no general reactions available for the single-step synthesis of various bridged bicyclo[m.n.2] ring systems from acyclic precursors. Here, we report an unusual type II intramolecular (3+2) dipolar cycloaddition strategy for the syntheses of various bridged bicyclo[m.n.2] ring systems. This rhodium-catalysed cascade reaction provides a relatively general strategy for the direct and efficient regioselective and diastereoselective synthesis of highly functionalized and synthetically challenging bridged medium-sized polycyclic systems. Asymmetric total synthesis of nakafuran-8 was accomplished using this method as a key step. Quantum mechanical calculations demonstrate the mechanism of this transformation and the origins of its multiple selectivities. This reaction will inspire the design of the strategies to make complex bioactive molecules with bridged medium-sized polycyclic systems.
Similar content being viewed by others


Introduction
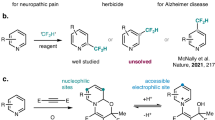
Molecules with bridged medium-sized ring systems1,2 are advantageous for their pharmacological activity and for their selective and tight binding to biological targets. Bridged medium-sized bicyclo[m.n.1] and bicyclo[m.n.2] ring systems are strained and widely found in natural products with important biological activities (such as medicines Taxol, Picato, and Artemisinin; Fig. 1a)3,4. In contrast to the myriad approaches for the creation of bicyclo[m.n.1] ring systems, chemistry for bicyclo[m.n.2] ring systems remains underdeveloped. Bridged bicyclo[m.n.2] ring systems pose a great synthetic challenge and have prompted considerable interest from the synthetic community, leading to several remarkable syntheses5,6,7,8,9,10,11. However, so far there are no general reactions available for the single-step synthesis of various bridged bicyclo[m.n.2] ring systems from acyclic precursors. Thus, developing efficient reactions for achieving these bridged ring systems is very important.

a Bridged medium-sized ring systems are highly valued in many compounds, including in some natural products that also serve as medicines. b Type II IMDA cycloaddition is used to make a few of bicyclo[m.3.1] ring systems but rarely used for the syntheses of bicyclo[m.2.2] ring systems, and all these bridged ring systems have a highly strained bridgehead olefin. c Type II (3+2) cycloaddition reaction (this work), their use in creating bicyclo[m.n.2] ring systems, and their potential competitive reactions. Direct generation of bridged ring systems by an intramolecular cycloaddition is called type II cycloaddition. Bridged ring systems are highlighted in red. Bz benzoxyl, Ac acetyl, Ts tosyl.
Intramolecular cycloaddition reactions are very significant for efficiently creating polycyclic systems. The intramolecular Diels–Alder (IMDA) cycloaddition reaction is one of the most extensively used reactions for making ring systems12,13,14,15. IMDA cycloadditions are classified into type I and type II according to how the dienophile motif is linked to the diene. Type I IMDA cycloadditions (linked at the 1-position of the diene) are great for synthesizing fused bicyclo[m.4.0] ring systems. The pioneering Shea type II IMDA cycloadditions (linked at the 2-position of the diene) are powerful for the preparation of a few of bridged bicyclo[m.3.1] ring systems15,16,17; however, these are very rarely used for the creation of all-carbon bicyclo[m.2.2] ring systems18, because the formation of such bicyclo[m.2.2] ring systems (m = 3, 4, or 5) also with an unfavorable strained bridgehead olefin19 (Bredt’s rule)20 are usually more challenging than their regioisomeric products21 (Fig. 1b). Particularly, there have been no reports of type II IMDA reactions being used to make bicyclo[5.2.2], bicyclo [4.2.2], and bicyclo[3.2.2] ring systems. In addition, other type II intramolecular cycloadditions22,23 (including innovative Davies-[4+3]24, remarkable Wender-[4+4]25, and our [5+2]26,27) are unknown for the synthesis of bicyclo[m.n.2] ring systems. Currently, few intramolecular cycloaddition reactions are available for the direct and efficient synthesis of various bicyclo[m.n.2] ring systems. An absence of direct procedures has impeded the in-depth evaluation of their potential pharmaceutical value. Therefore, it is still highly desirable to develop new and efficient strategies for constructing these attractive bridged bicyclo[m.n.2] ring systems.
The 1,3-diploar cycloaddition of transient carbonyl ylides generated from carbene is a very well-established reaction and it is also as an attractive strategy toward the synthesis of complex natural products28,29,30. Stimulated by the challenges of previous type II cycloadditions, we posited that an unusual rhodium-catalyzed type II intramolecular (3+2) dipolar cycloaddition might be achieved with different types of substrates to synthesize bicyclo[m.n.2] ring systems without strained bridgehead olefins (Fig. 1c). Specifically, we considered an N-sulfonyl-1,2,3-triazole moiety and an unactivated alkene both tethered at the α-position of the carbonyl group of an aldehyde with various tether lengths (A, Fig. 1c). Treatment of N-sulfonyl-1,2,3-triazole A with rhodium catalysts would generate the rhodium-iminocarbene B31,32. The iminocarbene B would give reactive 1,3-dipole C. The 1,3-dipole and alkene group in C would undergo the desired (3+2) dipolar cycloaddition via intermediate D, to provide various useful and highly functionalized bridged bicyclo[m.n.2] ring systems (e.g., bicyclo[5.2.2], bicyclo[4.2.2], bicyclo[3.2.2], and azabicyclo[3.2.2] ring systems). The anticipated cycloaddition described herein is different from the previous examples because it does not produce anti-Bredt double bond. This is the reason why smaller rings could be accessed compared to the other type II cycloadditions. Remarkably, these bridged skeletons contain medium-sized ring systems1,2, which have high strain energy. It is very difficult to form such ring systems and doing so depends on the reactivities of the corresponding acyclic precursors and their steric effects because of unfavorable transannular interactions and enthalpic and entropic effects33,34. The carbenes derived from the corresponding N-sulfonyltriazoles have been used in cycloaddition reactions to synthesize fused all-carbon ring systems35,36,37. So far, there have been no reports of intramolecular dipolar cycloaddition reactions for the synthesis of all-carbon bicyclo[m.n.2] ring systems38. Particularly, the activation free energy of type II (3+2) cycloadditions is higher than that of type I cycloadditions, because of the strain inherent to the formation of the bridged ring systems15,22. Furthermore, alkene groups could potentially undergo competitive intramolecular cyclopropanation39 with the reactive rhodium-carbene in intermediate B to give unexpected products. Meanwhile, the rhodium-carbene in B can also undergo intramolecular insertion into the C-H bond of alkanes40,41. In addition, the alkene group in C can competitively undergo intramolecular (5+2) cycloaddition involving the carbonyl ylide and the N-tosylimine. Similar to type II IMDA cycloadditions, the 1,3-dipole and alkene group in C can also undergo regioisomeric (3+2) cycloaddition to give bicyclo[m.n.1] ring system through intermediate E. All these competitive reactions make the desired type II (3+2) cycloaddition reaction more challenging. Furthermore, it was uncertain as to which diastereomer would form and if the cycloaddition would follow a concerted or stepwise pathway. Herein we report rhodium-catalyzed type II intramolecular (3+2) cycloadditions of N-sulfonyl-1,2,3-triazoles. These unique reactions provide a new strategy for the direct and efficient synthesis of various functionalized bridged medium-sized polycyclic systems and enable asymmetric total synthesis of nakafuran-8.
Results and discussion
Reaction development
To test our hypothesis, initially, the 1,2,3-triazole 1a (Fig. 2) was synthesized as the model substrate to identify feasible reaction conditions [see Supporting Information (SI) for details]. Treatment of 1a with Rh2(Oct)4 (5 mol%) in 1,2-dichloroethane (DCE) at 85 °C for 3 h yielded the corresponding imine product 2a. Compound 2a was unstable to chromatography on both silica gel and aluminum oxide and was directly reduced by LiAlH4 in a one-pot sequence to give the desired bicyclo[4.2.2] ring system 3a as a single diastereomer in 81% isolated yield. The structure of 3a was confirmed by X-ray diffraction. Notably, this cycloaddition constructed three new chemical bonds (highlighted in blue in 2), two carbocyclic rings (highlighted in red in 3) and one heterocyclic ring, and three new stereogenic centers, including one bridgehead all-carbon quaternary stereogenic center in a single step. This result is particularly significant, because installation of a bridgehead quaternary stereogenic center within a bridged bicycle is very challenging and is highly valuable in synthesis42,43. This reaction affords the opportunity to efficiently construct these bridgehead all-carbon quaternary stereogenic centers in natural products, such as nakafuran-8, PF-1018, tubingensin B, and calyciphylline C (C* in Fig. 1a).

Rhodium-catalyzed type II (3+2) cycloaddition reaction using various substrates to give highly functionalized bridged medium-sized ring systems. Bn benzyl, Piv pivaloyl, TMS trimethylsilyl.
Substrate scope
With the optimized conditions in hand, the generality of this strategy was examined with various substrates, as summarized in Fig. 2. First, we varied the R1 groups and R2 substituents on the internal position of the alkenes, affording bicyclo[4.2.2] ring systems 3b–r in moderate-to-excellent yields (61–90%). The reactions proceeded well for aromatic substituents on the internal positions of the alkenes with electron-withdrawing or electron-donating groups in para or meta position of aryl groups, forming 3b–h with 67–80% yields. Pleasingly, the methyl-substituted substrate 1i delivered the desired product 3i in 61% yield. Notably, the bridged bicyclo[4.2.2] ring system 3o (R1 = H, R2 = Me) was obtained in an acceptable 61% yield. This finding shows that this cycloaddition probably does not rely on the Thorpe–Ingold effect, which is a considerable advantage. Remarkably, substrates 1s–z with a fused four- or five-membered ring, all led to formation of 3s–z with diverse stereochemistry in good yields, respectively. Particularly, the mono-substituted alkenes 1x and 1z yielded 3x and 3z, respectively. It is worth mentioning that the functionalized bridged ring system 3z is a potential advanced intermediate for the total synthesis of triphyllenol (Fig. 1a). Pleasingly, heterocycle-substituted products 3p–r, which are good handles for further functionalization, were prepared in 75–90% yields. Rewardingly, several protecting groups, including triisopropylsilyl (TIPS), pivaloyl, benzyl, and trimethylsilyl groups, were tolerated very well. Furthermore, the pivaloyl group was potentially removed under LiAlH4 conditions. Consequently, the imine products 2j–l were hydrolyzed with wet basic alumina to give the corresponding aldehydes 3j–l, respectively, in good yields, offering potential for further functional group transformations.
Notably, two substrates containing a phenyl ring or a nitrogen as the tether were also compatible (Fig. 2), and the desired phenyl-fused bicyclo[4.2.2] 3aa or azabicyclo[4.2.2] 3ab were obtained. Interestingly, treatment of 1ac (R1 = CH2OTIPS, R2 = isopropenyl) with Rh2(Oct)4 (5 mol%) at 85 °C, followed by a wet basic alumina and tetrabutylammonium fluoride gave the desired bicyclo[4.2.2] ring system 3ac and the unexpected bicyclo[5.2.2] ring system 3ad in yields of 46 and 22%, respectively. Compound 3ac with a bridgehead isopropenyl group has good potential for further modification. Furthermore, the highly strained bicyclo[5.2.2] ring system 3ae with a difficult-to-form medium-sized 9-membered ring was also obtained, albeit with a 25% yield. The successful formation of bicyclo[5.2.2] ring systems 3ad and 3ae will provide useful ideas for designing the synthetic route of subincanadine E (Fig. 1a). It is noteworthy that the type II IMDA reaction conditions for unactivated dienophiles were usually very harsh (400–500 °C, gas phase)15,16,17; our type II (3+2) cycloaddition conditions for unactivated alkene dipolarophiles are much milder, which will be a significant advantage for their further application.
Encouraged by the successful syntheses of bicyclo[4.2.2] ring systems, we set out to investigate the feasibility of using the current strategy to make other bicyclo[m.2.2] skeletons with various tether lengths. As shown in Fig. 2, the corresponding precursors 1af–aj with different tether lengths could undergo the rhodium-catalyzed type II (3+2) cycloaddition reaction, thus delivering a series of unique, functionalized bicyclo[3.2.2] skeletons 3af–aj. Alkenes with various R2 groups, including phenyl, furanyl, thienyl, and methyl groups, could take part in the cycloaddition. The desired bridged bicyclo[3.2.2] ring systems 3af–ah with different functional groups were obtained in 77–83% yields. To our delight, the chiral azabicyclo[3.2.2] product 3ai and the highly functionalized oxabicyclo[3.2.2] ring system 3aj were obtained in good and acceptable yields, respectively. These results are particularly important, because the synthetically challenging azabicyclo[3.2.2] and oxabicyclo[3.2.2] ring systems are also found in some natural products, such as calyciphylline C and briarellin J, respectively (Fig. 1a). The structures of 3b, 3d, 3f–h, 3s, and 3af–ah and the derivatives of 3p, 3t, 3w, and 3x were unambiguously confirmed by X-ray diffraction (see SI for details). To our knowledge, this work represents the first example of an intramolecular (3+2) dipolar cycloaddition for constructing bicyclo[3.2.2], bicyclo [4.2.2], and bicyclo[5.2.2] ring systems.
Asymmetric total synthesis of nakafuran-8
After establishing the type II (3+2) cycloaddition reaction to make various bridged medium-sized ring systems, we moved toward the asymmetric total synthesis of nakafuran-8 (Fig. 3). Nakafuran-8 is an antifeedant against common reef fishes44 and has a remarkable inhibitory activity against human protein tyrosine phosphatase 1B, an enzyme involved in insulin signal regulation45. Structurally, nakafuran-8 has a unique bicyclo[4.2.2]decadiene skeleton with a furan ring and a bridgehead all-carbon quaternary stereocenter, which presents a formidable synthetic challenge. In addition, its absolute configuration has not been determined. Construction of the desired bicyclo[4.2.2] ring system showed very poor regioselectivity in the previous synthesis5,6. To date, asymmetric synthesis of nakafuran-8 has not been achieved.

DIPEA N,N-diisopropylethylamine, DMP Dess–Martin periodinane, LDA lithium diisopropylamide, DIBAL diisobutylaluminium hydride, KHMDS potassium bis(trimethylsilyl)amide, LiHMDS lithium bis(trimethylsilyl)amide.
The readily available chiral compound 446 underwent a diastereoselective aldol reaction with aldehyde 547 followed by TIPS protection of the resulting hydroxyl group to give compound 6. Reduction of 6 with NaBH4 and subsequent oxidation provided 7 in 55% overall yield (20 g scale). The alkyne group in 7 reacted with TsN3 to give 8 in 85% yield (10 g scale). The rhodium-catalyzed type II (3+2) cycloaddition of 8 with a N-sulfonyl-1,2,3-triazole moiety, an unactivated alkene and an aldehyde, provided the desired product 9, which underwent hydrolysis and reduction in a one-pot sequence to give alcohol 10 in 50% overall yield (gram scale). Compound 10 was converted to the corresponding iodide followed by cleavage of the C-O bond to generate alkene 11 in 63% overall yield (gram scale). Subsequent oxidation of 11 with Dess–Martin periodinane (DMP) gave the expected ketone. Diastereoselective and regioselective methylation at the α-position of the resultant ketone group with lithium diisopropylamide and MeI and subsequent reduction provided 12 in 60% overall yield over 3 steps (gram scale). The absolute configurations of 11 and 12 were confirmed by the X-ray diffraction analysis of their derivatives (see SI). Accordingly, compound 12 underwent standard Barton deoxygenation smoothly and then TIPS deprotection to give 13 in 62% overall yield (gram scale). DMP oxidation of 13 followed by the formation of furanone and chemoselective olefin isomerization developed by Shenvi48 produced compound 14. Treatment of 14 with DIBAL gave nakafuran-8 in 65% yield, completing asymmetric total synthesis of nakafuran-8. Thus, the absolute configuration of nakafuran-8 was unambiguously established by our total synthesis.
Quantum mechanical study of mechanisms and selectivities
We carried out a computational exploration of the regioselectivity, chemoselectivity, and diastereoselectivity of the (3+2) cycloaddition between an alkene dipolarophile and a carbonyl ylide 1,3-dipole. Quantum mechanical calculations (Fig. 4) were carried out with density functional theory and solvation energies computed at the ωB97X-D/6-311 + G(d,p),CPCM(DCE) level of theory. Conformational analysis was carried out with CREST. All (3+2) cycloadditions were found to proceed through concerted but highly asynchronous mechanisms.

a Optimized structures of the two favored transition states. b FMO interactions contribute to the stabilization of the TS for the major product. c Energetics of (3+2) and (5+2) cycloadditions and charge distribution of the carbonyl ylide.
The three-dimensional structures of the lowest energy transition states (TSs) are shown in Fig. 4a. Both involve typical intramolecular carbonyl ylide cycloadditions to produce functionalized bridged ring systems. The favored cycloaddition leads to a bicyclo[4.2.2] ring system (highlighted in red) in 15. In this reaction, the most nucleophilic unsubstituted terminus of the alkene attacks the most electrophilic terminus of the carbonyl ylide. The disfavored cycloaddition leads to the bicyclo[4.3.1] ring system (highlighted in red) in 16, with a mismatch of electrophilic and nucleophilic sites in the TS. Both have the connecting linker anti to the O of the carbonyl ylide (exo). Although the favored TS is highly asynchronous, no intermediate could be located. As expected, the disfavored regioisomeric TS is less asynchronous because of more balanced, but overall weaker, frontier molecular orbital (FMO) interactions49,50,51,52.
The FMOs of the carbonyl ylide and the alkene dipolarophile are shown in Fig. 4b. These calculations show that the strongest interaction is between the π bonding orbital on the olefin (highest occupied molecular orbital (HOMO) − 1) and the π* on the carbonyl ylide (lowest unoccupied molecular orbital (LUMO)), which have an energy difference of 7.90 eV. The allyl anion-like (HOMO) of the carbonyl ylide and the π* orbital on the olefin (LUMO + 4) interact less strongly with an energy difference of 9.16 eV between the two orbitals. As expected, the HOMO of the carbonyl ylide resembles a normal allyl anion-like HOMO, with a node at O and highly polarized toward the terminus bearing the electron-withdrawing substituent. The LUMO is strongly polarized in the opposite direction. The major FMO interaction is between the carbonyl ylide LUMO and the alkene HOMO, which has the largest coefficient at the unsubstituted terminus, consequently the most nucleophilic site. Shown in blue in Fig. 4c are the Hirshfeld charges corresponding to each of these atoms. Most evident is the significant negative charge on the terminal methylene of the olefin (−0.12) and the positive charge on the carbonyl ylide carbon (0.20). The electrostatic interaction between these two carbons in the TS further influences the regioselectivity of this reaction.
Reactants, TSs, and products were optimized for the four possible (3+2) cycloadditions and for two of the forbidden (5+2) cycloadditions. These results are also shown in Fig. 4c. The major exo-product 15 discussed above and its corresponding TS barrier were calculated, revealing an extremely exothermic transformation with a barrier of only 16.9 kcal/mol and overall Gibbs free energy change of −40.8 kcal/mol. The diastereomer 15-dia in which the two methyl groups are trans to one another, resulting from the opposite facial selectivity, is geometrically impossible and was not found to be a minimum on the potential energy surface. The activation barrier and the overall free energy change of the regioisomeric exo-product 16 were computed to be 19.3 kcal/mol and −43.9 kcal/mol, respectively. This 2.4 kcal/mol higher activation barrier means that only a few percent of this adduct should be formed. The alternate diastereomer 16-dia with trans methyl groups is geometrically highly contorted but could be located with an energy barrier of 41.2 kcal/mol and an overall exothermicity of only −0.5 kcal/mol. Finally, the products 17 and 18 of the (5+2) reaction involving the carbonyl ylide and the N-tosylimine were calculated to be relatively unstable with overall Gibbs free energy changes of −14.3 kcal/mol and −26.2 kcal/mol for the two regioisomers. We did not attempt to find TSs for these highly disfavored reactions.
Summary
In summary, we have developed a rhodium-catalyzed type II (3+2) intramolecular cycloaddition cascade reaction of a N-sulfonyl-1,2,3-triazole, involving a carbonyl group and a simple unactivated alkene. This unusual reaction will be an approach complementary to type II IMDA cycloaddition to access bridged polycyclic systems. The reaction proceeds chemoselectively, regioselectively, and diastereoselectively, thus establishing a new and relatively general strategy for the efficient and straightforward synthesis of various functionalized and synthetically challenging bridged medium-sized ring systems (e.g., bicyclo[5.2.2], bicyclo[4.2.2], azabicyclo[4.2.2], bicyclo[3.2.2], azabicyclo[3.2.2], and oxabicyclo[3.2.2] ring systems). Particularly, this work represents, to our knowledge, the first example of a rhodium-catalyzed intramolecular (3+2) cycloaddition to make synthetically challenging eight-membered ring systems53 and represents the first example of an intramolecular dipolar cycloaddition for constructing all-carbon bicyclo[m.2.2] ring systems. To the best of our knowledge, the first asymmetric total synthesis of nakafuran-8 was achieved using this method. Quantum mechanical calculations show the nature of the TSs for these reactions and the origins of regioselectivity and stereoselectivity. We believe that the reaction developed here will inspire the design of new strategies to make complex natural products and other bioactive molecules with bridged medium-sized polycyclic systems.
Methods
General experimental procedure
An oven-dried tube was charged with triazole compound 1 (0.1 mmol, 1.0 equiv.), 3 Å MS (30 mg), and Rh2(OCt)4 (4 mg, 0.005 mmol, 0.05 equiv.). The tube was evacuated and backfilled with argon (repeated three times). Then DCE (2 mL, 0.05 M/L) was added into the reaction via syringe. The reaction mixture was stirred at 85 °C for 3 h. The solution was then cooled to 25 °C. Tetrahydrofuran (THF; 4 mL) was added into the reaction via syringe. The solution was cooled to 0 °C. LiAlH4 (1.0 M in THF, 0.2 mL, 0.2 mmol, 2.0 equiv.) was added dropwise and the solution was stirred at 0 °C for 1.5 h. Following quenching with saturated aqueous potassium sodium tartrate (6 mL), the reaction mixture was extracted by EtOAc (30 mL × 3), the combined organic layers were washed with saturated brine (10 mL), dried over Na2SO4, concentrated in vacuum, and purified by column chromatography on silica gel to give the products.
Data availability
The data supporting the findings of this study are available in the paper and its Supplementary Information; further data are available from the corresponding author on request. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers CCDC 2065575 (3a), CCDC 2068651 (3b), CCDC 2053925 (3d), CCDC 2053921 (3f), CCDC 2053927 (3g), CCDC 2053929 (3h), CCDC 2053972 (3p’), CCDC 2054598 (3s), CCDC 2054000 (3t’), CCDC 1920650 (3w’), CCDC 1920646 (3x’), CCDC 2053961 (3af), CCDC 2053962 (3ag), CCDC 2053964 (3ah), CCDC 2054002 (11′), CCDC 2054003 (12′). These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
References
Molander, G. A. Diverse methods for medium ring synthesis. Acc. Chem. Res. 31, 603–609 (1998).
Yet, L. Metal-mediated synthesis of medium-sized rings. Chem. Rev. 100, 2963–3008 (2000).
Nicolaou, K. C. & Montagnon, T. Molecules that Changed the World (Wiley-VCH, 2008).
Nicolaou, K. C., Dai, W.-M. & Guy, R. K. Chemistry and biology of taxol. Angew. Chem. Int. Ed. 33, 15–44 (1994).
Uyehara, T., Sugimoto, M., Suzuki, I. & Yamamoto, Y. Total synthesis of (±)-nakafuran-8, a marine metabolite with antifeedant properties, based on formal bridgehead substitution of a bicyclo[2.2.2]oct-5-en-2-one system. J. Chem. Soc., Chem. Commun. 23, 1841–1842 (1989).
Uyehara, T., Sugimoto, M., Suzuki, I. & Yamamoto, Y. Rearrangement approaches to cyclic skeletons. part 8. total synthesis of (±)-nakafuran-8, a marine metabolite with antifeedant properties, on the basis of bridgehead substitution of a bicyclo[2.2.2]oct-5-en-2-one system. J. Chem. Soc. Perkin Trans. 1, 1785–1788 (1992).
Liu, W.-C. & Liao, C.-C. The first total synthesis of (±)-pallescensin B. Chem. Commun. 2, 117–118 (1999).
Snyder, S. A., Breazzano, S. P., Ross, A. G., Lin, Y. & Zografos, A. L. Total synthesis of diverse carbogenic complexity within the resveratrol class from a common building block. J. Am. Chem. Soc. 131, 1753–1765 (2009).
Corsello, M. A., Kim, J. & Garg, N. K. Total synthesis of (−)-tubingensin B enabled by the strategic use of an aryne cyclization. Nat. Chem. 9, 944–949 (2017).
Holmbo, S. D. & Pronin, S. V. A concise approach to anthraquinone-xanthone heterodimers. J. Am. Chem. Soc. 140, 5065–5068 (2018).
Quintela, V. H., Jamieson, C. S., Shao, Q., Houk, K. N. & Trauner, D. Bioinspired synthesis of (−)-PF-1018. Angew. Chem. Int. Ed. 59, 5263–5267 (2020).
Brieger, G. & Bennett, J. N. The intramolecular Diels–Alder reaction. Chem. Rev. 80, 63–97 (1980).
Nicolaou, K. C., Snyder, S. A., Montagnon, T. & Vassilikogiannakis, G. The Diels−Alder reaction in total synthesis. Angew. Chem. Int. Ed. 41, 1668–1698 (2002).
Juhl, M. & Tanner, D. Recent applications of intramolecular Diels−Alder reactions to natural product synthesis. Chem. Soc. Rev. 38, 2983–2992 (2009).
Bear, B. R., Sparks, S. M. & Shea, K. J. The type II intramolecular Diels-Alder reaction: synthesis and chemistry of bridgehead alkenes. Angew. Chem. Int. Ed. 40, 820–849 (2001).
Shea, K. J. & Wise, S. Intramolecular Diels-Alder reactions. A new entry into bridgehead bicyclo[3.n.1]alkenes. J. Am. Chem. Soc. 100, 6519–6521 (1978).
Shea, K. J. et al. Applications of the intramolecular Diels–Alder reaction to the formation of strained molecules. Synthesis of bridgehead alkenes. J. Am. Chem. Soc. 104, 5708–5715 (1982).
Schreiber, S. L. & Kiessling, L. L. Further investigations of the type II Diels-Alder route to the bicyclic core of esperamicin/calichemicin reveal a regiochemical misassignment: meta vs. para selectivity. Tetrahedron Lett. 30, 433–436 (1989).
Liu, J., Liu, X., Wu, J. & Li, C.-C. Total synthesis of natural products containing a bridgehead double bond. Chem 6, 579–615 (2020).
Bredt, J. Steric hindrance in the bridge ring (Bredt’s rule) and the meso-trans-position in condensed ring systems of the hexamethylenes. Liebigs Ann. Chem. 437, 1–13 (1924).
Cleary, L., Mak, V. W., Rychnovsky, S. D., Shea, K. J. & Sizemore, N. Origins of regio- and stereochemistry in type 2 intramolecular N-acylnitroso Diels-Alder reactions: a computational study of tether length and substituent effects. J. Org. Chem. 78, 4090–4098 (2013).
Min, L., Hu, Y.-J., Fan, J.-H., Zhang, W. & Li, C.-C. Synthetic applications of type II intramolecular cycloadditions. Chem. Soc. Rev. 49, 7015–7043 (2020).
Hou, S.-H. et al. Enantioselective type II cycloaddition of alkynes via C-C activation of cyclobutanones: rapid and asymmetric construction of [3.3.1] bridged bicycles. J. Am. Chem. Soc. 142, 13180–13189 (2020).
Davies, H. M. L., Calvo, R. & Ahmed, G. Type II intramolecular annulations between vinylcarbenoids and furans. Tetrahedron Lett. 38, 1737–1740 (1997).
Wender, P. A. & Snapper, M. L. Intramolecular nickel catalyzed cycloadditions of bis-dienes: 3 approaches to the taxane skeleton. Tetrahedron Lett. 28, 2221–2224 (1987).
Mei, G. J., Liu, X., Qiao, C., Chen, W. & Li, C.-C. Type II intramolecular [5+2] cycloaddition: facile synthesis of highly functionalized bridged ring systems. Angew. Chem. Int. Ed. 54, 1754–1758 (2015).
Min, L., Liu, X. & Li, C.-C. Total synthesis of natural products with bridged bicyclo[m.n.1] ring systems via type II [5+2] cycloaddition. Acc. Chem. Res. 53, 703–718 (2020).
Padwa, A. Domino reactions of rhodium(II) carbenoids for alkaloid synthesis. Chem. Soc. Rev. 38, 3072–3081 (2009).
Padwa, P. Intramolecular cycloaddition of carbonyl ylides as a strategy for natural product synthesis. Tetrahedron 67, 8057–8072 (2011).
Davies, H. M. L. & Alford, J. S. Reactions of metallocarbenes derived from N-sulfonyl-1,2,3-triazoles. Chem. Soc. Rev. 43, 5151–5162 (2014).
Chuprakov, S., Hwang, F. W. & Gevorgyan, V. Rh-catalyzed transannulation of pyridotriazoles with alkynes and nitriles. Angew. Chem. Int. Ed. 46, 4757–4759 (2007).
Horneff, T., Chuprakov, S., Chernyak, N., Gevorgyan, V. & Fokin, V. V. Rhodium-catalyzed transannulation of 1,2,3-triazoles with nitriles. J. Am. Chem. Soc. 130, 14972–14974 (2008).
Illuminati, G. & Mandolini, L. Ring closure reactions of bifunctional chain molecules. Acc. Chem. Res. 14, 95–102 (1981).
Galli, C. & Mandolini, L. The role of ring strain on the ease of ring closure of bifunctional chain molecules. Eur. J. Org. Chem. 2000, 3117–3125 (2000).
Spangler, J. E. & Davies, H. M. L. Catalytic asymmetric synthesis of pyrroloindolines via a Rhodium (II)-catalyzed annulation of indoles. J. Am. Chem. Soc. 135, 6802–6805 (2013).
Li, Y., Zhang, Q. Y., Du, Q. C. & Zhai, H. B. Rh-Catalyzed [3+2] cycloaddition of 1-sulfonyl-1,2,3-triazoles: access to the framework of aspidosperma and kopsia indole alkaloids. Org. Lett. 18, 4076–4079 (2016).
Yuan, H., Gong, J. X. & Yang, Z. Stereoselective synthesis of oxabicyclo[2.2.1]heptenes via a tandem dirhodium(II)-catalyzed triazole denitrogenation and [3 + 2] cycloaddition. Org. Lett. 18, 5500–5503 (2016).
Padwa, A. & John, P. W. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products (Wiley-VCH, 2003).
Chuprakov, S., Kwok, S. W., Zhang, L., Lercher, L. & Fokin, V. V. Rhodium-catalyzed enantioselective cyclopropanation of olefins with N-sulfonyl 1,2,3-triazoles. J. Am. Chem. Soc. 131, 18034–18035 (2009).
Liao, K., Negretti, S., Musaev, D. G., Bacsa, J. & Davies, H. M. L. Site-selective and stereoselective functionalization of non-activated C–H bonds. Nature 533, 230–234 (2016).
Liao, K. et al. Site-selective and stereoselective functionalization of non-activated tertiary C–H bonds. Nature 551, 609–613 (2017).
Christoffers, J. & Baro, A. (eds) Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis (Wiley-VCH, 2005).
Hu, P. F. et al. Quaternary-center-guided synthesis of complex polycyclic terpenes. Nature 569, 703–707 (2019).
Schulte, G., Scheuer, P. J. & McConnell, O. J. Two furanosesquiterpene marine metabolites with antifeedant properties. Helv. Chim. Acta 63, 2159–2167 (1980).
Huang, X.-C., Li, J., Li, Z.-Y., Shi, L. & Guo, Y.-W. Sesquiterpenes from the hainan sponge dysidea septosa. J. Nat. Prod. 71, 1399–1403 (2008).
Vintonyak, V. V. & Maier, M. E. Synthesis of the core structure of cruentaren A. Org. Lett. 9, 655–658 (2007).
Parsons, D. E. & Frontier, A. J. Noncanonical cation−π cyclizations of alkylidene β-ketoesters: synthesis of spiro-fused and bridged bicyclic ring systems. Org. Lett. 21, 2008–2012 (2019).
Crossley, S. W. M., Barabé, F. & Shenvi, R. A. Simple, chemoselective, catalytic olefin isomerization. J. Am. Chem. Soc. 136, 16788–16791 (2014).
Houk, K. N. The frontier molecular orbital theory of cycloaddition reactions. Acc. Chem. Res. 8, 361–369 (1975).
Houk, K. N. Regioselectivity and reactivity in the 1,3-dipolar cycloadditions of diazonium betaines (diazoalkanes, azides, and nitrous oxide). J. Am. Chem. Soc. 94, 8953–8955 (1972).
Houk, K. N., Sims, J., Duke, R. E. Jr, Strozier, R. W. & George, J. K. Frontier molecular orbitals of 1,3-dipoles and dipolarophiles. J. Am. Chem. Soc. 95, 7287–7301 (1973).
Houk, K. N., Sims, J., Watts, C. R. & Luskus, L. J. The origin of reactivity, regioiselectivity, and periselectivity in 1,3-dipolar cycloadditions. J. Am. Chem. Soc. 95, 7301–7315 (1973).
Hu, Y.-J., Li, L.-X., Han, J.-C., Min, L. & Li, C.-C. Recent advances in the total synthesis of natural products containing eight-membered carbocycles (2009–2019). Chem. Rev. 120, 5910–5953 (2020).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 21971105), the Shenzhen Nobel Prize Scientists Laboratory Project (C17783101), Science and Technology Key Project of Guangdong Province (2020B1111110004), and Research Projects of Universities of Guangdong Province (2019KZDXM005). K.N.H. is also grateful to the National Science Foundation (CHF-1764328) for financial support. Calculations were performed on the Hoffman2 cluster at the University of California, Los Angeles, and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (Grant OCI-1053575).
Author information
Authors and Affiliations
Contributions
C.-C.L. conceived of and directed the project. B.-L.H., N.L. and Y.-Q.W. performed the synthetic experiments, developed the reactions, and finished the asymmetric total synthesis of nakafuran-8. J.J.W. performed the computational experiments and provided mechanism analysis. K.N.H. directed the computational calculations and mechanism analysis. All authors analyzed and discussed the results. K.N.H. and C.-C.L. prepared the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hou, BL., Wong, J.J., Lv, N. et al. Facile generation of bridged medium-sized polycyclic systems by rhodium-catalysed intramolecular (3+2) dipolar cycloadditions. Nat Commun 12, 5239 (2021). https://doi.org/10.1038/s41467-021-25513-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-25513-7
This article is cited by
-
Deep learning for development of organic optoelectronic devices: efficient prescreening of hosts and emitters in deep-blue fluorescent OLEDs
npj Computational Materials (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
