
Abstract
This monocentric study included fifteen children under a year old in intensive care with suspected monogenic conditions for rapid trio exome sequencing (rES) between April 2019 and April 2021. The primary outcome was the time from blood sampling to rapid exome sequencing report to parents. All results were available within 16 days and were reported to parents in or under 16 days in 13 of the 15 individuals (86%). Six individuals (40%) received a diagnosis with rES, two had a genetic condition not diagnosed by rES. Eight individuals had their care impacted by their rES results, four were discharged or died before the results. This small-scale study shows that rES can be implemented in a regional University hospital with rapid impactful diagnosis to improve care in critically ill infants.
Similar content being viewed by others
Introduction
In severely ill children hospitalized in the neonatal intensive care unit (NICU) or pediatric intensive care unit (PICU), a quick etiological diagnosis is key to proper care management. The phenotypic and genetic variability of severe early genetic conditions and the lack of developmental data make a phenotype-first approach challenging. Exome (ES) and genome sequencing (GS) have been used in research studies for decision-making and care of critically ill patients with a proof-of-principal in 2012 [1] then in larger studies when the turn-around time from blood sampling to results became compatible with the time constraints of the NICU/PICU [2,3,4,5,6].
In France, current genetic testing with no phenotypic diagnosis includes as first-tier testing array-comparative genomic hybridization array (CGH-array). Further testing is carried out using gene panels, ES or GS. This lengthy process does not suit the timeline of critical care. The aim of this study was to evaluate the possibility of on-site rES in a French regional university hospital of getting a rapid genetic diagnosis in critically ill infants.
Methods
Patients
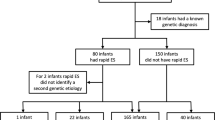
The inclusion criteria were: children under one year admitted to intensive care who would benefit from an early diagnosis as evaluated by the NCIU/PCIU team, suspicion by a consultant of the clinical genetics team of a monogenic condition (i.e.,: multi-systemic involvement, severity of symptoms, consanguinity), both biological parents available (Fig. 1). Patients were excluded if a genetic diagnosis was clinically recognizable, if a chromosomal anomaly was thought more likely or if another non-genetic condition could explain the phenotype.

BWS Beckwith Wiedemann syndrome, CMV Cytomegalovirus.
Study design and ethics
This prospective single-center study recruited patients from April 2019 to April 2021 who were admitted in the NICU or PICU of the University hospital of Montpellier. Informed signed consent was obtained from both parents. This study was accepted by the ethical committee of Montpellier university hospital (CNRIPH 18.07.17.33927).
Outcomes
The primary outcome was the number of results reported to parents within 16 days from blood sampling. Secondary outcomes were the number of diagnosis, the number of cases where rES led to a change in medical care, as defined by a treatment, test, action performed because of the rES results.
Clinical data and concomitant genetic testing
Clinical data was collected from medical records and phenotype was described using the human phenotype ontology (HPO) [7] terms and detailed in Table 1. Patients were clinically reevaluated at 3 months and a year after the inclusion. Other genetic tests, including array-CGH if indicated, were performed as part of standard genetic testing.
ES processing and interpretation
DNA was extracted from a venous blood sample. We used the SureSelect QTX Library prep (Agilent technologies) enrichment kit and SureSelect Human All Exons V7 (Agilent technologies) following the manufacturer’s instruction. Sequencing was performed on a NextSeq 500 (Illumina). Bioinformatics were performed following Broad institute GATK best practice guidelines [8]. Pipeline for variant calling and prioritization is reported in literature [9].
rES data was jointly interpreted by molecular biologists and clinical geneticists in a multi-disciplinary approach. Classification of variants followed the recommendations of the American College of Medical Genetics and Genomics (ACMG) [10]. The report was written by the molecular biologist. Variants were confirmed with Sanger sequencing and a new report issued.
Results
Population
Fifteen patients were included in this study. The median age at blood sampling was 38 days (extremes: 4–272; interquartile range: 8–82). Phenotypes of patients are summarized in Table 1; results and impact on care in Table 2. Prenatal invasive genetic testing was carried out in 5 mothers of included infants.
Primary outcome
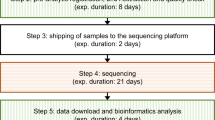
Results were reported to parents within 16 days from blood sampling in 13/15 cases (86%), with a median of 14 days (extremes 9–145; interquartile range: 13–15; Table 2). All rES results were available within 16 days. For individual 4, the parents declined the scheduled genetic consultation, because of their child’s death. For individual 7, the parents were not available for a consultation before day 16. Of the 15 children, two died before the results.
Secondary outcome
Number of diagnoses after rES analysis
A diagnosis was made by rES in 6 individuals (40%, Table 2). Concomitant tests are summarized in Table 1. In individual 13 diagnosis was made by array-CGH (concomitant genetic test) who had a diagnosis of complex chromosome 18 rearrangement. A variant of unknown significance (VUS) was found in LAMA1 (Laminin alpha-1, MIM 150320) is in trans of an interstitial deletion due to the complex chromosomal rearrangement which could partly explain the phenotype [11]. Individual 5 had a diagnosis of Beckwith Wiedemann syndrome with mosaic paternal uniparental disomy. In individual 11, because of the severity of the phenotype, a double diagnosis (genetic and a cytomegalovirus infection) was suspected. rES overturned this hypothesis.
rES impact in patient’s care
The diagnosis changed patients’ care in patients where rES was positive in four individuals. In individual 1 with a pathogenic variant in the potassium voltage-gated channel subfamily Q member 2, (KCNQ2, MIM 602235), antiepileptic drugs were switched to carbamazepine, rapidly stopping the seizures. For individual 2, as rES excluded a severe metabolic disorder, the ventricular septal defect was repaired. Individual 3 who had a myotubularin gene variant (MTM1, MIM 300415) had his muscular biopsy cancelled and was evaluated for a clinical trial of gene therapy (Clinical trial “ASPIRO”, NCT03199469). In individual 15, the WT1 variant (WT1 transcription factor, MIM 607102) explained the terminal renal failure. Because of the increased tumor risk, she underwent bilateral gonadectomy and nephrectomy with a diagnosis of nephroblastoma at pathological examination. Caryotype further revealed 46, XY chromosomes.
Negative results were also useful in three individuals. For individual 5, rES eliminated differential diagnoses so active care was continued as Beckwith Wiedemann syndrome generally has a good prognosis when properly managed [12]. For individuals 9 and 10, the negative results helped the pediatric team to offer invasive surgical procedures that would not have been considered had there been a severe genetic disease diagnosed by rES.
In individual 8 a VUS was found in ATP7A (ATPase Copper Transporting Alpha, MIM 300011), the gene responsible for Menkes syndrome. After review by the clinical genetics team and the molecular biologists, this variant was reported, because further biochemical testing was necessary with a potential treatment [13].
A VUS was also found in ABCD1 (ATP Binding Cassette Subfamily D Member 1, MIM 300371, c.896A > G;p.His299Arg) responsible for adrenoleukodystrophy [14] in individual 15. Very long fatty chain screening showed slightly increased C26 at 1.38 µmol/L (normal 0.3–1.2). Familial segregation showed that two maternal great-uncles carry the variant with normal very long fatty acid chain screening re-classifying this variant as likely benign, reassuring for her (male karyotype), and family counselling.
Discussion
This monocentric study shows the experience of a French regional university hospital, very close to what can be implemented in clinical routine care. rES was usefull in early diagnosis and medical care. In this study, 6 individuals (40%) received a diagnosis with rES. This yield is comparable to larger studies who have found a variable diagnostic yield (21–72% [15]). Some symptoms have remained unexplained: macroglossia in individual 1 and seizures in individual 7. Seizures have occasionally been reported in individuals with cardio-facio-cutaneous syndrome which is a RASopathy [16] but is not reported as a usual feature of Noonan syndrome including in individuals with LZTR1 pathogenic variants. A negative exome is usefull while discussing the possibility of palliative care: a poor genetic prognosis can direct towards palliative care and negative rES to continue active invasive care. The usefulness of a negative result has also been previously reported [6]. A positive result limits the number of diagnostic invasive tests and cost [17, 18].
Teams considering this strategy as a first-tier genetic test in critically ill children should implement a protocol for dealing with incidental findings of the ACMG list [19], heterozygosity for a genetic disorder with a high rate of heterozygosity in the general population, and VUS. In the case of VUS, further tests can be needed to specify the pathogenicity of the variant. VUS in individuals 8 and 15 highlights the difficulties arising with the uncertainty of the clinical consequences of VUS, the medical and familial stress generated by the subsequent delay of the additional tests. In the diagnostic clinical setting, teams should have a protocol for reporting VUS and incidental findings [19].
Limits to this study include its small size sample. Other studies have reported quicker turnaround time with the shortest of 3.3 days [6]. In their study, 86% (ultra-rES) and 78% (rES) of parents had the result before their child was discharged or died.
The rapid evolution of genetic sequencing technologies as well as the acceptability of costs involved by health services might be hurdles to a quick introduction of the latest sequencing technologies in routine patient care [6]. Concerning funding of rapid pan-genomic sequencing, rES reduces unnecessary, costly, and sometimes invasive procedures as well as the time to diagnosis. It also allows genetic counselling as well as proper care management of the child according to their diagnosed condition [18]. Parental acceptance is good with little adverse reactions [20]. Hence, the medical, psychological, and monetary benefits of rES outweigh the risks.
In conclusion, rES is feasible in critically ill infants in a French regional university hospital with a good diagnostic yield. It can be offered in individuals for whom a genetic condition is suspected where a diagnosis could help in their care. New studies show a possible switch to rGS to diagnose monogenic diseases but also chromosomal abnormalities. Sequencing technology and costs as well as evolution of data storage will make GS more accessible and may soon become the first-tier genetic test in critically ill children with suspected genetic disorders.
Data availability
All data generated or analyzed during this study are included in this published article.
References
Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med. 2012;4:154ra135–154ra135.
Willig LK, Petrikin JE, Smith LD, Saunders CJ, Thiffault I, Miller NA, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015;3:377–87.
Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 2017;171:e173438.
French CE, Delon I, Dolling H, Sanchis-Juan A, Shamardina O, Mégy K, et al. Whole genome sequencing reveals that genetic conditions are frequent in intensively ill children. Intensive Care Med. 2019;45:627–36.
Kingsmore SF, Cakici JA, Clark MM, Gaughran M, Feddock M, Batalov S, et al. A randomized, controlled trial of the analytic and diagnostic performance of singleton and trio, rapid genome and exome sequencing in ill infants. Am J Hum Genet. 2019;105:719–33.
Australian Genomics Health Alliance Acute Care Flagship. Feasibility of ultra-rapid exome sequencing in critically ill infants and children with suspected monogenic conditions in the Australian Public Health Care System. JAMA. 2020;323:2503–11.
Köhler S, Gargano M, Matentzoglu N, Carmody LC, Lewis-Smith D, Vasilevsky NA, et al. The human phenotype ontology in 2021. Nucleic Acids Res. 2021;49:D1207–17.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinforma. 2013;43:11.10.1–11.10.33.
Yauy K, Baux D, Pegeot H, Van Goethem C, Mathieu C, Guignard T, et al. MoBiDiC prioritization algorithm, a free, accessible, and efficient pipeline for single-nucleotide variant annotation and prioritization for next-generation sequencing routine molecular diagnosis. J Mol Diagn JMD. 2018;20:465–73.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med J Am Coll Med Genet. 2015;17:405–24.
Micalizzi A, Poretti A, Romani M, Ginevrino M, Mazza T, Aiello C, et al. Clinical, neuroradiological and molecular characterization of cerebellar dysplasia with cysts (Poretti–Boltshauser syndrome). Eur J Hum Genet. 2016;24:1262–7.
Shuman C, Beckwith JB, Weksberg R, Beckwith-Wiedemann Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, et al., editors. GeneReviews® . Seattle (WA): University of Washington, Seattle; 1993. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1394/.
Kaler SG, Holmes CS, Goldstein DS, Tang J, Godwin SC, Donsante A, et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med 2008;358:605–14.
Raymond GV, Moser AB, Fatemi A, X-Linked Adrenoleukodystrophy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, et al., editors. GeneReviews® . Seattle (WA): University of Washington, Seattle; 1993. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1315/.
Stark Z, Ellard S. Rapid genomic testing for critically ill children: time to become standard of care? Eur J Hum Genet EJHG. 2022;30:142–9.
Rauen KA, Cardiofaciocutaneous Syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, et al., editors. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1186/.
Dimmock D, Caylor S, Waldman B, Benson W, Ashburner C, Carmichael JL, et al. Project Baby Bear: Rapid precision care incorporating rWGS in 5 California children’s hospitals demonstrates improved clinical outcomes and reduced costs of care. Am J Hum Genet. 2021;108:1231–8.
Chung CCY, Leung GKC, Mak CCY, Fung JLF, Lee M, Pei SLC, et al. Rapid whole-exome sequencing facilitates precision medicine in paediatric rare disease patients and reduces healthcare costs. Lancet Reg Health-West Pac. 2020;1:100001.
Miller DT, Lee K, Gordon AS, Amendola LM, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med J Am Coll Med Genet. 2021;23:1391–8.
Cakici JA, Dimmock DP, Caylor SA, Gaughran M, Clarke C, Triplett C, et al. A prospective study of parental perceptions of rapid whole-genome and -exome sequencing among seriously ill infants. Am J Hum Genet. 2020;107:953–62.
Acknowledgements
We thank all the families for their involvement in the study. We also thank Dr Marie Christine Picot for her help in the methodology and Dr Florence Masson, Dr Floriane Hemery, Dr Odile Plan for their involvement in patient care.
Funding
Funding for this study was obtained thanks to internal research credits of the university hospital of Montpellier (“Appel d’offre interne jeune chercheur”).
Author information
Authors and Affiliations
Contributions
CFW, KY, GB, MBH and MW participated in drafting the manuscript and correction of the manuscript. GB, MT, MF, GC, TG, were involved in correction of the manuscript. PB, LP, CC, GC, JB, OP, MBH, CM, GC, RM, MD were involved in the curation of data. KY, OA, GB, NRP, DM, CW, MBH, VR, MT, KY, OA, TG DG and MW participated in the curation and interpretation data. MW, MD, DG were implicated in the grant applications.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was accepted by the ethical committee of Montpellier university hospital (CNRIPH 18.07.17.33927). All parents signed a consent to participate to the study for themselves and their child.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wells, C.F., Boursier, G., Yauy, K. et al. Rapid exome sequencing in critically ill infants: implementation in routine care from French regional hospital’s perspective. Eur J Hum Genet 30, 1076–1082 (2022). https://doi.org/10.1038/s41431-022-01133-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01133-7
This article is cited by
-
Role of next generation sequencing in diagnosis and management of critically ill children with suspected monogenic disorder
European Journal of Human Genetics (2024)
-
Rapid genomic sequencing for genetic disease diagnosis and therapy in intensive care units: a review
npj Genomic Medicine (2024)
-
2022: the year that was in the European Journal of Human Genetics
European Journal of Human Genetics (2023)
-
Guidelines, guidelines everywhere—and still I’m not sure what to do
European Journal of Human Genetics (2022)
