
Abstract
Purpose
As exome sequencing (ES) is increasingly used as a diagnostic tool, we aimed to compare ES with status quo genetic diagnostic workup for infants with suspected genetic disorders in terms of identifying diagnoses, survival, and cost of care.
Methods
We studied newborns and infants admitted to intensive care with a suspected genetic etiology within the first year of life at a US quaternary-referral children’s hospital over 5 years. In this propensity-matched cohort study using electronic medical record data, we compared patients who received ES as part of a diagnostic workup (ES cohort, n = 368) with clinically similar patients who did not receive ES (No-ES cohort, n = 368).
Results
Diagnostic yield (27.4% ES, 25.8% No-ES; p = 0.62) and 1-year survival (80.2% ES, 84.8% No-ES; p = 0.10) were no different between cohorts. ES cohort patients had higher cost of admission, diagnostic investigation, and genetic testing (all p < 0.01).
Conclusion
ES did not differ from status quo genetic testing collectively in terms of diagnostic yield or patient survival; however, it had high yield as a single test, led to complementary classes of diagnoses, and was associated with higher costs. Further work is needed to define the most efficient use of diagnostic ES for critically ill newborns and infants.
Similar content being viewed by others

INTRODUCTION
Evaluation of patients with suspected genetic disorders is important to pediatric and neonatology practice due to the incidence of single-gene disorders and chromosomal abnormalities that manifest at birth or soon after.1,2 A leading cause of mortality in infancy, genetic disorders afflict more than one quarter of level IV neonatal intensive care unit (NICU) patients who die before age 5.3,4 During the neonatal period, diagnostic workup for a suspected genetic etiology and genetic diagnoses are associated with longer hospital admissions that cost on average $77,000 more than those not associated with genetic disorders.5,6,7 Timely and accurate identification of a genetic diagnosis can impact a patient’s hospital course and health outcome.8,9
Given the prevalence of genetic conditions and resource intensive nature of NICUs, the clinical and economic impact of exome sequencing (ES) may be greatest in neonatal critical care.10,11 ES has demonstrated high diagnostic yield, and evidence of its clinical utility, while currently sparse, is building.12,13,14 Although clinical guidelines for ES application have not yet been developed due in part to a lack of robust utility data,15 clinical uptake is increasing.16,17 Real-world data to assess the impact of ES compared with the current standard genetic diagnostic workup, including chromosomal microarray (CMA) and targeted gene sequencing,18,19,20 on health outcomes and cost of care are extremely limited, especially within US health-care system context.21,22,23,24
We studied the clinical application of ES over more than 5 years at a large US children’s hospital. Variation in clinical practice allowed comparison of diagnostic approaches in contemporaneous cohorts of patients with similar clinical presentations. Primary outcome measures were diagnostic yield and survival, and we explored cost of care as a secondary outcome. The objectives of this study were to (1) describe a population of critically ill patients who had a suspected genetic etiology in the first year of life and underwent ES during diagnostic testing; and (2) compare outcomes for contemporaneous, clinically similar patients who did not have ES. We combined administrative and electronic medical record (EMR) data to analyze outcomes over the year following the initial inpatient genetics consultation.
MATERIALS AND METHODS
Study design
We examined ES use in patients with a suspected genetic etiology admitted to an intensive care unit (ICU) at Texas Children’s Hospital (TCH) within the first year of life. We employed a propensity-matched retrospective cohort study design with ES as the exposure factor to compare clinically similar patients who did (ES cohort) and did not have ES (No-ES cohort) as part of a diagnostic workup. Baylor Genetics (BG) laboratory performed all TCH ES. ES cohort patients had one of three ES forms: sequencing of the patient only (proband ES), sequencing of the patient and both parents (trio ES), or sequencing of the patient and both parents with expedited turnaround time (critical trio ES). Patients in the No-ES cohort had a status quo genetic diagnostic workup that did not include ES but may have included CMA, fluorescence in situ hybridization (FISH), single-gene or gene panel sequencing, deletion/duplication analysis, and methylation studies. A clinician may have ordered ES for patients in the No-ES cohort without it being performed for reasons such as lack of parental consent or cancellation by lab due to insufficient blood sample; we refer to these patients as the ES-recommended subgroup.
Documentation of an inpatient genetics consultation indicated suspected genetic etiology in the patient. We defined the index admission as the admission during which the first genetics consult was ordered and the index year as the 365 days after that consultation. The one-year time horizon allowed estimation of ES impact on outcomes even if results were not returned before index admission discharge. The Baylor College of Medicine Institutional Review Board approved this study.
Identification of eligible patients
We combined hospital and diagnostic laboratory data to identify eligible patients who met the following criteria: (1) ICU admission within the first year of life, (2) inpatient genetics consultation documented in EMR, (3) admission and genetics consultation occurred between 1 December 2011 and 30 June 2017, and (4) no definitive clinical diagnosis. The ES cohort included all eligible patients who also had ES in the first year of life. To construct the No-ES cohort, each patient in the ES cohort was propensity score matched to a patient who did not receive ES. Propensity scores were calculated based on clinical characteristics and Human Phenotype Ontology (HPO) terms generated from the text of the genetics consult note (Supplementary Materials and Methods). Our identification strategy assumes that in the absence of clinical guidelines for ES application, ordering behavior of the consulting geneticist determined, at least in part, whether a given patient had ES instead of status quo genetic testing. Variability in the number of consults in which ES was ordered as a proportion of total consults by an attending geneticist supports this assumption (Figs. S1–S2).
The study team (H.S.S.) reviewed EMRs and abstracted information from administrative and clinical notes on admission characteristics, demographics, consultation, and clinical outcomes. EMR review was completed in January 2019 such that a minimum of one year of clinical follow-up data was available for each patient. We obtained hospital administrative data on diagnostic-related investigations and costs for each included patient.
Measurement and comparison of outcomes
Primary outcomes were establishment of a molecular diagnosis and survival. Molecular diagnosis was defined as identification of a specific genetic change interpreted as causal or probably causal of the patient’s clinical presentation. Results of all genetic tests ordered in the year following initial genetics consultation were individually reviewed and interpretation was verified in clinical notes. We considered ES cases as diagnosed if reported by the laboratory as “solved” or “probably solved.” We tallied changes in medical management of ES patients based on documentation at the time of ES return of results, regardless of when result return occurred.
Statistical analysis
We calculated descriptive statistics on patient and index admission characteristics. We tested for differences between cohorts using chi-square and Wilcoxon rank-sum tests and for differences in outcomes by ES form using analysis of variance (ANOVA). All hypothesis tests were two-sided and used an alpha level of 0.05. We produced Kaplan–Meier survival curves to visualize survival to 28 days and one year of age. We used logistic regression to estimate impact of ES on odds of diagnosis and of being alive at 28 days and one year.
We conducted exploratory cost analyses from the hospital’s perspective over the timeframe of the index admission and the index year for total, diagnostic pathway (Supplementary Materials and Methods), and genetic test costs. We performed post hoc subgroup analyses for the ES-recommended group and patients with the same ultimate diagnosis. To account for skewed cost distribution, we used nonparametric tests of comparison and natural log of cost as the dependent variable in ordinary least squares regression. All costs were adjusted to 2017 USD using the Consumer Price Index for All Urban Consumers (CPI-U). Data analysis was performed using Stata/IC 15 (StataCorp, College Station, TX).
RESULTS
Outcomes
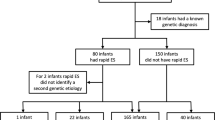
A total of 368 patients comprised the ES cohort. Of 936 eligible patients who did not have ES, 368 were matched to ES patients to comprise the No-ES cohort (Fig. 1). Propensity score matching successfully reduced differences in covariates between ES and No-ES patients such that the ES and No-ES cohorts had similar distributions for patient characteristics (Table 1). Patients represented a diverse population with home addresses in 361 unique zip codes from across the United States. Overall, more patients were male, non-Hispanic, and white. The majority of patients were in the NICU for the initial genetics consultation, which most frequently took place within the first quarter year of life. ES patients had longer length of stay (LOS) than No-ES patients (Table 1, p = 0.008; Fig. S3). Form of ES was proband for 227 (61.7%) patients, trio for 54 (14.7%) patients, and critical trio for 87 (23.6%) patients. ES results were returned before discharge from the index admission for 107 (29.1%) patients.

ES exome sequencing, EMR electronic medical record, HPO Human Phenotype Ontology, MRN medical record number.
Diagnostic yield and survival
Overall, 205 of 736 (27.9%) patients received a molecular diagnosis from any genetic test (Table 2). Yield of ES alone in the ES cohort (27.4%) was no different from collective yield of other modalities in the No-ES cohort (25.8%; p = 0.62). Most ES cohort patients (328, 89.1%) also had a CMA, and nine patients with nondiagnostic ES were diagnosed by CMA (n = 8) and cSNP (n = 1). Diagnoses and tests with which they were identified are listed in Tables S1–S2. In the No-ES cohort, 71.6% (68/95) of diagnoses were made via chromosomal analyses (CMA, FISH, and karyotype).
Hispanic patients had 41.0% lower odds of diagnosis than non-Hispanic patients (Table S3, p = 0.003), and in the ES cohort only, odds of diagnosis for Hispanic patients were approximately half that for non-Hispanic patients (Table S4, p = 0.01). By form of ES, there was no difference in diagnostic yield or survival (Table S5). Diagnostic yield was lower in the ES-recommended subgroup (2/36, 5.6%, p = 0.004) compared with the ES cohort (Table S6).
A geneticist recommended at least one change in clinical care based on ES results for 22% of patients in the ES cohort, including those diagnosed (n = 49) and undiagnosed by ES (n = 32; Table S7). In undiagnosed patients, subspecialist referrals (n = 19) and screening recommendations (n = 10) accounted for most management changes. There was no difference in mean age at death between cohorts. The proportion of patients alive at 28 days (94.8% ES cohort; 91.6% No-ES cohort, p = 0.08, Table 2; Table S8 adjusted odds ratio [OR] = 2.42, p = 0.11) and at one year (80.2% ES cohort; 84.8% No-ES cohort, p = 0.099; Table S9 adjusted OR = 1.07, p = 0.85) was not significantly different between cohorts. However, based on visual inspection of the survival curves in Fig. 2a, deaths tended to occur earlier in the No-ES cohort while the death rate was steadier in the ES cohort. Diagnosed patients in both cohorts had the lowest survival compared with patients who remained undiagnosed (Fig. 2b). Among hospital units, survival was lowest among patients in the pediatric intensive care unit (Fig. S4).

(a) illustrates survival in the ES cohort (red) and the No-ES cohort (blue). (b) illustrates survival of ES cohort patients who received a diagnosis (blue) and did not receive a diagnosis (red), alongside survival of No-ES cohort patients who received a diagnosis (green) and did not receive a diagnosis (orange). ES exome sequencing. Lines at 28 days and 100 days represent benchmarks for comparisons with other studies.
Costs
Mean index admission total, diagnostic pathway, and genetic test costs were higher in the ES cohort than No-ES cohort (Table 3; Fig. S5). The same was true for costs during the index year (Fig. S6). Mean cost of the diagnostic pathway per diagnosis made was $60,869 (95% bootstrapped confidence interval [CI] $49,749–$71,953) for the ES cohort and $24,763 ($19,956–$29,569) for the No-ES cohort. Controlling for length of stay and other features of hospitalization, ES was associated with approximately 17.4% higher total cost of admission (p < 0.001, Table S10). Total cost of genetic tests during the index admission and year differed between groups of patients who had different forms of ES (Table S11). Diagnoses of four conditions in 23 patients were made in both cohorts (Table S12): CHARGE syndrome (ES = 3, No-ES = 4), Noonan syndrome (ES = 9, No-ES = 3), Walker–Warburg syndrome (ES = 1, No-ES = 1), and Gaucher disease (ES = 1, No-ES = 1).
DISCUSSION
We studied critically ill newborns and infants with suspected genetic disease to compare outcomes for patients who did and did not have ES as part of a diagnostic workup. Genetic testing identified a diagnosis in 27.9% of patients overall. As an individual test to detect monogenic disorders, ES demonstrated high diagnostic yield that was similar to that of the status quo toolbox of genetic modalities collectively. Even in the context of matched phenotypes, however, we observed a high incidence of chromosomal abnormalities detected by CMA. These findings highlight the importance of continuing to assess ES alongside array-based tools for patients with severe clinical presentation in the first year of life.
Odds of being alive at 28 days and one year of age were not significantly different between cohorts. Survival was lower among diagnosed patients in both cohorts compared with those who remained undiagnosed, which is consistent with other studies of genomic sequencing in the NICU.9 Between cohorts, our finding of lower odds of one-year survival in the ES cohort is unsurprising. It may indicate the severity of diagnosed conditions and lack of available treatments and therapeutics for those conditions. Our findings regarding timing of death, however, are somewhat unexpected. Sequencing has been predicted to increase 28-day mortality, driven by redirection of care to comfort care only following confirmation of severe, untreatable disease.10 We find No-ES patients died sooner after birth than ES patients, likely driven by diagnoses of chromosomal abnormalities not compatible with survival.
A strength of our study is that it assesses effectiveness of ES using real-world data from application in a clinical setting, rather than efficacy of ES in a controlled environment as evaluated in most prior studies.25 Our approach enabled us to study a large patient population over more than five years, ensure follow-up time of at least one year over which to measure outcomes, and compare groups of patients with similar observable clinical characteristics. Although others have suggested propensity score matching,26 this is the first analysis, to our knowledge, to use this approach to select phenotypically matched controls who were contemporaneously hospitalized to draw comparisons against a counterfactual cohort of patients. After matching, observed patient characteristics were well-balanced between the two cohorts.
The lack of guidelines regarding a standard position of ES in the diagnostic pathway leaves the workup for a suspected genetic disorder largely at the discretion of geneticists caring for a particular patient. Observed variability in ES uptake at the clinician level supports our assumption that whether or not ES was ordered for a given patient may be due, in part, to propensity of the attending geneticist to order ES. It prevents us, however, from being able to draw conclusions about optimal placement of ES within the diagnostic pathway. Guideline development could be advanced by defining categories of patients most likely to benefit clinically from ES. This would require analysis of more nuanced details than we were able to capture retrospectively through HPO terms. In a clinical area with such rapid evolution of technology as genetics has seen over the past decade, conducting timely analyses to inform practice guidelines and educate clinicians is a challenge.
While CMA is first-tier diagnostic standard care and is therefore an appropriate status quo comparator to measure the value of ES,11 the diagnoses made in each cohort were qualitatively different based on the types of changes identifiable by tests used in each group (i.e., structural changes in the No-ES cohort versus single-gene sequence variants in the ES cohort). The importance of both CMA and ES in contributing to nonoverlapping diagnoses may point to the eventual implementation of genome sequencing for both detecting structural changes and identifying Mendelian diagnoses that ES cannot.27 The ES-recommended subgroup that did not have ES due to external factors had lower diagnostic yield than in the ES cohort. This finding supports the hypothesis that ES might be most cost-effective when applied in patients who are difficult or impossible to diagnose with other modalities.28,29 Amid increased emphasis on addressing the limited diversity of ethnic group participation in genomic research,30 our finding that Hispanic patients had half the odds of diagnosis compared with non-Hispanic patients in the ES cohort suggests further study is warranted.
Total costs of care in the ES cohort were higher than the No-ES cohort. Because use of ES was a clinical decision, albeit one with variability, we are not able to draw conclusions about cost-effectiveness. Reduction of number of days in the NICU has driven some of the largest estimated cost savings in hypothetical counterfactual cases when rapid sequencing results are returned while patients are admitted (median 23 days from order).8 ES patients had a longer LOS than No-ES patients and the majority did not have ES results returned during the index admission, meaning there was little potential for impact on care provision during the index admission although result return occurred within the expected turnaround time. We analyzed a one-year follow-up period to capture potential impact of return of results on longer-term care. If knowledge of ES information impacted care so as to avert costs that would otherwise have accrued in the future, then we would expect the within-cohort difference between the cost of the index admission and the cost of the index year to be smaller for the ES cohort than the No-ES cohort. Instead, the ES cohort had a greater increase in cost between index admission and index year. Further examination of the drivers of costs in both populations is needed.
Several limitations deserve mention. We were not able to account for deaths not recorded in the EMR or care received at other institutions, meaning that we potentially underestimate the cost of the index year. We recognize potential indication bias and cannot rule out the possibility that ES was applied in overall more complex patients. There may be features relevant to clinician decision-making and outcomes for which we are unable to control. Although we tried to capture important clinical and phenotypic qualities of patients, our matching process relied on documentation in the EMR, and individual HPO terms may not capture the full range of features that an astute geneticist would detect upon clinical exam. Relatedly, HPO term generation depended upon phenotypic description in clinical notes, which differed from patient to patient in detail and extent.
This study focused on diagnostic yield as a measure of clinical utility. While diagnostic yield is the most commonly used outcome measure in sequencing studies,23 it is not a complete measure of clinical utility and does not capture the broad range of health and nonhealth outcomes that may arise from ES for the patient and family.31,32 Apart from clinical implications, families value genetic information for personal purposes such as reassurance regarding a transition to comfort care only measures, help with coping, risk information for other family members, and reproductive planning.33,34,35,36,37,38 Clinicians also perceive a broad range of outcomes to be important results of ES.39 There is an immediate need for more systematic outcomes measurement, along with valuation of various outcomes, to advance health services research and economic evaluation of genomic sequencing.23,24,40 Future work should incorporate family preferences with clinical outcomes to move toward a more holistic evaluation of the utility of genomic sequencing.
Conclusion
In a broad population of newborns and infants with suspected genetic disease, ES demonstrated important diagnostic utility. Due to the contribution of large chromosome structural rearrangements and gene copy-number variations present in this population, status quo genetic diagnostics including chromosomal analyses identified a diagnosis in a similar percentage of patients and were associated with lower costs. Further work is needed to define the most efficient use of ES in a genetic diagnostic pathway for critically ill newborns and infants.
References
Lu X-Y, Phung MT, Shaw CA, et al. Genomic imbalances in neonates with birth defects: high detection rates by using chromosomal microarray analysis. Pediatrics. 2008;122:1310–1318.
Baird PA, Anderson TW, Newcombe HB, Lowry RB. Genetic disorders in children and young adults: a population study. Am J Hum Genet. 1988;42:677–693.
Xu J, Murphy SL, Kochanek KD, Bastian B, Arias E. Deaths: final data for 2016. Natl Vital Stat Rep. 2018;67:1–76.
Wojcik MH, Schwartz TS, Yamin I, et al. Genetic disorders and mortality in infancy and early childhood: delayed diagnoses and missed opportunities. Genet Med. 2018;20:1396–1404.
Gonzaludo N, Belmont JW, Gainullin VG, Taft RJ. Estimating the burden and economic impact of pediatric genetic disease. Genet Med. 2019;21:1781–1789.
McCandless SE, Brunger JW, Cassidy SB. The burden of genetic disease on inpatient care in a children’s hospital. Am J Hum Genet. 2004;74:121–127.
Marshall DA, Benchimol EI, MacKenzie A, et al. Direct health-care costs for children diagnosed with genetic diseases are significantly higher than for children with other chronic diseases. Genet Med. 2019;21:1049–1057.
Farnaes L, Hildreth A, Sweeney NM, et al. Rapid whole-genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genomic Medicine. 2018;3:10.
Willig LK, Petrikin JE, Smith LD, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015;3:377–387.
Petrikin JE, Willig LK, Smith LD, Kingsmore SF. Rapid whole genome sequencing and precision neonatology. Semin Perinatol. 2015;39:623–631.
Friedman JM, Bombard Y, Cornel MC, et al. Genome-wide sequencing in acutely ill infants: genomic medicine’s critical application? Genet Med. 2019;21:498–504.
Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–1511.
Meng L, Pammi M, Saronwala A, et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr. 2017;171:e173438.
Soden SE, Saunders CJ, Willig LK, et al. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med. 2014;6:265ra168.
Burke W, Clayton EW, Wolf SM, et al. Improving recommendations for genomic medicine: building an evolutionary process from clinical practice advisory documents to guidelines. Genet Med. 2019;21:2431–2438.
Phillips KA, Deverka PA, Marshall DA, et al. Methodological issues in assessing the economic value of next-generation sequencing tests: many challenges and not enough solutions. Value Health. 2018;21:1033–1042.
Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet. 2018;19:253.
Manning M, Hudgins L. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet Med. 2010;12:742.
Lalani SR. Current genetic testing tools in neonatal medicine. Pediatr Neonatol. 2017;58:111–121.
Malam F, Hartley T, Gillespie MK, et al. Benchmarking outcomes in the neonatal intensive care unit: cytogenetic and molecular diagnostic rates in a retrospective cohort. Am J Med Genet A. 2017;173:1839–1847.
Stark Z, Schofield D, Alam K, et al. Prospective comparison of the cost-effectiveness of clinical whole-exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genet Med. 2017;19:867–874.
Dragojlovic N, Elliott AM, Adam S, et al. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genet Med. 2018;20:1013–1021.
Smith HS, Swint JM, Lalani SR, et al. Clinical application of genome and exome sequencing as a diagnostic tool for pediatric patients: a scoping review of the literature. Genet Med. 2019;21:3–16.
Schwarze K, Buchanan J, Taylor JC, Wordsworth S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet Med. 2018;20:1122–1130.
Franklin JM, Glynn RJ, Martin D, Schneeweiss S. Evaluating the use of nonrandomized real‐world data analyses for regulatory decision making. Clin Pharmacol Ther. 2019;105:867–877.
Wordsworth S, Doble B, Payne K, et al. Using “big data” in the cost-effectiveness analysis of next-generation sequencing technologies: challenges and potential solutions. Value Health. 2018;21:1048–1053.
Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2017;20:435.
Johnston J, Lantos JD, Goldenberg A, et al. Sequencing newborns: a call for nuanced use of genomic technologies. Hastings Cent Rep. 2018;48:S2–S6.
van der Sluijs PJ, Aten E, Barge-Schaapveld DQ, et al. Putting genome-wide sequencing in neonates into perspective. Genet Med. 2019;21:1074.
Landry LG, Ali N, Williams DR, Rehm HL, Bonham VL. Lack of diversity in genomic databases is a barrier to translating precision medicine research into practice. Health Aff (Millwood). 2018;37:780–785.
ACMG Board of Directors. Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2015;17:505–507.
Regier DA, Weymann D, Buchanan J, Marshall DA, Wordsworth S. Valuation of health and nonhealth outcomes from next-generation sequencing: approaches, challenges, and solutions. Value Health. 2018;21:1043–1047.
Makela NL, Birch PH, Friedman JM, Marra CA. Parental perceived value of a diagnosis for intellectual disability (ID): a qualitative comparison of families with and without a diagnosis for their child’s ID. Am J Med Genet A. 2009;149A:2393–2402.
Carmichael N, Tsipis J, Windmueller G, Mandel L, Estrella E. “Is it going to hurt?”: the impact of the diagnostic odyssey on children and their families. J Genet Couns. 2015;24:325–335.
Tarini BA, Singer D, Clark SJ, Davis MM. Parents’ interest in predictive genetic testing for their children when a disease has no treatment. Pediatrics. 2009;124:e432–e438.
Pereira S, Robinson JO, Gutierrez AM, et al. Perceived benefits, risks, and utility of newborn genomic sequencing in the BabySeq Project. Pediatrics. 2019;143 Suppl 1:S6–S13.
Kohler JN, Turbitt E, Lewis KL, et al. Defining personal utility in genomics: a Delphi study. Clin Genet. 2017;92:290–297.
Lewis MA, Stine A, Paquin RS, et al. Parental preferences toward genomic sequencing for non-medically actionable conditions in children: a discrete-choice experiment. Genet Med. 2018;20:181–189.
Smith HS, Russell HV, Lee BH, Morain SR. Using the Delphi method to identify clinicians’ perceived importance of pediatric exome sequencing results. Genet Med. 2020;22:69–76.
Payne K, Nicholls S, McAllister M, MacLeod R, Donnai D, Davies LM. Outcome measurement in clinical genetics services: a systematic review of validated measures. Value Health. 2008;11:497–508.
Acknowledgements
At the time of this research, HSS was a Research Associate in the Department of Molecular and Human Genetics at Baylor College of Medicine. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from Baylor Genetics.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Smith, H.S., Swint, J.M., Lalani, S.R. et al. Exome sequencing compared with standard genetic tests for critically ill infants with suspected genetic conditions. Genet Med 22, 1303–1310 (2020). https://doi.org/10.1038/s41436-020-0798-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0798-1
Keywords
This article is cited by
-
Rapid genomic sequencing for genetic disease diagnosis and therapy in intensive care units: a review
npj Genomic Medicine (2024)
-
Genomic testing and molecular diagnosis among infants with congenital heart disease in the neonatal intensive care unit
Journal of Perinatology (2024)
-
Genomic medicine in neonatal care: progress and challenges
European Journal of Human Genetics (2023)
-
Role of genomic medicine and implementing equitable access for critically ill infants in neonatal intensive care units
Journal of Perinatology (2023)
-
Rapid exome sequencing as a first-tier test in neonates with suspected genetic disorder: results of a prospective multicenter clinical utility study in the Netherlands
European Journal of Pediatrics (2023)
