
Abstract
Somatic gene editing (SGE) holds great promise for making genetic therapy possible for many monogenic conditions very soon. Is our current system of European market authorization and reimbursement ready for the expected tsunami of gene therapies? At a recent workshop of the Netherlands ZonMw consortium on ethical, legal, and social implications of personalized medicine, we discussed the current possibilities for bringing new gene therapies to the clinic. In Europe, it is not yet clear whether the route via the European medicines agency as an advanced therapy medicinal product is the most appropriate for evaluation of highly personalized SGE applications, although this may optimally guarantee safety and effectiveness. Compassionate use may ensure faster access than the centralized procedure but does not stimulate the commercial development of products. Prescription to named patients may only provide adequate access for single patients. Temporary authorization of use may allow access to medication half a year before formal market authorization has been granted, but may also have large budget impacts. Magistral compounding under a hospital exemption may be an attractive solution for rare, tailor-made applications at an acceptable price. To approve local experimental use of a therapy on a case-by-case basis may be fast, but does not guarantee optimal safety, effectiveness, and broad implementation. We argue that alternative routes should be considered for products developed for a market of large groups of patients versus unique personalized treatments. A balance between scientific evidence for safety and effectiveness, affordability, and fast access may demand a range of alternative solutions.
Similar content being viewed by others

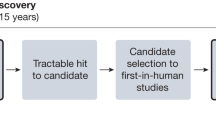
Introduction
In the past, monogenic disorders were considered incurable and few treatment options were available. The emergence of somatic gene therapy (SGT) has created new hopes for treatment and might enable life-saving cures [1,2,3]. While current gene therapies deliver an additional DNA product in the cell, recently developed gene-editing tools, such as clustered regularly interspaced short palindromic repeats–CRISPR-associated proteins (CRISPR-Cas), have the potential to truly repair disease-causing variants. Somatic gene editing (SGE) could theoretically be used to repair thousands of different variants in thousands of genes in a highly personalized way.
The clinical potential of genome editing depends on safety, effectiveness, and delivery, but also on navigating the road to market: legal regulation and authorization, distribution, availability, costs, and reimbursement. For different parts of the world, market authorization and availability of treatment are governed by different legislative systems, for example, the food and drug administration (FDA) in the USA and the European medicines agency (EMA) in the European Union (EU) [4]. These systems aim to regulate treatments as products, while for genome editing it may be needed to optimize the clinical procedure, both for ex vivo and in vivo editing. This would include the use of tailored guide-RNAs, certified laboratory equipment, and trained staff.
SGE does not neatly fall into any clear category for regulation by current legislative systems. Since monogenic disorders are rare, the number of patients needing specific treatments may be very small, and therefore the commercial development of a product may not be very attractive for drug manufacturers. Financial incentives already exist for orphan medicinal products [5]. However, due to the relatively small market, costly development, and market exclusivity, the price of some orphan drugs is still extremely high, limiting the affordability and thus available for patients.
Doudna et al. state that per-patient cost and fair access of gene therapies should be addressed not solely for application in patients with rare diseases, but also with consideration of the possibility that sometimes a standardized product may be suitable for a large, international population [1]. For example, a CRISPR-Cas product that is now under development for sickle cell disease and beta-thalassemia (CTX001) might serve a relatively large number of patients [6]. Would this CRISPR-Cas-based gene therapy become available globally in a fast and affordable manner?
Gupta strategists estimated that the average R&D costs per new molecular entity for an orphan disease could be around 0.5 bln USD [7]. More than 7% of total pharmaceutical expenditure in 2017 was for orphan medicinal products, and this percentage has not stopped increasing since [8]. This indicates a shift towards expenditure in higher cost, lower volume patient populations, and a shift in drug development towards more specialized targeting of diseases for higher unmet needs [8].
EMA’s Committee for medicinal products for human use lists six advanced therapy medicinal products (ATMPs) on its list of applications for new human medicines for 2021 [9]. Clinicaltrials.gov, however, lists 4702 studies for “gene therapy” as of February 2021. The expected quick rise in applications for gene editing, in the face of possible patch-work regulation by member states and hurdles to accessing, leads to the following questions: is the current system of market authorization and reimbursement in Europe ready for the potential tsunami of gene therapies? How can patient access to gene therapy be ensured in the years to come? In brief, there is friction between the current legislative approach to gene therapy as a medicinal product and the clinical approach to gene therapy as a personalized procedure to cure the patient (Fig. 1).

The flow emphasizes which aspects of quality control might need optimization. Aspects include the selection of tailored products (i.e., guide RNAs for the specific mutation of the patient in combination with a template for repair); certified laboratory equipment; and well-trained staff working according to clinical protocols.
The consortium ethical, legal, and social issues of personalized medicine (ELSI-PM) organized a workshop on the fifth of March 2020 to discuss potential ways forward. ELSI-PM is supported by ZonMw (the Netherlands Organization for Health Research and Development).
Using input from the workshop, we will briefly describe the current situation of market approval for SGT, including SGE, in Europe via the EMA. Next, we will discuss the pros and cons of potential alternative routes for patient access, market authorization, and reimbursement in the EU for SGE specifically, including a few examples of accelerated patient access to very innovative treatments. We follow the order starting with the regular European route to national to local solutions, ending with very personal (and therefore local) solutions.
Market approval as ATMPs through the EMA
When a new therapy is being developed, several steps must be taken before patients can receive it as part of regular health care. Product developers typically submit a dossier to the EMA, which carries out a thorough assessment of the data including safety, efficacy, and legislative requirements before granting market authorization. Whereas on the one hand, this protects patients from adverse effects, on the other hand, the development and evaluation of the dossier takes significant time and resources and may delay patient access to effective drugs for unmet medical needs. In Europe, special legislation is in place for ATMPs. ATMPs benefit from a single evaluation and authorization procedure for all member states and include biological medicines such as gene therapy medicinal products (GTMPs) [10]. Note, however, that it has been questioned whether CRISPR-Cas therapies would be covered by European legislation for GTMPs, which are defined as consisting of “recombinant nucleic acid” since the DNA alterations in CRISPR-Cas9 gene therapy products are not produced by recombinant DNA technology [11].
Market approval is not only labor-intensive for both the pharmaceutical company and the regulatory agency, it can also come with specific requirements for the products after approval such as risk-mitigating measures. The EMA’s authorization of an ATMP is valid in the entire EU jurisdiction, suggesting broad accessibility. Yet, to ensure that the new technology is offered as high-quality care, it may be desirable to centralize expertise and implement it in a limited number of centers. For example, the market authorization of Yescarta™, a CAR T-cell product, stipulates that prior to the launch of the product in each member state, the company must make arrangements with the national competent authority about educational materials, training, and a guidance document for health care providers and an educational program for patients to explain the risks, such as cytokine storms, requiring that they remain in the proximity of the location where they received the infusion for at least 4 weeks [12]. At the same time, these extra risk-mitigating measures which require action at the national level effectively limit the number of centers where the new product can be administered. Besides the EMA’s stipulations, national reimbursement authorities may also want to limit the number of treatment centers in order to foster expertise [13]. Consensus on and organization of such a center of expertise might further extend the time it takes before patients have access to treatment. Furthermore, the price of some of these products is extremely high, limiting the affordability and thus available for patients. An example of an ATMP, designated as an orphan gene therapy product that was conditionally approved by EMA in 2020, is Zolgensma™. At $2.1 million, it is the most expensive drug worldwide [14, 15]. In general, annual treatment cost per non-oncological rare disease patient may be ~300 k€, much higher than often cited thresholds for cost-effectiveness, but the budgetary impact still is limited [16].
While CRISPR-Cas is “efficient, simple and cheap” according to some, a labor-intensive route for marketing authorization of orphan drugs for thousands of variants in thousands of genes may lead to unnecessary delays, and thus treatments may effectively not be affordable and accessible [14]. A summary of characteristics for the ATMP/EMA route and alternative routes to market access is provided in Table 1. The following sections will further elaborate on the pros and cons of the different alternative routes.
Compassionate use
In the period before a market authorization is granted, patients can sometimes get access to a product via a compassionate use program. The committee for medicinal products for human use of the EMA has the legal authority to advise national agencies to permit compassionate use programs, which are valid for one year. Compassionate use programs provide medicines to patients “free of costs”, for example after they enrolled in a clinical trial. This brings about a societal quid pro quo. The company is able to build a dossier for the EMA on the basis of a clinical trial. Afterward, patients who benefitted from the treatment retain access to the therapy, and the company benefits from publicity and “customer loyalty”, while the medicine is not yet generating income. Compassionate use designation might effectively bridge the time between a clinical trial and a product entering the market. However, it does not incentivize early or large-scale production and access.
At the Dutch ELSI-PM workshop, a particular problem for pharmaceutical companies was mentioned, namely that consistent manufacturing of large batches of the product cannot be guaranteed in this phase when the product is not yet generating income. Thus companies may have limited products available for patients outside clinical trials. Selecting countries for compassionate use programs and then selecting patients eligible for these products can be a challenge. Therefore, a company recently provided some doses for free to children with spinal muscular atrophy who were selected by lottery [17]. However, the perceived inequity is ethically problematic [18].
Named patients (doctor’s declaration)
Like compassionate use, named-patient prescriptions, sometimes also referred to as doctor’s declaration, exist for medicines that are not marketed for the specific indication in the country. In the Netherlands, this route is often used for rare diseases, though not for ATMPs. There are two disadvantages: the permission is only valid for an individual, and medicines provided via doctor’s declaration are not necessarily reimbursed [19].
Temporary authorization for use (ATU)
In France, the ATU system encompasses aspects of both compassionate use and named-patient use, but with a different system of reimbursement prior to market approval [20]. During the ATU validity period, products are entirely reimbursed by the French National Health Insurance, potentially having a large budget impact on the French healthcare system [21]. Manufacturers are required to pay back the total revenue difference between ATU pricing and the final negotiated price after market access in the case of a lower post-launch price. However, in the opposite case, manufacturers are compensated for every sold product to the level of the determined final product price [22]. For SGT, the ATU system may provide an incentive for early patient access to innovative medication during the market approval procedure.
Local institutional review board approval
Some innovative gene therapies have been provided in a research setting after local institutional review board (IRB) approval. In Spain, for example, local IRB approval was given for the application of hematopoietic gene therapy for four Fanconi anemia patients [23]. Similarly, Kim et al. performed an “N-of-1” study for the development of milasen—a patient-customized oligonucleotide therapy—for the treatment of a life-threatening form of Batten’s disease [24]. Financing for the production of milasen was crowd-funded. This raises questions of equity and sustainability: will (the parents of) every patient with a rare disease be able to raise funding for his own treatment? Would society consider this funding model fair and just?
The IRB route has clear ties to the framework of research. On the one hand, the examples illustrate fast translation to patient care. On the other hand, this is not a sustainable solution to guarantee safety and effectiveness for all patients eligible for such treatments. In a large-scale clinical framework, it may not be feasible to assess effectiveness and safety per patient. Furthermore, the experience needed for such assessment may be better guaranteed by (inter)national agencies.
Compounding under a hospital exemption
Compounding by (hospital) pharmacies has been historically used to tailor dose and form of otherwise standardized pharmaceutical products, e.g., if a patient cannot swallow a pill the pharmacist may provide the drug as a liquid. When novel expensive treatments such as SGE become available, a possibility may be to compound the treatment in hospital pharmacies at a significantly lower price. Recent legislation in the Netherlands has established that a compounded drug may also be reimbursed if it is equivalent to a registered product that was deemed to be effective but not cost-effective or if its budget impact is too high [25].
“Hospital exemption” is a similar pathway specific for treatments that meet the criteria for ATMPs. The ATMP must be produced on prescription and it must be made to order “for a specific patient”. In fact, production and dispensing for up to a maximum of five patients is considered production “for a specific patient”. The ATMP must be used under the exclusive professional responsibility of a physician. There are also procedural rules for the dispensing pharmacist. Permission to produce an ATMP under hospital exemption can only be granted if the ATMP is produced and used in the same country. A system for pharmacovigilance (drug safety) is required for all ATMPs, including those which are produced under hospital exemption. A disadvantage of the hospital exemption route is that there is currently a lack of overall transparency. If treatments are carried out under hospital exemption but outside the scope of clinical trials, the data are not publicly available. A group of European research universities has called for a voluntary, open-access registry of hospital exemption uses. This would facilitate access to strategies and data, provide evidence on (in)efficacy of treatments, and help identify which treatment-related costs need to be considered for reimbursement [26].
Combining European regulation and local oversight: the Bio-Nespresso model?
Some gene editing with CRISPR-Cas may serve large patient numbers, and products may be brought to the market via the main EMA route for ATMPs. However, ultra-rare disease patients with rare gene variants may need personalized treatments, for which regimens similar to compounding and hospital exemption may be more suitable. A central question is how to ensure fast and equitable access while simultaneously guaranteeing optimal safety and effectiveness and reimbursement of costs of development and treatment.
Some medical treatments such as stem cell transplantation and surgery are regulated as health services, which require adequately trained personnel, monitoring, and evaluating results according to standard operational procedures. Quality control is organized at the level of the health service. CRISPR-Cas-based repair of specific disease-causing variants could be done in centers experienced in stem cell transplantation as well as proceedings related to CRISPR-Cas: drawing blood, isolating stem cells, modifying these cells ex vivo, and transfusing them back to the individual patient. The dispensing might resemble compounding. Such a model could circumvent many of the financial challenges of precision medicine. Also, bedside development under hospital exemption could be an affordable, safe and flexible regulatory context for CRISPR-Cas-based treatments. Could CRISPR-Cas as a technique be regulated as an ATMP, with individual products being prepared at the local hospital? Currently, this is not an option under EU Regulation 1394/2007.
A relevant analogy is the concept of a “Bio-Nespresso” machine for monoclonal antibodies, which do not fall under the ATMP regulation [27]. Its design goal is small-scale, timely production of monoclonal antibodies, according to the clinical needs of individual patients. It has been envisioned for the hospital pharmacy setting and would thus fall under the paradigm of compounding “which does not have to comply with the regulations commercially produced medicine have to” [27]. Similarly, if production-specific supplies are available in the hospital pharmacy or CRISPR-Cas facility, a tailored solution for every patient could be prepared at a “small-scale manufacturing unit for personalized medicine production” [27]. These therapies would have to be produced under good manufacturing practice (GMP) using standardized protocols that have to be adapted for each specific variant causing a specific genetic condition with controls and tests adapted to the individual patient and/or product. The system to guarantee the safety of these techniques at the CRISPR-facility remains to be developed. Novel oversight mechanisms are also needed for injections of guide-RNA, Cas-protein, and template, or if cells modified ex vivo by personalized CRISPR were given back to the patient.
Towards coverage by basic health care insurance
Apart from having market access, SGE products also need to be financially accessible. Preferably, a funding model would also support the implementation of successful research findings into healthcare. As stated earlier, compassionate use is paid for by manufacturers. Named-patient use and compounded products may be covered by health insurers but at their discretion. Perhaps most relevant for CRISPR-Cas is an example of how commercial high-cost medicines are processed in the Netherlands. Most in-hospital treatments (including medicines) are automatically covered by the basic healthcare insurance package, without governmental assessment for relative effectiveness. But if a commercially produced medicine is at risk for high-budget impact of questionable cost-effectiveness, the Minister of Health can place it in the so-called “waiting room” or “sluice”. Such products can only be accepted into the insured package after assessment of its efficacy, and if the Ministry, after negotiations, can arrive at a reasonable financial arrangement with the supplier [28]. It is likely that commercially produced CRISPR-Cas products will be assessed in this way, but it is unclear whether and how compounded CRISPR-Cas treatments will be assessed for relative effectiveness. The guidance clearly also needs to be in place for compounding.
Discussion
Different routes to market access can be considered for SGE therapies. In Europe, it is not yet clear whether the route via the EMA as an ATMP is the most appropriate, considering the highly personalized nature of most of the products, clinical procedures, and associated timetables, although this may optimally guarantee safety and effectiveness. SGE for thousands of specific variants involved in monogenic conditions requires a more flexible, personalized, and less labor-intensive route, especially if the patient groups are very small. Compassionate use may be fast for an individual patient, but it does not stimulate the commercial development of products. Prescription to named patients may only be a solution for single patients. Temporary authorization of use may allow access to medication half a year before formal market access has been granted, but it leads to financial uncertainty. Compounding under a hospital exemption may be an attractive solution for rare tailor-made applications at an acceptable price, but quality systems will need to be developed. Experimental therapy developed for research purposes and approved by local IRB is fast and adequate for small groups of patients but does not guarantee optimal safety, effectiveness, and broad implementation.
Perhaps a hybrid model of the existing routes is most appropriate to grant patients with very rare disorders relatively fast access to innovative SGE therapies while ensuring ensure safety and efficacy. It is conceivable that similar challenges and solutions may be relevant for innovative cancer drugs, making the development and implementation of new or adapted routes and attuning between stakeholders even more urgent. Economic aspects need to be considered to warrant affordable and equitable access.
Conclusion
Gene therapies are becoming available at a rapid pace. Alternative routes to market access need to be considered, all with their own benefits and challenges. In order to realize safe, effective, and affordable therapies, scientists, clinicians, and bioethicists will need to collaborate with healthcare economists and regulators [1]. For CRISPR-Cas products that serve large patient populations, a route via EMA as ATMP may be most applicable, but for individualized therapies, regimens similar to compounding under a hospital exemption should be considered.
References
Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578:229–36.
Cornel MC, Howard HC, Lim D, Bonham VL, Wartiovaara K. Moving towards a cure in genetics: what is needed to bring somatic gene therapy to the clinic? Eur J Hum Genet. 2019;27:484–7.
Wilbie D, Walther J, Mastrobattista E. Delivery aspects of CRISPR/Cas for in vivo genome editing. Acc Chem Res. 2019;52:1555–64.
Teixeira T, Kweder SL, Saint-Raymond A. Are the European medicines agency, US food and drug administration, and other international regulators talking to each other? Clin Pharmacol Therap. 2020;107:507–13.
European Medicines Agency (EMA). Orphan designation: overview. June, 2020. https://www.ema.europa.eu/en/human-regulatory/overview/orphan-designation-overview.
Vertex Pharmaceuticals Incorporated. A safety and efficacy study evaluating CTX001 in subjects with severe sickle cell disease. 2018. https://ClinicalTrials.gov/show/NCT03745287.
Gupta Strategists. The cost of opportunity: a study on pharmaceutical R&D costs. 2019. https://gupta-strategists.nl/storage/files/The-cost-of-opportunity-Gupta-Strategists.pdf.
Mestre-Ferrandiz J, Palaska C, Kelly T, Hutchings A, Parnaby A. An analysis of orphan medicine expenditure in Europe: is it sustainable? Orphanet J Rare Dis. 2019;14:287.
European Medicines Agency (EMA). Applications for new human medicines under evaluation by the Committee for Medicinal Products for Human Use. 2021; March. https://www.ema.europa.eu/en/documents/report/applications-new-human-medicines-under-evaluation-chmp-january-2021_en.pdf.
European Medicines Agency (EMA). Advanced therapy medicinal products: overview. April 2020. https://www.ema.europa.eu/en/human-regulatory/overview/advanced-therapy-medicinal-products-overview.
Mourby M, Morrison M. Gene therapy regulation: could in-body editing fall through the net? Eur J Hum Genet. 2020;28:979–81.
European Medicines Agency (EMA). Yescarta. European public assessment report. 2018; May. https://www.ema.europa.eu/en/documents/assessment-report/yescarta-epar-public-assessment-report_en.pdf.
Zorginstituut Nederland [National Health Care Institute]. Pakketadvies voretigene neparvovec (Luxturna®) bij de behandeling van visusverlies door erfelijke retinale dystrofie met bi-allelische RPE65-mutaties. December 2020. https://www.zorginstituutnederland.nl/publicaties/adviezen/2020/02/17/pakketadvies-voretigene-neparvovec-luxturna.
Hesselgrave B Zolgensma: The $2.1 million drug. Drug Topics. 2019; April 2020; 163. https://www.drugtopics.com/drug-price/zolgensma-21-million-drug.
European Medicines Agency (EMA). New gene therapy to treat spinal muscular atrophy [press release]. 2020. https://www.ema.europa.eu/en/news/new-gene-therapy-treat-spinal-muscular-atrophy-corrected.
Schlander M, Dintsios CM, Gandjour A. budgetary impact and cost drivers of drugs for rare and ultrarare diseases. Value Health. 2018;21:525–31.
Grant K. Canadian parents of babies with rare deadly disease look to Novartis treatment lottery. The Globe and Mail. 2020. https://www.theglobeandmail.com/canada/article-parents-of-babies-with-deadly-rare-disease-desperate-to-win-drug-maker/.
Roscamp JA, D’Cruz DP. The funding lottery for potentially life-threatening rare diseases. Rheumatology. 2017;57:1–2.
Health and Youth Care Inspectorate of the Ministry of Health, Welfare and Sport (the Netherlands). Supply of unregistered pharmaceutical products: FAQ’s. 2018; May 2020. https://english.igj.nl/documents/publication/2017/07/21/supply-of-unregistered-pharmaceutical-products-faqs.
Bourdoncle M, Juillard-Condat B, Taboulet F. Patient access to orphan drugs in France. Orphanet J Rare Dis. 2019;14:47.
Haering M. French early access compassionate use program ATU: still an eternal flame? 2019.
Agence Nationale De Sécurité du Médicament et des Produits de Santé (ANSM). Notice to applicants for marketing for temporary authorisation for use (ATU). 2015. p. 4.
Rio P, Navarro S, Guenechea G, Sanchez-Dominguez R, Lamana ML, Yanez R. et al. Engraftment and in vivo proliferation advantage of gene-corrected mobilized CD34(+) cells from Fanconi anemia patients. Blood. 2017;130:1535–42.
Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A, et al. Patient-customized oligonucleotide therapy for a rare genetic disease. N Engl J Med. 2019;381:1644–52.
Ministerie van Volksgezondheid, Welzijn en Sport [Ministry of Health, Welfare and Sport]. Besluit van 1 oktober 2019, houdende wijziging van het besluit zorgverzekering in verband met het zorgpakket Zvw. 2020. stb-2019-314 ed2019. https://zoek.officielebekendmakingen.nl/stb-2019-314.html.
Meij P, Canals JM, Scott M. Advances therapy medicinal products. Brief Pap. 2019;3(May):2020. LERU [Internet]
Bisnajak A. The design of a small-scale manufacturing unit for personalized medicine production. Delft University of Technology. 2018.
Zorg Instituut Nederland (ZIN) [Dutch National Healthcare Institute]. Advising on and clarifying the contents of the standard health care benefit package. April 2020. https://english.zorginstituutnederland.nl/about-us/tasks-of-the-national-health-care-institute/advising-on-and-clarifying-the-contents-of-the-standard-health-care-benefit-package.
Weinreich SS, Vrinten C, Kuijpers MR, Lipka AF, Schimmel KJM, van Zwet EW, et al. Aggregated N-of-1 trials for unlicensed medicines for small populations: an assessment of a trial with ephedrine for myasthenia gravis. Orphanet J Rare Dis. 2017;12:88.
Jørgensen J, Hanna E, Kefalas P. Outcomes-based reimbursement for gene therapies in practice: the experience of recently launched CAR-T cell therapies in major European countries. J Mark Access Health Policy. 2020;8:1715536.
Cosset D, Legros C, Haugen J, Meddeb L. ATU in France: the case for early consideration in local and european P&MA strategy. Simon Kucher & Partners. 2020. Available from: https://www.simon-kucher.com/nl/node/5099.
Sheldon T. Dutch hospital resumes production of rare drug after sourcing purer ingredients. BMJ. 2020;368:m401.
European Association of Hospital Pharmacists (EAHP). Compounding. 2019; April 2020. https://www.eahp.eu/practice-and-policy/compounding.
Health and Youth Care Inspectorate of the Ministry of Health, Welfare and Sport (The Netherlands). Vragen en antwoorden over de hospital exemption voor ATMP’s [Questions and answers about the hospital exemption for ATMPs]. 2010; April 2020. https://www.igj.nl/zorgsectoren/geneesmiddelen/documenten/publicaties/2010/08/05/vragen-en-antwoorden-over-de-hospital-exemption-voor-atmp%E2%80%99s.
Acknowledgements
We would like to thank all participants to our expert meeting for their contribution to the discussion. We thank our colleagues from the Consortium on Ethical Legal and Social Implication of Personalized Medicine (ELSI-PM) for both their input in the preparation of the meeting and the discussion on the implications afterward.
Funding
This work was funded by ZonMw (Netherlands Organization for Health Research & Development). (ELSI-PM Consortium, grant number 846003102).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical approval
The Medical Ethical Committee of VU University Medical Center Amsterdam considered the study exempt from the Law Medical Scientific Research (decision #2018.455, 6 September 2018). All participants signed an informed consent form prior to the meeting.
Conflict of interest
The authors declare that they have no conflict of interest. The views expressed in this article are the personal views of the authors and may not be understood nor quoted as being made on behalf of, or reflecting the position of the National Health Care Institute.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41431_2021_877_MOESM1_ESM.mp4
Tessel Rigter, PhD, explains in four minutes that very personalized medication such as somatic gene editing needs flexible routes to patient access, including a route for tailor made medication under local quality control next to the ATMP/EMA route.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rigter, T., Klein, D., Weinreich, S.S. et al. Moving somatic gene editing to the clinic: routes to market access and reimbursement in Europe. Eur J Hum Genet 29, 1477–1484 (2021). https://doi.org/10.1038/s41431-021-00877-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00877-y
This article is cited by
-
Therapeutic base editing in the adult liver
Nature Reviews Gastroenterology & Hepatology (2021)
