
Abstract
Previously, intragenic CAMTA1 copy number variants (CNVs) have been shown to cause non-progressive, congenital ataxia with or without intellectual disability (OMIM#614756). However, ataxia, intellectual disability, and dysmorphic features were all incompletely penetrant, even within families. Here, we describe four patients with de novo nonsense, frameshift or missense CAMTA1 variants. All four patients predominantly manifested features of ataxia and/or spasticity. Borderline intellectual disability and dysmorphic features were both present in one patient only, and other neurological and behavioural symptoms were variably present. Neurodevelopmental delay was found to be mild. Our findings indicate that also nonsense, frameshift and missense variants in CAMTA1 can cause a spastic ataxia syndrome as the main phenotype.
Similar content being viewed by others


Introduction
Genetic ataxias and hereditary spastic paraplegias (HSP) are both clinically and genetically heterogeneous conditions. Genetic ataxias are an important etiological group within the cerebellar ataxias. The incidence is estimated at >1 in 15,000 worldwide [1]. Genetic ataxia can be inherited in an autosomal dominant, autosomal recessive, or X-linked fashion [2]. The locus heterogeneity is exemplified by the fact that over a hundred different genes have already been linked to ataxia. The spectrum of molecular defects includes repeat expansions, single, or multiple nucleotide variants, and more rarely copy number variants (CNV’s). HSP is characterized by progressive lower limb spasticity and weakness, with great clinical heterogeneity, owing to the fact that currently ~60 HSP genes have been identified. HSP can be inherited in autosomal dominant, autosomal recessive or X-linked fashion [3], and has an estimated prevalence of 1.8 per 100,000 [1]. Collectively, genetic ataxia and HSP affect ~1 in 10,000 people worldwide [1]. Increasingly, an overlapping syndrome of spastic ataxia is being recognized, with shared phenotypes, genotypes, and mechanisms between genetic ataxia and HSP (i.e., SPG7 and ARSACS) [4, 5]. Furthermore, a number of genes have been associated with syndromes presenting a combination of genetic ataxia and intellectual disability. As such, variants in some of the autosomal recessive (SNX14, VWA3B, TDP2), or the autosomal dominant (ITPR1, KCND3) cerebellar ataxia genes give rise to more complex neurodevelopmental phenotypes, characterized by cerebellar ataxia, delayed psychomotor development, childhood-onset intellectual disability, and poor or absent speech [6,7,8].
CNVs of one or multiple exons of CAMTA1 can also cause cerebellar ataxia with intellectual disability as shown by Shinawi et al. and Thevenon et al. (OMIM#614756 [9, 10]). In these families, 8 out of 16 subjects were described as having either an ataxic or “widely based” gait. All 14 tested subjects had intellectual disability, seven of which were also delayed in speech or language development. In addition, 14 of the 16 subjects had a facial dysmorphia characterized by a long face with a pointed chin, bulbous nose with anteverted nostrils, a long philtrum, and a thick lower lip with down-turned corners of the mouth. Pleiotropy and incomplete penetrance were suggested to cause the heterogeneity in clinical features even within families carrying the same CAMTA1 variant.
The CAMTA1 gene codes for a member of the calmodulin-binding transcription activator family (OMIM 611501). The CAMTA1 protein is mainly expressed in the brain [9, 11, 12], and has been shown to be involved in several processes, including embryonic development, growth control, sensory mechanisms [11], and memory [12]. It was therefore proposed that CAMTA1 haplo-insufficiency was the cause of ataxia with or without intellectual disability in the families reported [9, 10]. CAMTA1 knock-out mice show severe ataxia with Purkinje cell degeneration and cerebellar atrophy, partially resembling the ataxia phenotype in humans [13]. Here, we report de novo nonsense, frameshift and missense variants in CAMTA1 in four unrelated patients with clinical manifestations of ataxia, spasticity or both, variably accompanied by other neurological features.
Methods
Patients cohort
Clinical exome sequencing is performed in our departments for various disorders, including movement disorders and ID. Filtering of the exome sequencing data is done on basis of one or multiple gene panels, and both the movement disorders and intellectual disability gene panels list CAMTA1. Patients with ID or with a (suspected) genetic movement disorder are counseled by (pediatric) neurologists and/or clinical geneticists before undertaking exome sequencing. Results were discussed in the expert centers for movement disorders (in Nijmegen or Groningen). Clinical exome sequencing was approved by the Medical Review Ethics Committee, Region Arnhem-Nijmegen, Number 2011/188. All human subjects provided informed consent for this study. Typically, only one patient (proband) per family is included for exome sequencing.
For patients 1, 2, and 4 trio-based exome sequencing and subsequent analysis of the movement disorders gene panel (patients 1 and 2) or the intellectual disability gene panel (patient 4) was performed. Patient 3 underwent singleton (no parents were included) exome sequencing and analysis of the movement disorders gene panel.
Confirmation of the presence of the variants, as well as confirmation of their de novo occurrence was revealed by targeted analysis (Sanger sequencing).
Exome sequencing and data analysis
Exome sequencing was performed as previously described [14, 15] for patients 1–3. Briefly, capture of exons was done using an Agilent SureSelect Human All Exon 50 Mb Kit (Santa Clara, CA, USA). Sequencing was performed using an Illumina Hiseq 2000 or 4000 (San Diego, CA, USA). Read mapping and variant calling were done using BWA and GATK, respectively. A filter for a ‘movement disorders’ gene panel or ‘intellectual disabilities’ gene panel was applied. The movement disorders gene panel consists of ∼200 genes implicated in various forms of genetic ataxias, HSP and hyperkinetic movement disorders. Only genes with substantial evidence (multiple families, functional evidence, and/or literature reports) were included in this panel. Genes with repeat expansions as the only molecular mechanism were not included either. The genes in this panel and coverage statistics can be found at www.genomediagnosticsnijmegen.nl/exome. Variants were prioritized based on the following criteria: frequency (<5% dbSNP, <1% in-house database of >20,000 exomes), nucleotide and amino acid conservation (based on alignments), relation of the gene to disease (per family), and inheritance pattern. CNV calling was performed using CoNIFER (http://conifer.sourceforge.net/) [16]. CNVs with an absolute Z-score greater than 1.7 were considered for analysis. Annotation of variants was done using in-house pipelines previously described [17].
For patient 4, a gene panel for ID comprising ~800 genes was analyzed in exome data (available upon request). Sequence variants were filtered by using Cartagenia Next-Generation Sequencing–Bench Laboratory software (Agilent, Santa Clara, CA) by using an automated filtering tree. The variants remaining after this filtering step were manually evaluated. The CAMTA1 variants described in this manuscript were submitted to LOVD (https://databases.lovd.nl/shared/genes/CAMTA1). Only de novo variants in autosomal dominant genes and compound heterozygous or homozygous variants in autosomal recessive genes, besides X-linked variants, were classified according the ACMG guidelines [18]. No CNV calling was done in the exome data. CNV’s above 150 kb or including OMIM-disease related genes were excluded with SNP array.
Predicting variant pathogenicity
Missense variants were examined using multiple techniques. Nucleotide conservation was calculated using PhyloP. Amino acid conservation was determined by comparing the human sequence with eight species (chimpanzee, rat, mouse, dog, chicken, zebrafish, fruit fly, and C. elegans). Physiochemical differences between canonical and patient amino acid sequence were determined using the Grantham matrix score, CADD, PolyPhen-2 HUMVAR, and AlignGVGD. MutationTaster and SIFT were also used to estimate the impact of the variant on DNA and protein level.
Results
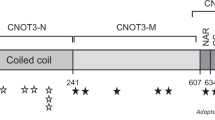
Clinical exome sequencing was done for ~1500 patients with a movement disorder. This had resulted in the discovery of four CAMTA1 variants that were likely pathogenic. Two were nonsense variants, one was a missense variant, and one resulted in a frameshift due to a deletion of two bases. Patient 1 has a premature stop codon closely after the CG1 DNA-binding domain, which is located near the beginning of the protein, resulting in loss of all functional domains after CG1 (Fig. 1). The premature stop codon in patient 2 is located in the CG1 domain of the protein, resulting in a loss of all functional domains, including the TIG domain, Ankyrin repeats, and IQ-motifs. Similarly, patient 4 was found to have a deletion of two bases in the CG1 domain, resulting in a premature stop codon near the end of this domain. As with patients 1 and 2, the protein produced by patient 4 therefore also loses all functional domains including TIG, Ankyrin repeats, and the IQ-motifs. In patient 3, a de novo missense was detected that affects an evolutionary conserved amino acid in the Camta1 protein (Fig. 2). No (intragenic) rearrangements or deletions were identified in our exome sequencing cohort. Parental analyses, either by trio-exome sequencing or subsequent Sanger sequencing, as well as paternity tests, revealed de novo occurrence of all four CAMTA1 variants.

The full-length Camta1 protein consists of 1673 amino acids and contains a CG1 DNA-binding domain (position 67–183), transcription factor immunoglobulin-like DNA-binding domain (TIG) domain (position 873–952), Ankyrin repeats for oligomerization (positions 1064–1086; 1109–1129; 1143–1172), and IQ motifs involved in calmodulin binding (positions 1547–1576; 1577–1599; 1600–1622). Arrows indicate where the patient variants are located. Bars below the image indicate the locations of CNVs as described by Thevenon et al. [9] and Shinawi et al. [10]. Exons are numbered as in NG_053148.1.

Alignments show very high conservation of glutamate 135 residue in nine species (conserved in all metazoans), which contains a variant in patient 3 (E > G).
All four patients had ataxia, spasticity or both, but additional neurological features were variably present. One patient was also diagnosed with borderline intellectual disability, and another two by language delay. Extensive clinical details are listed in Table 1 and expanded upon below.
Patient 1
Patient 1 is a 20-year-old male, born to healthy non-consanguineous parents. In his first year of life, it became apparent that his motor skills were underdeveloped. At school age, he developed a tremor of the hands. He is currently on Propranolol. Speech has always been unclear, with an element of fatigability. There was also dysphagia, partly also related to limited tongue movements. IQ was found to be 70, indicative of borderline intellectual disability. His behavior is characterized by irritability. Very recently, he developed brief jerks of the neck and right arm, which may occur a couple of times a day, without altered consciousness. On examination at age 20 years, there is borderline microcephaly (head circumference 52 cm, −2 SD); no dysmorphic features; jerky ocular pursuit; dysarthria and limited tongue movements; normal muscle tone; reduced dexterity and general slowness; stiff gait with no swinging of the arms, which are kept abducted; increased tendon reflexes and upgoing plantars; postural and intention tremor of the hands; and difficult tandem gait. MRI showed mild microcephaly and a small periventricular hyperintensity adjacent to posterior horn of the lateral ventricle in the right hemisphere. Chromosomal and metabolomic assays were unremarkable. A de novo CAMTA1 nonsense variant NM_015215.3:c.576 C > A; p.(Cys192*) was detected by exome sequencing (LOVD Variant ID: 0000461180).
Patient 2
Patient 2 is a 6-year-old female child, one of dizygotic twins born to healthy non-consanguineous parents. Her twin brother and older sister are both healthy. She was born prematurely at 34 weeks and 2 days. She received phototherapy for jaundice. In the first 2 years she had feeding difficulties, frequent cystitis with normal kidneys, and upper airway infections. The latter was treated by adenotomy, tonsillectomy, and ear tubes. She could walk unsupported from age 16 months. Speech development was normal, but swallowing difficulties were often observed. She was referred at age 2.5 years because of unexplained ataxia and delay in motor development. No dysmorphic features were present besides a broad nasal bridge (ICD p97). On examination, there was a suggestion of jerky ocular pursuit but normal saccades; nasal, slurred speech; mild ataxia of upper extremities and gait ataxia; distal upper limb myoclonus; normal tendon reflexes with downgoing plantars. Behaviorally, there are indications of an autism spectrum disorder. Cognitive ability appears average but was not formally tested. She is attending a regular primary school. MRI of the brain revealed no abnormalities. Over time, her broad-based ataxic gait improved, but the ataxia still led to regular falls and the necessity to use a wheelchair for longer distances; however, she did respond to rehabilitation intervention. Main problems currently include fatigue and short staring episodes during which she does not react, with eyes turning outwards and increased tremor. An EEG was normal. With exome sequencing, a de novo nonsense variant was detected in CAMTA1, NM_015215.3:c.430 G > T; p.(Gly144*) (LOVD Variant ID: 0000461179).
Patient 3
Patient 3 is a 3-year-old male and the first-born child to healthy, unrelated parents. He was born in week 41 after a normal pregnancy and delivery. He had bilateral restricted abduction in his hips that were treated with a Frejka pillow for 3 weeks. Calcaneovalgus positioning of the left foot was treated with stretching and a splint for 3 weeks. He was initially also followed for 2 months because of a torticollis. He has had chewing difficulties and a drooling tendency since birth. Psychomotor development has been mildly delayed, but there have been no signs of arrest or regression. His movements were described as stiff in the morning with frequent wobbling and falling when fatigued. Upon the last follow-up at 2 years 11 months of age, eye movements were normal. He walked and ran with a mild ataxia and spoke 4–5 words sentences with dysarthria. There was mild titubation. Upon testing of fine motor activities, there was a mild dysmetria and tremor. Muscle tone was slightly decreased but tendon reflexes were increased in the lower limbs with bilateral extensor plantars and left-sided ankle clonus. MRI of the brain performed at 2 years 11 months of age was considered normal. Routine laboratory investigations including CK, thyroid function, lactate, ammonia, alpha-fetoprotein, CDT, and lipoproteins were normal as were CSF analyses regarding cells, lactate, glucose, and electrophoresis. A de novo CAMTA1 missense variant was identified: NM_015215.3:c.404 A > G; p.(Glu135Gly) (LOVD Variant ID: 0000461181).
Patient 4
Patient 4 is a 3-year-old female, born to healthy non-consanguineous parents, and has a healthy older sister. Her nasal tip is somewhat broad, with a trace of epicanthal folds. She has a flat nasal bridge and mildly deep-set eyes. She was found to have a developmental delay regarding motor development and speech, being able to sit upright at 11 months of age, and crawl at 14,5 months. At age 20 months, she could walk freely but with frequent falls. Currently, at age 3 years, she is able to speak ~30 words, sometimes in two-word sentences, but pronunciation is unclear. Her understanding of words appears to have improved, and hearing and vision appear to be normal. There is increased distractibility. On examination, there were mainly balance difficulties, with locking of the knees and frequent falls; normal ocular movements; and no muscle weakness. The child-neurologist at age 20 months detected hypotonia in arms and legs, no ataxia of the hands, symmetrical vivid reflexes, and downgoing plantar sign. Her balance is immature. There was borderline hypermobility of the lower extremities. Metabolic investigations were normal. A SNP array performed in 2017 was found to be normal. An MRI was not made prior to exome sequencing. Using exome sequencing, a de novo frameshift variant in CAMTA1 was detected, as a result of deletion of two bases: NM_015215.3 c.379_380delAG; p.(Arg127Glufs*152) (LOVD Variant ID: 0000461182).
Discussion
Our results indicate that not only CNVs, but also nonsense, frameshift and missense variants in CAMTA1 can cause a spastic ataxia syndrome with variable other neurological features. Ataxia and/or spasticity were the dominant and consistent features in the four patients. Only one patient was shown to have borderline intellectual disability, while other neurological problems, i.e., speech/language disorder (n = 2), tremor (n = 2), myoclonus (n = 1), or torticollis (n = 1) were also variably present. A facial dysmorphism (a broad nasal bridge) was observed in only one patient, and borderline microcephaly was observed in another.
Gait and balance problems, labeled as ataxia, spasticity, or unsteady gait, were documented for all patients with CNV’s of CAMTA1 as well as for those in this cohort. Ataxia was unequivocally present in 4 out 4 patients in this cohort and in 8 of 16 patients with CNV’s, while the others had a spastic or unsteady gait ([9, 10]; Table 1). Patient 1 had spastic paraplegia as the main phenotype, with a suggestion of an ataxic element in his gait, whereas patient 3 had a clear combination of ataxia and pyramidal features. Synofzik et al. [4] already stated that, although ataxia and spastic paraplegia were traditionally designated as separate clinicogenetic classifications, next generation sequencing has indicated that genes previously associated with only one of the disorders, can be causative for both ataxia and spastic paraplegia. We suggest that CAMTA1 can be added to this list of genes for childhood presentations of a spastic ataxic phenotype.
Behavioural abnormalities constated in our patients include mild features of the autism spectrum as indicated in Table 1. In the families with CNV’s, there were variable indications for ADHD and autism spectrum disorder. In the patients with CNV’s, mild to severe intellectual disability (IQ < 70) was described in most patients [9, 10], whereas intellectual disability was not observed in our patients with nonsense, frameshift or missense CAMTA1 variants. Only two of our patients had a borderline IQ and others were not formally tested. None of the other patients showed signs of a probable severe intellectual disability (i.e., lack of contact, difficulties understanding, etc); however, these patients were too young to formally test. Therefore, intellectual disability cannot be excluded for patients 3 and 4. Language delay was found in two patients. Similarly, a long face with a pointed chin, bulbous nose with anteverted nostrils, a long philtrum, and a thick lower lip with down-turned corners of the mouth were common in patients with CNV’s, but not observed in the four patients presented here.
Finally, individuals with CAMTA1 variants, both CNV’s and other mutation types, with quite divergent phenotypes are present in publicly available datasets (DDD study [19] and Decipher (https://decipher.sanger.ac.uk/search?q=camta1#consented-patients/results)), suggesting that the syndrome associated with CAMTA1 variants may be variable. However, these datasets are difficult to interpret, since many individuals have candidate variants in multiple genes, the presence of the CAMTA1 variants is not always validated by another test, and segregation is often not performed. Thus, future studies of larger cohorts of patients with disease causing CAMTA1 variants are needed to determine whether the CAMTA1-related syndrome constitutes a broader phenotypic spectrum. The majority of the intragenic CAMTA1 CNVs resulted in frameshifts and likely nonsense-mediated decay, although the latter was not proven due to low expression of CAMTA1 in fibroblasts [9]. The premature stop codons in our patients 1, 2, and 4 are also likely to induce nonsense-mediated decay of the mRNA, since they are not present in the penultimate or ultimate exon. However, even in the absence of nonsense-mediated decay these variants would result in drastically shortened polypeptide chains (Fig. 1). Thus, these nonsense and frameshift variants are considered loss-of-function variants, which is in agreement with haplo-insufficiency as the underlying genetic mechanism in the CAMTA1-related disorder. This is corroborated by the finding of only a single loss-of-function variant (different from the ones described here) in large control populations (ExAC), where 51 would have been expected on basis of random chance, resulting in a probability of loss-of-function intolerance (pLI) of 1.00 [20].
The mechanism by which the de novo missense variant p.(Glu135Gly) in patient 3 may be pathogenic is speculative. This variant is located in the CG1 DNA-binding domain of the protein (Fig. 1) and is highly conserved between orthologues of Camta1 in different species (up to C. elegans, Fig. 2), while a glutamate to glycine change is likely to have a moderate physiochemical difference. In silico pathogenicity scores by multiple programs (scaled CADD: 27.3, SIFT: 0; PolyPhen-2 HUMVAR: 0.996, Align GVGD: C0, mutation taster: 1) predict that this variant may be pathogenic. In addition, the p.(Glu135Gly) variant was absent from control populations ExAC and gnomAD [20].
In our cohort of patients with movement disorders or intellectual disability, no CAMTA1 CNVs were identified. A possible explanation would be that our method to detect CNVs using Conifer [17] detects variants with a size of three or more exons only. The described rearrangements and deletions in the literature involve one exon (two cases), two exons (one case), and three or more exons (another two cases). Of these five, only two would have been detected using our analyses with Conifer. Further testing of our cohort by other methods, such as MLPA or SNP arrays or analyses of the WES data by more sensitive methods than Conifer, are needed to detect putative small CNVs.
In summary, we describe four patients with features of ataxia and/or spasticity due to nonsense, frameshift and missense variants in CAMTA1. Similar to the patients described with intragenic CNV’s, other neurological and behavioral presentations are variably present. In contrast, our patients do not have facial characteristics, nor do they have an overt intellectual disability, which were observed in most patients with intragenic deletions of the CAMTA1 gene. With the relatively small number of patients with CAMTA1 variants, however, it is impossible to infer genotype—phenotype correlations at this stage.
References
Ruano L, Melo C, Silva MC, Coutinho P. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. 2014;42:174–83.
Akbar U, Ashizawa T. Ataxia. Neurologic Clin. 2015;33:225–48.
Klebe S, Stevanin G, Depienne C. Clinical and genetic heterogeneity in hereditary spastic paraplegias: From SPG1 to SPG72 and still counting. Rev Neurologique. 2015;171:505–30.
Synofzik M, Schule R. Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways. Mov Disord. 2017;32:332–45.
de Bot ST, Willemsen MA, Vermeer S, Kremer HP, van de Warrenburg BP. Reviewing the genetic causes of spastic-ataxias. Neurology. 2012;79:1507–14.
Gómez-Herreros F, Schuurs-Hoeijmakers JHM, McCormack M, Greally MT, Rulten S, Romero-Granados R, et al. TDP2 protects transcription from abortive topoisomerase activity and is required for normal neural function. Nat Genet. 2014;46:516.
Thomas Anna C, Williams H, Setó-Salvia N, Bacchelli C, Jenkins D, O’Sullivan M, et al. Mutations in SNX14 cause a distinctive autosomal-recessive cerebellar ataxia and intellectual disability syndrome. Am J Hum Genet. 2014;95:611–21.
Kawarai T, Tajima A, Kuroda Y, Saji N, Orlacchio A, Terasawa H, et al. A homozygous mutation of VWA3B causes cerebellar ataxia with intellectual disability. J Neurol Neurosurg Psychiatry. 2016;87:656–62.
Thevenon J, Lopez E, Keren B, Heron D, Mignot C, Altuzarra C, et al. Intragenic CAMTA1 rearrangements cause non-progressive congenital ataxia with or without intellectual disability. J Med Genet. 2012;49:400–8.
Shinawi M, Coorg R, Shimony JS, Grange DK, Al-Kateb H. Intragenic CAMTA1 deletions are associated with a spectrum of neurobehavioral phenotypes. Clin Genet. 2015;87:478–82.
Finkler A, Ashery-Padan R, Fromm H. CAMTAs: calmodulin-binding transcription activators from plants to human. FEBS Lett. 2007;581:3893–8.
Huentelman MJ, Papassotiropoulos A, Craig DW, Hoerndli FJ, Pearson JV, Huynh K-D, et al. Calmodulin-binding transcription activator 1 (CAMTA1) alleles predispose human episodic memory performance. Hum Mol Genet. 2007;16:1469–77.
Long C, Grueter CE, Song K, Qin S, Qi X, Kong YM, et al. Ataxia and Purkinje cell degeneration in mice lacking the CAMTA1 transcription factor. Proc Natl Acad Sci USA. 2014;111:11521–6.
Neveling K, Feenstra I, Gilissen C, Hoefsloot LH, Kamsteeg EJ, Mensenkamp AR, et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat. 2013;34:1721–6.
Herkert JC, Abbott KM, Birnie E, Meems-Veldhuis MT, Boven LG, Benjamins M, et al. Toward an effective exome-based genetic testing strategy in pediatric dilated cardiomyopathy. Genet. Med. 2018;20:1374–86.
Krumm N, Sudmant PH, Ko A, O’Roak BJ, Malig M, Coe BP, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22:1525–32.
Pfundt R, Del Rosario M, Vissers L, Kwint MP, Janssen IM, de Leeuw N, et al. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genet Med. 2017;19:667–75.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature. 2017;542:433–8.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Acknowledgements
The authors would like to thank MAJ Boon from the Department of Neurology, University Medical Centre Groningen, for her neurological evaluation of patient 2.
Funding
BvdW is supported by research grants from ZonMW and Hersenstichting. ND is financed by grants from the Swedish state under the agreement between the Swedish government and the county councils, the ALF-agreement (76200).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wijnen, I.G.M., Veenstra-Knol, H.E., Vansenne, F. et al. De novo variants in CAMTA1 cause a syndrome variably associated with spasticity, ataxia, and intellectual disability. Eur J Hum Genet 28, 763–769 (2020). https://doi.org/10.1038/s41431-020-0600-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0600-5
