
Abstract
Thirty percent of all inherited retinal disease (IRD) is accounted for by conditions with extra-ocular features. This study aimed to establish the genetic diagnostic pick-up rate for IRD patients with one or more extra-ocular features undergoing panel-based screening in a clinical setting. One hundred and six participants, tested on a gene panel which contained both isolated and syndromic IRD genes, were retrospectively ascertained from the Manchester Genomic Diagnostics Laboratory database spanning 6 years (2012–2017). Phenotypic features were extracted from the clinical notes and classified according to Human Phenotype Ontology; all identified genetic variants were interpreted in accordance to the American College of Medical Genetics and Genomics guidelines. Overall, 49% (n = 52) of patients received a probable genetic diagnosis. A further 6% (n = 6) had a single disease-associated variant in an autosomal recessive disease-relevant gene. Fifty-two percent (n = 55) of patients had a clinical diagnosis at the time of testing. Of these, 71% (n = 39) received a probable genetic diagnosis. By contrast, for those without a provisional clinical diagnosis (n = 51), only 25% (n = 13) received a probable genetic diagnosis. The clinical diagnosis of Usher (n = 33) and Bardet–Biedl syndrome (n = 10) was confirmed in 67% (n = 22) and 80% (n = 8), respectively. The testing diagnostic rate in patients with clinically diagnosed multisystemic IRD conditions was significantly higher than those without one (71% versus 25%; p value < 0.001). The lower pick-up rate in patients without a clinical diagnosis suggests that panel-based approaches are unlikely to be the most effective means of achieving a molecular diagnosis for this group. Here, we suggest that genome-wide approaches (whole exome or genome) are more appropriate.
Similar content being viewed by others


Introduction
Inherited retinal disease (IRD) refers to a group of clinically and genetically heterogeneous conditions affecting the retina [1]. IRD is usually confined to the eye but up to 30% of affected individuals are considered syndromic as they have one or more associated extra-ocular features [2], some of which require health surveillance or specific clinical management measures to be put in place. Making a timely definitive diagnosis in IRD cases complicated by extra-ocular features is important, therefore, but is often challenging [3, 4]. More than 260 genes have been associated with IRD; over 100 of these have been linked to conditions of IRD with extra-ocular features. These disorders are associated with autosomal dominant (AD), autosomal recessive (AR) and X-linked inheritance (RetNet, http://www.sph.uth.tmc.edu/RetNet/).
There are a small number of well known, relatively prevalent multisystemic conditions that feature IRD as a prominent feature; including Bardet–Biedl syndrome (BBS) and Usher syndrome. The diagnostic pick-up rate of molecular genetic testing for these syndromes is high as the majority of disease-associated genes have been discovered. Variants in at least 24 genes are found in ~80% of individuals affected with BBS, a ciliopathy that impacts multiple body systems including the eye and the kidney [5, 6]. Usher syndrome, a ciliopathy manifesting with IRD and sensorineural hearing impairment (SNHI), is caused by variants in 15 genes (RetNet). Two other such conditions where a clinical diagnosis can be made and chances of finding a diagnosis is high are Cohen syndrome (VPS13B) and Alström syndrome (ALMS1). Genes related to these conditions are sometimes included in an IRD NGS testing approach.
Genomic testing provides a fast, cost-effective and reliable way of interrogating multiple genes at one time [7]. Over the past decade, this technology has revolutionised the diagnosis of genetically heterogeneous conditions like IRD [8]. Importantly, there is a lack of studies that have examined the yield of genetic testing in syndromic IRD in general as most reports focus on either isolated IRD, or on specific conditions such as Usher syndrome [7,8,9,10,11,12]. A panel-based NGS test for IRD genes was designed by the Genomic Diagnostics Laboratory within Manchester Centre for Genomic Medicine (MCGM) in 2012 to genetically test for a custom group of genes known to be causative for both isolated and syndromic IRD conditions. Our hypothesis was that cases with distinctive multisystemic presentations would more likely benefit from this genetic testing strategy compared with those in which such a diagnosis had not been formulated clinically. Aiming to gain further insight into this, we have assessed the clinical utility of our panel-based genetic testing approach in individuals with assumed syndromic IRD. In addition, we have described the range of genes and genomic variants in this cohort and compared them with the literature.
Subjects and methods
Recruitment and phenotype data collection
The cohort was assembled retrospectively from a database of patients referred for genetic testing to the Genomic Diagnostics Laboratory within MCGM; (Manchester, UK) between January 2012 and December 2017. Before genetic testing, all study participants were examined by a clinical geneticist and a paediatric and/or genetic ophthalmologist within the Manchester University NHS Foundation Trust (Manchester, UK), and then subsequently referred for genetic testing at the Genomic Diagnostics Laboratory at MCGM. Only patients referred with an IRD with at least one non-ocular feature were included in this study. For the purpose of this study, an individual is considered to have a syndromic IRD if they have ophthalmic signs and symptoms suggestive of retinal disease (including those with inherited retinal dystrophies and retinal dysplasia) and at least one feature identified by a clinical geneticist to be potentially an associated manifestation of a multisystem disorder. This might be, for example, a structural malformation, hearing loss, or a significant growth or developmental disorder. Informed consent was obtained from all patients prior to panel-based clinical genetic testing and the study protocol observed the tenets of the Declaration of Helsinki.
The clinical notes were reviewed and all relevant phenotypic features were collected and converted into Human Phenotype Ontology (HPO) terms using the HPO Browser (http://compbio.charite.de/hpoweb/ (accessed between February and July 2018) [13]. The clinical diagnosis of a specific syndromic condition was provisionally made when the phenotype fulfilled predefined diagnostic criteria (Supplementary Table S1) or when there was a high index of suspicion for a specific diagnosis. We used the diagnostic criteria set by Beales et al. (1999) to define a clinical diagnosis of BBS [14] (Supplementary Table S1). Also, for Senior–Loken syndrome (SLS), a high index of suspicion was recorded in an individual with apparently isolated IRD because a family history of SLS was suspected.
Genetic testing and bioinformatics analysis
IRD panel testing and data analysis were performed at the Genomic Diagnostics Laboratory within MCGM (Clinical Pathology Accredited no. 4015). Sequencing and bioinformatics analyses have been described previously [15, 16]. Briefly, panel tests were custom designed to include the coding regions (±50 bp of flanking intronic sequence) of either 105 genes (41 samples tested between January 2012 and June 2014) or 176 genes (65 samples tested between July 2014 and December 2017) that have previously been associated with isolated and/or syndromic IRD; lists of the genes included in each panel can be found in Supplementary Tables S2 and S3. Notably, the 105 gene panel included 14 BBS-associated genes and 8 Usher syndrome associated genes, while the 176 gene panel included 22 BBS-associated genes and 11 Usher syndrome associated genes.
Samples were processed using the Agilent SureSelectXT (Agilent Technologies, Santa Clara, USA) target enrichment chemistry, and sequenced on an Illumina HiSeq 2000/2500 or NextSeq500 system (Illumina Inc, San Diego, USA) according to the manufacturers’ protocols. Sequence reads were de-multiplexed using CASAVA software version 1.8.2 (Illumina) and aligned using the Burrows Wheeler Aligner (BWA-short version 0.62) to the hg19 reference genome [17]. Duplicate reads were removed and the Genome Analysis Tool Kit (GATK-lite version 2.0.39) was used for single-nucleotide variant and insertion–deletion discovery [18]. Copy number variants (CNVs) were detected from high-throughput sequencing read data using ExomeDepth version 1.1.6 as described in references [19, 20].
Variant validation and classification
Clinical interpretation of genetic variants was performed by UK National Healthcare Service (NHS) clinical scientists within the Genomic Diagnostics Laboratory at MCGM. Only variants with a minor allele frequency of <1% in Exome Variant Server (NHLBI GO Exome Sequencing Project (ESP), Seattle, WA were considered. Release ESP6500SI-V2. http://evs.gs.washington.edu/EVS/) and dbSNP were considered. Variant interpretation was supported by in silico pathogenicity predictions (i.e. from SIFT, PolyPhen2 and AlignGVGD), extensive evaluation of the scientific literature, and the patients’ clinical referral, in line with the American College of Medical Genetics and Genomics best practice guidelines [21]. Segregation studies were also performed where clinically appropriate, when relevant samples were available for analysis. For the purpose of this study, molecular genetic diagnoses were classified into three groups using a previously described strategy [16]: (1) Probable genetic diagnosis: a clearly or likely disease-associated variant(s) identified in a gene relevant to the patient’s phenotype, which is present in an apparently disease-causing state (i.e. biallelic variants in AR disease-associated genes and monoallelic variants in AD disease-associated genes); (2) possible genetic diagnosis: a single clearly or likely disease-associated variant identified in a recessive disease gene that is known to cause a spectrum of phenotypic features that match the patients clinical presentation; it may be reasoned that the patient harbours a second change that could not be detected by the chemistry/technology of the test used for diagnosis; (3) unknown genetic diagnosis: no likely or causal disease-associated variant(s) detected. Variants that were concluded likely to contribute to a patients’ molecular diagnosis were confirmed by an alternative method (i.e. Sanger sequencing) and have been submitted to the ClinVar database [https://www.ncbi.nlm.nih.gov/clinvar/] [22] under the searchable submission name: (SyndromicIRD_variants), if not already done so during our previous work (ClinVar accession numbers: SCV000259090.1, SCV000259091.1, SCV000259087.1, SCV000259094.1, SCV000259095.1, SCV000259101.1, SCV000259102.1, SCV000282652.1, SCV000282640.1, SCV000259083.1, SCV000259082.1, SCV000259084.1, SCV000259085.1, SCV000493124.1, SCV000493116.1, SCV000493114.1) (REF: [15, 19, 23]). On occasions where more than one ‘probable’ or ‘possible’ disease-associated variant was identified in disease-causing state, family history, the patients phenotypic presentation, and evidence of variant pathogenicity were considered in detail by a multidisciplinary team. Although all variants are detailed in clinical reports issued by the diagnostic laboratory, only the variant(s) considered most likely to be causal (and/or that had been confirmed to segregate with disease within a family) was included for the purpose of our analysis. It is also of note that ‘probable’ or possible’ disease-associated variants identified in carrier state were clinically reported as carrier findings.
Statistical analysis
A comparison of the genetic pick-up rate between the two groups was performed using chi-squared test of independence to determine the dependence of a genetic diagnosis by the panel test on having a provisional clinical diagnosis, the statistical significance was defined as α = 0.001.
Results
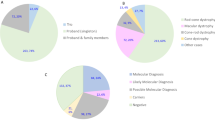
A total of 106 patients were included in our analysis, 61 females and 45 males. Age at referral ranged between 0 days and 69.5 years with a median age of 21.9 years; 43% of study participants were 16 years or younger. Patients fell into one of the two categories (Fig. 1):
-
(i)
Fifty-two percent (55/106) had a provisional clinical diagnosis of a specific syndromic condition (e.g. BBS or Usher syndrome) at the time of referral for genetic testing or,
-
(ii)
forty-eight percent (51/106) referred as probably syndromic IRD, with no clinically recognised diagnosis.

Of the 55/106 (52%) patients who were referred with a provisional clinical diagnosis, 71% received a probable genetic diagnosis compared with 25% of those patients with no known clinical diagnosis (51/106).
There was a wide range of extra-ocular phenotypes; the most common included hearing impairment (any type/severity), developmental delay (DD)/intellectual disability (ID) (any type, specific/global), polydactyly, microcephaly, obesity, abnormal facial shape (dysmorphic) and kidney/renal abnormalities; a list of HPO terms generated from the clinical notes of the study population can be found in Supplementary Table S4.
A probable genetic diagnosis was made in 49% (52/106) of cases; 6% (6/106) of patients received a possible genetic diagnosis (i.e. they were found to be heterozygous for a variant in an AR gene relevant to the patient’s phenotype). The test failed to detect any likely disease-associated variants in 45% (48/106). Notably, of the 55 patients referred with a specific clinical diagnosis, 39 (71%) received a probable, and confirmed, genetic diagnosis, and 5 (9%) a possible genetic diagnosis. For the 51 patients referred without a specific clinical diagnosis, 13 (25%) received a probable diagnosis and 1 patient received a possible genetic diagnosis (2%) (Fig. 1). The difference was significant, χ2 (2, N = 106) = 31.8264, p < 0.001.
Referrals with a provisional clinical diagnosis
Usher syndrome
Thirty-three patients, 25 (76%) females and 8 (24%) males, aged between 5 months and 58.5 years (median age of 29.3 years), had a provisional clinical diagnosis of Usher syndrome (Supplementary Table S5 and Supplementary Fig. S1). Additional atypical syndromic features were identified in three of these Usher syndrome patients: one patient who was found to have biallelic variants in PCDH15 had ID, two patients who did not receive a genetic diagnosis: one had global brain atrophy with ID and the other had seizures and migraines (Fig. 2).

Features in the orange and yellow boxes are the two main features of Usher syndrome that were used as the diagnostic criteria for Usher syndrome in this study. Types and age of onset of hearing impairment in the patients are shown as well as the type of retinal dystrophy and any unusual features found in these patients. The patients are divided by the genetic diagnosis received from the IRD NGS panel test: probable-, possible- and no genetic diagnosis.
Panel testing enabled a probable genetic diagnosis to be identified in 67 % (22/33) of individuals confirming the clinical diagnosis of Usher syndrome. A further 15% (5/33) received a possible genetic diagnosis and no causal variants were found in the remaining 18% (6/33) of patients with clinical Usher syndrome.
The diagnostic pick-up rate differed according to the age of onset of SNHI; post-lingual SNHI was associated with lower yield of genetic testing (Orange boxes in Fig. 2). Regarding family history, ten patients had a family history of a similar condition. Five had congenital/pre-lingual SNHI of which four received a probable genetic diagnosis and one a possible diagnosis. Three had post-lingual SNHI of which two received a probable diagnosis and one a possible diagnosis. Lastly, two patients had unknown ages of onset of SNHI and both received a probable genetic diagnosis (Supplementary Fig. S1).
Two patients with congenital SNHI (one with a family history of Usher and one without) who were found to have a single variant in a gene known to be mutated in Usher syndrome (possible genetic diagnosis), as well as one of the six individuals with no genetic diagnosis, were subject to further testing using whole genome sequencing (WGS). A second heterozygous disease-associated variant was identified in both patients (patients #25 and #26) with a possible genetic diagnosis, confirming the clinical diagnosis of Usher syndrome. In the one patient with no genetic diagnosis following testing of the 105 gene panel (patient #31), subsequent WGS revealed a homozygous deletion of exons 1–7 of MERTK gene (NM_006343.2:c.[(?_-1)_(1144+1_1145-1)del];[(?_-1)_(1144+1_1145-1)del]). This deletion has been previously identified in heterozygous state in another in-house patient with isolated rod-cone dystrophy [23]. A 91 kb deletion including this region is a common founder variant in the Faroe Islands, associated with non-syndromic AR rod-cone dystrophy without hearing impairment [24]. No variants in genes associated with hearing impairment were identified in this individual who was diagnosed with hearing impairment in his late teenage years.
Overall, 42 different disease-associated variants were found in 6 known Usher syndrome genes. Of these, 14 have not previously been associated with Usher syndrome. The genes involved and variant types are shown in Supplementary Fig. S2.
Bardet–Biedl Syndrome
Ten patients were referred with a high index of suspicion for BBS (Fig. 3). Four were male and six female, and age at referral ranged between 3 and 41 years (median was 16.8 years) (Supplementary Table S6). Eight of these ten patients received a probable genetic diagnosis of BBS after panel testing; no genetic diagnosis was made in the remaining two study subjects. Seven of the eight patients with a probable genetic diagnosis were homozygous for the disease-causing variant and one patient harboured a compound heterozygous variant.

BBS was diagnosed in 8/10 patients who fulfilled the diagnostic criteria or with high index of suspicion. In addition, the NGS panel was able to pick-up two early cases with only two features. BBS1 was causative in half of the diagnosed cases.
Interestingly, the panel test revealed a probable genetic diagnosis of BBS in two additional patients who were in the unknown clinical diagnosis group, reflecting the observation that the clinical spectrum of BBS is now recognised to be wide. The mutated genes and variant types found in all ten patients with a probable genetic diagnosis of BBS are shown in Supplementary Fig. S3.
Other IRD multisystemic conditions
One patient was referred with a clinical diagnosis of Joubert syndrome (Supplementary Table S7). Genetic testing revealed a previously reported homozygous nonsense variant in CEP290. NM_025114.3:c.5668G>T; p.(Gly1890*) confirming the diagnosis [25, 26].
Five patients, two male and three female, were referred with, or fulfilled the diagnostic criteria for, SLS. Median age at referral was 28.9 years (range 9.3–58.8 years). All five patients had rod-cone dystrophy and of the four study subjects who had renal insufficiency; nephronophthisis (a key indicator of SLS) was specifically noted in only one patient (Supplementary Table S8). One 29-year-old patient (#46 in Supplementary Table S8) did not have renal impairment but due to a family history of SLS, she was highly suspected to have SLS. Panel-based testing identified a probable molecular diagnosis in four of these five patients (80%) with presumed SLS, including patient #46. Six different disease-associated variants were identified in three different SLS genes (CEP290, IQCB1 and NPHP4).
Five patients were referred with Cohen syndrome; three male and two female. Age at referral ranged between 3.3 years and 49.2 years with a median age of 18 years. All five patients fulfilled previously outlined diagnostic criteria [27] (Supplementary Table S9). A probable genetic diagnosis was made in three of five patients (60%); no disease-associated variants or CNVs were identified in the remaining two individuals. Each genetically diagnosed patient had different compound heterozygous variants in VPS13B, resulting in six different variants in total, one of which has not been previously reported. The three genetically confirmed Cohen syndrome patients had four features in common: characteristic facial dysmorphism, microcephaly, a form of DD, and an ocular phenotype.
One male patient was referred with Norrie disease at the age of 13 months (Supplementary Table S10). The panel test found a heterozygous missense variant NM_012193.3:c.313A>G, p.(Met105Val) in FZD4, which has been associated with AD familial exudative vitreoretinopathy [28, 29].
Referrals with unknown diagnoses
Fifty-one patients, 27 male and 24 female, with syndromic IRD were referred for IRD panel testing with no specific clinical diagnosis at the time of referral. Ages at referral ranged between 0 days and 69.5 years (median 14.2 years). Extra-ocular manifestations were highly variable, the most common being DD (global or specific), ID, hearing impairment and obesity. Twenty-five patients had one extra-ocular feature, 15 patients had two extra-ocular features, and 11 patients had 3–6 extra-ocular features (Supplementary Table S11).
Panel testing identified a probable genetic diagnosis in 13 of these 51 patients (25%) and a possible genetic diagnosis in 1 of these 51 (2%) patients; the remaining 37 of 51 patients (73%) received no genetic diagnosis. As mentioned, two patients who had not been suggested to have BBS—due to only having two primary features (one had rod-cone dystrophy and ID and the other had rod-cone dystrophy with polydactyly)—were in fact found to have homozygous variants in BBS1. Five patients received a probable genetic diagnosis due to the identification of likely disease-associated variants underlying one of the following syndromes: two patients with homozygous or compound heterozygous variants in MVK which is associated with the AR conditions mevalonic aciduria and hyper-IgD syndrome; one with a heterozygous change in OTX2 which is associated with AD syndromic microphthalmia type 5; one with a homozygous change in IFT140 which is associated with AR short-rib thoracic dysplasia 9 (with or without polydactyly); and one with a heterozygous change in KIF11 which is associated with AD microcephaly (with or without chorioretinopathy, lymphedema or mental retardation). Interestingly, six patients had probable disease-associated variants that only explained their retinal phenotype but not their extra-ocular manifestations (patients 64–69 in Supplementary Table S11; variants in the GNAT2, CNGA3, PROM1, PRPF3, EYS and USH2A genes).
Overall, in this unknown diagnosis group, panel testing identified 17 different disease-causing variants in 12 genes. Four of the variants are previously unreported and a further five have been exclusively reported by our group (Table 1) [16].
In one patient with congenital stationary night blindness and ID and a possible genetic diagnosis by panel testing, heterozygous missense variant NM_002420.5:c.2951G>A; p.(Arg984His) in TRPM1, previous array comparative genomic hybridisation screening identified a heterozygous 15q13.3 microdeletion in the trans allele which resulted in loss of the TRPM1 gene NM_002420.5:c.(?_−1)_(*1_?)del.
Discussion
The aim of this study was to determine the efficacy of providing a genetic diagnosis using panel-based testing in individuals with IRD and at least one potentially associated non-ocular feature. Although the panel-based strategy had proven useful in isolated ocular disease, we were more circumspect about whether this was the best way to investigate syndromic retinal disorders and in particular we hypothesised that cases with phenotypes typical of specific multisystemic conditions would have a higher rate of genetic diagnosis compared with those with phenotypes not aligning with any such condition. We also aimed to describe the range of genes and variants in this cohort and to compare these with other studies. A diverse population of 106 patients, of predominantly European origin, were included in our analysis. Our study included 55 cases with a provisional clinical diagnosis of a known condition and 51 cases without a clinical diagnosis of a specific condition at the time of testing. The diagnostic pick-up rate of genetic testing (probable genetic diagnosis) was significantly higher (71%) in the former when compared with the latter group (25%) (p < 0.001).
We identified biallelic or monoallelic variants in 67% and 15% of clinically diagnosed Usher syndrome patients, respectively. This is lower compared with the 93 and 6% pick-up rate quoted in a large cohort of 427 patients recruited in various European medical centres [30]. For BBS, we identified biallelic variants in eight of the ten cases referred with a provisional clinical diagnosis of BBS; this is similar to the frequently quoted figure of 80% yield of genetic testing in individuals with clinically diagnosed BBS [5, 31]. Given that the genetic architecture of these syndromes has, to a great extent, been deciphered we believe that any differences in diagnostic pick-up rate are most likely due to patient ascertainment factors, genetic test factors (quality and comprehensiveness of the test) or population structure factors.
We use Usher syndrome as an example: when we focused our analysis on individuals with Usher syndrome and pre-lingual SNHI (i.e. excluding mild/equivocal cases), we found that all 18 study subjects received a possible or probable genetic diagnosis; in contrast, the diagnostic yield in patients with presumed Usher syndrome and post-lingual SNHI was four out of nine patients (Fig. 2). It could also be noteworthy that regardless of Usher classification, family history of disease appears to have no bearing on the likelihood of a probable genetic diagnosis from panel-based testing (Supplementary Fig. S1). However, due to the relatively low numbers within each classification in our current cohort, it is not possible to draw a definitive conclusion at this time. Although there is an apparent difference in diagnostic yield between this study and the report by Bonnet et al., the latter employed a different testing strategy that included looking for possible large genomic rearrangements by genome-wide single-nucleotide variant array analysis, an approach that increased the final yield in that study by 9% [30]. In addition, their patient cohort consisted of only 2% with post-lingual SNHI compared with our 27%. Finally, a large study of 119 Usher syndrome patients from China reported an overall variant detection rate of 78%, a value that is lower than that in European cohorts [32, 33]. These points highlight the impact of having a high pre-genetic testing probability and of utilising comprehensive test strategies and analytical approaches that are appropriate for a given population.
Our cohort of patients with ciliopathies illustrates the genetic and phenotypic overlap observed previously in this group of diseases. We had two unrelated patients with different phenotypes but who were found to have the same homozygous nonsense variant NM_025114.3:c.5668G>T p.(Gly1890*) in CEP290 which had previously been described in Joubert syndrome patients. [25, 26, 34] (Fig. 4). It is well known that disease-associated CEP290 variants cause a spectrum of phenotypes that vary in organ involvement and severity. Although it remains unclear why the same homozygous nonsense variant results in variable phenotypes, it has recently been hypothesised that the presence of genetic modifiers, within CEP290 or its genetic interactors, could be one possible explanation [35].

Case study describing the pedigree and clinical features of two unrelated patients with different phenotypes but were found to have the same disease-associated homozygous variant in CEP290 gene causative of their conditions.
An intriguing finding in our study was that 6 of the 51 individuals who had extra-ocular features that did not fit with known clinical conditions were found to have variants in genes that had been linked to ocular-only disease (Table 1). All these six patients had a single extra-ocular manifestation each, including four patients with DD/ID, one with ataxia, one with skeletal dysplasia and one with significant obesity in childhood. There are two plausible explanations for this finding. One is that this represents expansion of the phenotype linked to specific non-syndromic IRD genes; notably, 23 of the 78 genes in our 176 gene panel that are linked to syndromic IRD have been also described in association with isolated retinopathy. Another explanation is that two distinct genetic conditions are present in a single individual. Notably, a previous retrospective analysis of 2076 patients with various disorders who received a molecular diagnosis revealed that 4.9% of them had had diagnoses that involved more than one disease locus [36] referred to more commonly now as a ‘blended’ phenotype.
Our findings suggest that panel-based testing such as was employed here is a highly successful molecular approach for providing a genetic diagnosis in patients with IRD and recognised features of a specific multisystemic condition such as BBS and Usher syndrome. One might expect this, as if a clinical diagnosis is already suspected, referring clinician can check that the relevant gene(s) are on the testing panel. In contrast, it can be argued that genome-wide approaches (exome or genome sequencing) are more appropriate initial molecular tests in complex cases where patients have multisystemic features but lack a specific clinical diagnosis. For example, Lionel et. al, have found that WGS had a better diagnostic yield in comparison with panel tests and WES, identifying the variants found by them and variants beyond their scope and increasing the yield from 24 to 41% [37]. In addition, over time, this will save time and cost, by avoiding the need to repeat genetic tests as new genes are discovered or new symptoms emerge. As we have found in this cohort, 15% (n = 10) of the 65 patients who were tested on 176 gene panel would not have been diagnosed using the 105 gene panel. However, this direct comparison is unfair as the types of patients referred to each panel differed according to the genes and their associated diseases. In practice, a hybrid approach, where a whole exome or genome is sequenced but virtual panels of genes are analysed, is likely to be a cost-effective strategy that offers a compromise between maximising the yield of genetic testing and addressing complicated issues surrounding secondary findings. This has the added advantage that one can go back to the initial data and analyse a further panel of genes if clinical signs and symptoms change over time and a clinical diagnosis becomes apparent.
References
Wright AF, Chakarova CF, El-Aziz MMA, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11:273–84.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809.
Sadagopan KA. Practical approach to syndromic pediatric retinal dystrophies. Curr Opin Ophthalmol. 2017;28:416–29.
Werdich XQ, Place EM, Pierce EA. Systemic diseases associated with retinal dystrophies. Semin Ophthalmol. 2014;29:319–28.
Forsythe E, Kenny J, Bacchelli C, Beales PL. Managing Bardet-Biedl Syndrome—now and in the future. Front Pediatrics. 2018;6:23.
Weihbrecht K, Goar WA, Pak T, Garrison JE, DeLuca AP, Stone EM, et al. Keeping an eye on Bardet-Biedl Syndrome: a comprehensive review of the role of Bardet-Biedl Syndrome genes in the eye. Med Res Arch. 2017;5:10.
Glockle N, Kohl S, Mohr J, Scheurenbrand T, Sprecher A, Weisschuh N, et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet. 2014;22:99–104.
Bravo-Gil N, Mendez-Vidal C, Romero-Perez L, Gonzalez-del Pozo M, Rodriguez-de la Rua E, Dopazo J. et al. Improving the management of inherited retinal dystrophies by targeted sequencing of a population-specific gene panel. Sci Rep.2016;6:10
Weisschuh N, Mayer AK, Strom TM, Kohl S, Glockle N, Schubach M, et al. Mutation detection in patients with retinal dystrophies using targeted next generation sequencing. PLoS ONE 2016;11:15.
Sanchez-Navarro I, da Silva LRJ, Blanco-Kelly F, Zurita O, Sanchez-Bolivar N, Villaverde C, et al. Combining targeted panel-based resequencing and copy-number variation analysis for the diagnosis of inherited syndromic retinopathies and associated ciliopathies. Sci Rep. 2018;8:11.
Aparisi MJ, Aller E, Fuster-Garcia C, Garcia-Garcia G, Rodrigo R, Vazquez-Manrique RP, et al. Targeted next generation sequencing for molecular diagnosis of Usher syndrome. Orphanet J Rare Dis. 2014;9:168.
Eandi CM, Dallorto L, Spinetta R, Micieli MP, Vanzetti M, Mariottini A, et al. Targeted next generation sequencing in Italian patients with Usher syndrome: phenotype-genotype correlations. Sci Rep. 2017;7:8.
Köhler S, Carmody L, Vasilevsky N, Jacobsen JOB, Danis D, Gourdine J-P, et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic acids Res. 2018;47:D1018–27.
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: results of a population survey. Journal of Medical Genetics. 1999;36:437–46.
Ellingford JM, Barton S, Bhaskar S, O’Sullivan J, Williams SG, Lamb JA, et al. Molecular findings from 537 individuals with inherited retinal disease. J Med Genet. 2016;53:761–7.
Taylor RL, Parry NRA, Barton SJ, Campbell C, Delaney CM, Ellingford JM, et al. Panel-based clinical genetic testing in 85 children with inherited retinal disease. Ophthalmology. 2017;124:985–91.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26:589–95.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy‐Moonshine A, et al. From FastQ data to high‐confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinforma. 2013;43:11.0.1–0.33.
Ellingford JM, Horn B, Campbell C, Arno G, Barton S, Tate C, et al. Assessment of the incorporation of CNV surveillance into gene panel next-generation sequencing testing for inherited retinal diseases. J Med Genet. 2018;55:114–21.
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–54.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8.
Ellingford JM, Campbell C, Barton S, Bhaskar S, Gupta S, Taylor RL, et al. Validation of copy number variation analysis for next-generation sequencing diagnostics. Eur J Hum Genet. 2017;25:719.
Ostergaard E, Duno M, Batbayli M, Vilhelmsen K, Rosenberg T. A novel MERTK deletion is a common founder mutation in the Faroe Islands and is responsible for a high proportion of retinitis pigmentosa cases. Mol Vision. 2011;17:1485–92.
Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, et al. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–5.
Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–81.
Chandler KE, Kidd A, Al-Gazali L, Kolehmainen J, Lehesjoki AE, Black GCM, et al. Diagnostic criteria, clinical characteristics, and natural history of Cohen syndrome. J Med Genet. 2003;40:233–41.
Kondo H, Hayashi H, Oshima K, Tahira T, Hayashi K. Frizzled 4 gene (FZD4) mutations in patients with familial exudative vitreoretinopathy with variable expressivity. Br J Ophthalmol. 2003;87:1291–5.
Milhem RM, Ben-Salem S, Al-Gazali L, Ali BR. Identification of the cellular mechanisms that modulate trafficking of Frizzled Family Receptor 4 (FZD4) missense mutants associated with familial exudative vitreoretinopathy. Investigative Ophthalmol Vis Sci. 2014;55:3423–31.
Bonnet C, Riahi Z, Chantot-Bastaraud S, Smagghe L, Letexier M, Marcaillou C, et al. An innovative strategy for the molecular diagnosis of Usher syndrome identifies causal biallelic mutations in 93% of European patients. Eur J Hum Genet. 2016;24:1730–8.
Forsythe E, Beales PL. Bardet-Biedl Syndrome. Eur J Hum Genet. 2013;21:8–13.
Sun T, Xu K, Ren Y, Xie Y, Zhang X, Tian L, et al. Comprehensive molecular screening in chinese Usher Syndrome patients. Investig Ophthalmol Vis Sci. 2018;59:1229–37.
Jouret G, Poirsier C, Spodenkiewicz M, Jaquin C, Gouy E, Arndt C, et al. Genetics of Usher Syndrome: new insights from a meta-analysis. Otol Neurotol. 2019;40:121–9.
Brancati F, Barrano G, Silhavy JL, Marsh SE, Travaglini L, Bielas SL, et al. CEP290 mutations are frequently identified in the oculo-renal form of Joubert syndrome-related disorders. Am J Hum Genet. 2007;81:104–13.
Lessieur EM, Song P, Nivar GC, Piccillo EM, Fogerty J, Rozic R, et al. Ciliary genes arl13b, ahi1 and cc2d2a differentially modify expression of visual acuity phenotypes but do not enhance retinal degeneration due to mutation of cep290 in zebrafish. PLoS ONE 2019;14:e0213960.
Posey JE, Harel T, Liu PF, Rosenfeld JA, James RA, Akdemir ZHC, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl J Med. 2017;376:21–31.
Lionel AC, Costain G, Monfared N, Walker S, Reuter MS, Hosseini SM, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2018;20:435–43.
Houten SM, Kuis W, Duran M, de Koning TJ, van Royen-Kerkhof A, Romeijn GJ, et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet. 1999;22:175–7.
Abulí A, Boada M, Rodríguez-Santiago B, Coroleu B, Veiga A, Armengol L, et al. NGS-Based Assay for the Identification of Individuals Carrying Recessive Genetic Mutations in Reproductive Medicine. Human Mutation. 2016;37:516–23.
M’hamdi O, Redin C, Stoetzel C, Ouertani I, Chaabouni M, Maazoul F, et al. Clinical and genetic characterization of Bardet-Biedl syndrome in Tunisia: defining a strategy for molecular diagnosis. Clinical Genetics. 2014;85:172–7.
Mykytyn K, Nishimura DY, Searby CC, Shastri M, Yen H-j, Beck JS, et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet. 2002;31:435–8.
Ashkinadze E, Rosen T, Brooks SS, Katsanis N, Davis EE. Combining fetal sonography with genetic and allele pathogenicity studies to secure a neonatal diagnosis of Bardet-Biedl syndrome. Clinical Genetics. 2013;83:553–9.
Hull S, Owen N, Islam F, Tracey-White D, Plagnol V, Holder GE, et al. Nonsyndromic Retinal Dystrophy due to Bi-Allelic Mutations in the Ciliary Transport Gene IFT140. Investigative Ophthalmology & Visual Science. 2016;57:1053–62.
Carss KJ, Arno G, Erwood M, Stephens J, Sanchis-Juan A, Hull S, et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. The American Journal of Human Genetics. 2017;100:75–90.
Zeitz C, Robson AG, Audo I. Congenital stationary night blindness: An analysis and update of genotype–phenotype correlations and pathogenic mechanisms. Progress in Retinal and Eye Research. 2015;45:58–110.
Pras E, Abu A, Rotenstreich Y, Avni I, Reish O, Morad Y, et al. Cone-rod dystrophy and a frameshift mutation in the PROM1 gene. Molecular vision. 2009;15:1709.
Habibi I, Chebil A, Falfoul Y, Allaman-Pillet N, Kort F, Schorderet DF, et al. (2016) Identifying mutations in Tunisian families with retinal dystrophy. Scientific Reports 6.
Arai Y, Maeda A, Hirami Y, Ishigami C, Kosugi S, Mandai M, et al. Retinitis Pigmentosa with Mutations Is the Most Prevalent Inherited Retinal Dystrophy in Japanese Populations. Journal of Ophthalmology. 2015;2015:1–10.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genetics in Medicine. 2015;17:253–61.
Eisenberger T, Neuhaus C, Khan AO, Decker C, Preising MN, Friedburg C, et al. Increasing the Yield in Targeted Next-Generation Sequencing by Implicating CNV Analysis, Non-Coding Exons and the Overall Variant Load: The Example of Retinal Dystrophies. PLoS ONE 2013;8:e78496.
Acknowledgements
The authors would like to acknowledge the support of the Manchester Academic Health Science Centre and the Manchester National Institute for Health Research Biomedical Research Centre.
The UK Inherited Retinal Dystrophy Consortium
Graeme Black, Georgina Hall, Stuart Ingram, Rachel Taylor, Forbes Manson, Panagiotis Sergouniotis, Andrew Webster, Alison Hardcastle, Michel Michaelides, Nikolas Pontikos, Michael Cheetham, Gavin Arno, Alessia Fiorentino, Chris Inglehearn, Carmel Toomes, Manir Ali, Martin McKibbin, Kamron Khan, Susan Downes, Jing Yu, Stephanie Halford, Suzanne Broadgate, and Veronica van Heyningen.
Funding
RLT is supported by an RCUK/UKRI Innovation Fellowship (MR/R024952/1) provided by the Medical Research Council (MRC). OAJ is funded by King Abdulaziz University. The views expressed are those of the authors, and not necessarily those of the NHS, the NIHR or the Department of Health. Funding bodies did not have any specific role in the design and conduct of the study.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jiman, O.A., Taylor, R.L., Lenassi, E. et al. Diagnostic yield of panel-based genetic testing in syndromic inherited retinal disease. Eur J Hum Genet 28, 576–586 (2020). https://doi.org/10.1038/s41431-019-0548-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0548-5
This article is cited by
-
Novel biallelic USH2A variants in a patient with usher syndrome type IIA- a case report
BMC Ophthalmology (2022)
-
Biallelic variants in coenzyme Q10 biosynthesis pathway genes cause a retinitis pigmentosa phenotype
npj Genomic Medicine (2022)
-
NGS and phenotypic ontology-based approaches increase the diagnostic yield in syndromic retinal diseases
Human Genetics (2021)
